
Abstract
Hepatocellular carcinoma (HCC) developed in non-alcoholic fatty liver disease (NAFLD) individuals presents substantial clinical and biological characteristics, which remain to be elucidated. Its occurrence in noncirrhotic patients raises issues regarding surveillance strategies, which cannot be considered as cost-effective given the high prevalence of obesity and metabolic syndrome, and furthermore delineates specific oncogenic process that could be targeted in the setting of primary or secondary prevention. In this context, the identification of a genetic heterogeneity modulating HCC risk as well as specific biological pathways have been made possible through genome-wide association studies, development of animal models and in-depth analyses of human samples at the pathological and genomic levels. These advances must be confirmed and pursued to pave the way for personalized management of NAFLD-related HCC.
Why we must characterize liver oncogenic process in non-alcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of chronic liver disease ranging from excessive cytoplasmic retention of triglyceride (steatosis) to inflammation and hepatocyte injury (nonalcoholic steatohepatitis [NASH]), then ultimately to hepatic fibrosis/cirrhosis. Liver-related mortality includes end-stage liver disease and hepatocellular carcinoma (HCC) [Citation1]. Its main driver is obesity, explaining why NAFLD has become one of the most frequent diseases in the world, affecting up to 40% of Western populations. Other contributors include features of the metabolic syndrome associated with impaired glucose regulation and insulin resistance, dyslipidemia and hypertension. Modeling approaches suggest that the prevalence of NAFLD and its potential deadly liver-related complications will continue to rise worldwide in the next decade [Citation2]. Nevertheless, despite this high prevalence, only a small subset of NAFLD patients (estimated as <15%) will progress toward severe forms liver disease and ultimately develop hepatic complication and/or HCC [Citation3]. Furthermore, HCC can develop in NAFLD patients without cirrhosis [Citation4–6]. It has been indeed estimated that nearly half of patients with NAFLD-related HCC may not have significant liver fibrosis [Citation7]. This clinical observation is a challenge for HCC surveillance. Indeed, all international liver societies recommend that patients with extensive fibrosis or cirrhosis should be included in HCC surveillance programs by means of semi-annual US examination [Citation8,Citation9], justified by an annual cancer incidence usually above 2%. As a consequence, HCC screening is not cost effective in NAFLD populations without cirrhosis given the frequency of this condition and expected low annual HCC incidence, which has been estimated as 0.8 per 1000 person-years in European populations aged 65–75 years with simple steatosis [Citation10]. In addition to this incidence insufficient to recommend screening [Citation11], the sensitivity of ultrasonography is notoriously challenging in obese individuals who often present with heterogeneous fatty livers [Citation12]. In this setting, more sophisticated imaging techniques such as magnetic resonance imaging (MRI) might be justified [Citation13,Citation14], but further increase cost–effectiveness imbalance.
For all these reasons, identifying NAFLD individuals who are more prone to develop HCC as well as the implicated underlying oncogenic process is pivotal. From a biological perspective, liver carcinogenic process in the setting of NAFLD has been up to now described regardless of fibrosis status: the extent to which different mechanisms may coexist as a function of the presence or not of cirrhosis is currently unknown. From a clinical standpoint, progress in knowledge might allow the implementation of dedicated behavioral or pharmacological interventions aimed at preventing and decreasing liver-related mortality as well as triggering personalized management of this growing population. The present review will focus on accumulated data enlightening the comprehensive mechanisms linking NAFLD to liver cancer development.
Characterizing the host: genetic susceptibility to HCC in NAFLD populations
Only a small fraction of NAFLD patients develop chronic inflammation, then fibrosis and ultimately HCC [Citation15]. Accumulating data suggest that genetics account for approximately half of the interindividual variability in all spectrums of NAFLD [Citation16]. Although genome-wide association studies (GWAS) have not yet been specifically dedicated to NAFLD-related HCC, unsuspected association between common genetic variants and the development or progression of fatty liver disease have been unraveled by such unsupervised approaches [Citation17]. Up to now, four genetic variations have been highlighted and include the following: patatin-like phospholipase domain-containing protein 3 (PNPLA3) p.I148M [Citation18]; transmembrane 6, superfamily member 2 (TM6SF2) p. E167K [Citation19]; membrane-bound O-acyltransferase domain containing 7 (MBOAT7) rs641738 C > T variant [Citation20]; and a splice variant (rs72613567) in hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13) [Citation21]. These four genes encode proteins involved in the regulation of hepatic lipid metabolism [Citation22]. These associations have prompted various teams to study the impact of these SNPs on NAFLD progression and their ability to refine HCC risk in these populations [Citation23]. Nevertheless, whether these variations impacting lipid turnover, alone or collectively, promote HCC by fostering a favorable microenvironment, or by specifically triggering specific oncogenic process, are currently unknown [Citation24]. In addition, yet unknown variations in other genes impacting different biological pathways might also be able to refine HCC risk stratification.
Evidence from association studies linking genetic variants & NAFLD-related HCC
The first genetic trait associated with NAFLD was identified in 2008 and corresponds to a common nonsynonymous variant rs738409 C > G (p. Ile148Met) located on chromosome 22 in the PNPLA3 gene also known as adiponutrin [Citation18]. The function of this protein is to hydrolyze triglycerides and retinyl esters, while the rs738409 C > G variant results in a loss of function. The latter favors the accumulation of triglycerides and retinyl esters in lipid droplets within both hepatocytes and hepatic stellate cells (HSCs), thus participating in fatty liver disease [Citation25,Citation26]. This association was replicated by others [Citation27–29], then was extended to more severe forms of fatty liver disease throughout the whole spectrum including NASH after adjustment of age, sex, BMI and insulin resistance [Citation30–32]. More recent GWAS conducted in larger sample sizes and/or using more recent chip arrays have also unraveled other loci associated with steatosis in NAFLD [Citation19,Citation31]. Among them was identified a variant in the TM6SF2 gene (rs58542926 C > T) resulting in a loss-of-function, which induces higher liver triglyceride content and lower circulating lipoproteins [Citation33]. Similarly, the rs641738 T allele in the MBOAT7 gene reduces its expression and has been shown to impact phosphatidyl-inositol plasma and hepatic composition, thus favoring hepatocellular fat accumulation and the production of inflammatory mediators [Citation20]. Finally, the rs72613567variant of HSD17B13 was recently identified as a protective factor of NAFLD progression leading to production of a truncated protein with a reduced enzymatic activity [Citation21]. Like PNPLA3, HSD17B13 also locates to lipid droplets, its expression being increased in NAFLD patients as compared with normal liver samples [Citation34]. All these polymorphisms were shown to influence all stages of NAFLD in association with PNPLA3 rs738409 C > G, suggesting an additive effect of these genetic variations on fatty liver disease progression [Citation35]. In this context, it was tempting to speculate that the same genetic traits might also promote liver NAFLD-related carcinogenesis.
Following the discovery of these variants, case–control studies were subsequently conducted in NAFLD patients, complicated or not by HCC. For instance, a two-stage case–control study performed in NAFLD patients [Citation36] compared genotype distributions between HCC patients and individuals from the general population and reported an odds ratio (OR) of 12.19. However, when controlling for the presence of cirrhosis, PNPLA3 rs738409[G] conferred a much smaller increase in HCC risk (OR = 2.26).
Another study performed in two European cohorts (Italy and the UK) studied the association between rs641738 C > T MBOAT7 variant and NAFLD-related HCC [Citation37]. The authors reported that each MBOAT7 rs641738 T allele conferred an approximately 80% increased risk of HCC in the Italian cohort, particularly in patients without advanced liver fibrosis, a finding which was replicated In the UK cohort. This finding suggests a stronger impact of the MBOAT7 on early steps of hepatocarcinogenesis, independently from fibrosis accumulation in the liver. As MBOAT7 is highly expressed in HSCs and inflammatory cells [Citation20], the deleterious effect of MBOAT7 variant could be in this setting decreased in more advanced stages of the fibrogenic process.
No direct association between between TM6SF2 and HSD17B1variants and NAFLD-HCC were clearly reported. However, both genetic traits were found to be linked with the presence of liver cancer in patients with alcoholic liver disease (ALD), alone or combined with PNPLA3 rs738409 [Citation38,Citation39]. Given the shared similarities between pathophysiological aspects of ALD and NAFLD progression characterized by lipid-accumulating liver disorder [Citation40] (as well as their frequent association into a ‘nonviral’ cause of chronic liver disease), the extent to which the promotion of hepatic carcinogenesis can be considered a direct consequence of fat accumulation and/or cancer-related molecular signaling in both diseases remains to be further characterized [Citation41].
Contribution of genetic traits to risk stratification & prediction of liver cancer in NAFLD: input of longitudinal cohorts & population studies
All aforementioned variants associated with NAFLD-related HCC can be incorporated, alone or combined, with other established risk factors for HCC (e.g., male gender, older age, obesity) into scoring systems aimed at to stratifying patients into different HCC risk classes. This has been particularly well-described in longitudinal cohorts of patients with extensive fibrosis or cirrhosis included in HCC surveillance programs [Citation42]. As previously suggested, in patients with nonviral cirrhosis, combining several of genetic risk factors with other phenotypic/clinical traits allowed a fair identification and stratification of patients according to their probability of HCC development [Citation43–45]. For instance, the rs378409 risk variant in PNPLA3 in combination with other clinical factors (BMI, age, gender) enabled HCC risk stratification into low-, intermediate- and high-risk groups as a function of HCC development [Citation46]. Using the same methodology, adding TM6SF2 genotypes further increased HCC prediction as the number of cases increased with carriage of both PNPLA3-G and TM6SF2-T risk alleles compared with carriage of only one risk allele in either polymorphism [Citation47]. Similar longitudinal observations were made when restricting studies to patients with NAFLD at various stages of liver disease. In this context, the impact of rs738409[G] on liver cancer occurrence in a prospective cohort of more than 3000 obese individuals highlights this important point [Citation48]. Similarly, the long follow-up of 471 consecutive Italian patients with a diagnosis of NAFLD based on histologic factors or a diagnosis of compensated NAFLD-related cirrhosis confirmed that PNPLA3 genotypes together with classic markers of liver function and portal hypertension were able to stratify the risk for HCC in such populations [Citation49].
Recently, the clinical significance of such genetic risk scores (GRS) for the prediction of cirrhosis and HCC development in NAFLD has been elegantly shown in large populations of 110,761 individuals from the Danish general population, 334,691 individuals from the UK Biobank in whom PNPLA3, TM6SF2 and HSD17B13 variants were assessed [Citation50]. Combining genotypes led to the creation of a scoring system ranging from 0 to 6 risk-increasing alleles. This GRS was found to be associated with up to 12-fold higher risk of cirrhosis and up to 29-fold higher risk of HCC. Interestingly, 94% of individuals had a score of 3 or lower not requiring further explorations, a finding highlighting the difficulty to select among large populations the small subset of individuals who should undergo dedicated work-up and periodical surveillance. Such approach combining both clinical and genetic data might help to overcome this issue and accurately allocate precious medical resources.
Overall, GRSs in combination with environmental risk factors have the potential to improve the prediction of NASH-related HCC and identification of high-risk individuals. Validation in large-scale prospective cohorts assessing their clinical utility and cost–effectiveness are now warranted to enter clinical practice [Citation51]. Deciding which NAFLD patients without cirrhosis should be included in HCC surveillance programs and refining screening periodicity and/or modality in those bearing the highest risk may then become feasible in the setting of precision medicine. Such implementation will facilitate a better translation of biological information in clinical practice to foster disease prevention and intervention dedicated to NAFLD-related HCC management.
Pathophysiology of NAFLD & liver carcinogenesis
Steatosis, one of the hallmarks of NALFD, is the consequence of alterations in lipid metabolism combining the accumulation of free fatty acids (FFA) derived from adipose tissue in hepatocytes, an increase in hepatic lipogenesis associated with a decrease in β-oxidation of fatty acids and a decrease in the output of hepatic triglycerides [Citation52]. The mechanisms underlying the transition from steatosis to NASH are complex and still poorly understood but depend on multiple parallel/sequential hits in which the immune system plays a major role. Prior to this robust immune response, hepatic lipid accumulation will lead to an induction of lipid-storing enzymes such as PPAR-g, which follow a strongly diurnal pattern. This lipid accumulation contributes to accumulation of lipotoxic metabolites, oxidative stress in addition to alteration of mitochondria, lysosomes and endoplasmic reticulum stress. It is finally responsible of hepatocellular injury and cell death through c-Jun N-terminal kinase (JNK) activation [Citation53]. Lipoapoptosis leads to the activation of innate system and the recruitment of adaptive immune cells. Sustained inflammation is one of the major features of NASH and is also triggered by the translocation of microbial compounds such as lipopolysaccharides (LPS) from intestine within the liver, which activate Toll-like receptors (TLRs) and induce a proinflammatory response. Circulating endotoxins have been found to be elevated in NASH patients and related to the gut leak phenomenon and to dysbiosis [Citation54]. The adipose tissue also plays a critical role in NAFLD progression through the release of adipokines, including adiponectin and leptin, and cytokines such as TNF and IL-6 leading to insulin resistance and a state of low-grade chronic systemic inflammation. Leptin is mainly found in the adipose tissue and contributes to energy homeostasis and neuroendocrine function such as appetite. An increased level of leptin inhibits both hepatic glucose production and de novo lipogenesis via FFA oxidation. However, chronic elevation of leptin seems to promote inflammation and fibrogenesis in NASH [Citation55]. Uncontrolled cytokine production leads to the activation and proliferation of myofibroblasts, including HSCs. Myofibroblasts acquire a fibrogenic myofibroblastic phenotype and produce inflammatory mediators, matrix molecules such as fibrillar collagens, which constitute the components of the fibrous septa and also contribute to deregulation of matrix remodeling mechanisms [Citation56]. HSC can also be directly activated by IGF-1 produced in excess due to insulin resistance [Citation57] and by LPS induced by dysbiosis through TLR4 [Citation58]. Gut microbiota also has a critical role in bile acid metabolism and indirectly modulate farnesoid X factor (FXR) function [Citation59]. FXR activation inhibits FFAs uptake and synthesis and also stimulates β-oxidation [Citation60]. In this line, obeticholic acid, an FXR agonist, has shown to improve histology in NASH patients [Citation61]. Ablation of FXR has been associated with higher enterohepatic bile acid levels and jet lag-induced HCC. Chronic jet lag has been associated with spontaneous HCC in wild-type mice due to altered circadian regulation of bile acid receptor and metabolic pathways that control the synthesis and metabolism of glucose, lipid and cholesterol [Citation62]. Moreover, sustained inflammation in association with accumulation of reactive oxygen species as well as JNK1 activation through FFA production and upregulation of the IGF pathway secondary to hyperinsulinemia promote cellular proliferation and inhibition of apoptosis promoting HCC occurrence [Citation63–66].
Animal models for the study of NAFLD-related HCC
Due to the limited access to liver tissue, human data on disease progression are sparse and usually limited to a single time point. Overall, most of the available data about pathophysiology of NAFLD are based on preclinical models (). An ideal preclinical model should be simple, triggered by the same causes as human disease (caloric excess), associated with the same risk factors (obesity, insulin resistance and dyslipidemia) and should reproduce all the major features of severe humans NAFLD, including similar patterns of genetic, epigenetic and histological changes. To date, various mice models are available such as dietary, genetic and toxic models, but mice rarely exhibit the entire pathological spectrum from steatosis to NASH with development of fibrosis and HCC.
Table 1. Animals models in NAFLD and hepatocellular carcinoma development.
Dietary models
The primary driver of NAFLD is Western diet (WD) and a sedentary lifestyle leading to increased weight and, ultimately, obesity. To mimic the natural history of NASH and have preclinical model for drug development, various diet-induced obesity models have been developed. In the last decades, methionine choline deficient (MCD) or choline alone (CD) diet has been commonly used for its simplicity and short duration with NASH and fibrosis observed at 3 and 5 weeks, respectively [Citation67]. However, MCD diet is associated with a loss of weight and no residual adipose tissue (by 8-week mice have lost 40% of their weight), with persistence of insulin sensitivity and no HCC occurrence [Citation68]. Consequently, MCD and CD diet are far from optimal to examine metabolic parameters of NAFLD [Citation69]. The semisynthetic choline-deficient L-amino acid-defined (CDAA) diet shares deficiency in choline. However, CDAA diet proteins are substituted with an equivalent and corresponding mixture of L-amino acids and mice do not experience the weight loss observed with the MCD diet. After 22 weeks, mice show an increase in body weight, dyslipidemia and insulin resistance [Citation70]. High-fat diet (HFD) composed more than 60% of saturated fat causes obesity, insulin resistance, steatosis but no fibrosis and HCC. Adding sucrose and cholesterol (usually 0.2%) to HFD, so-called WD increases liver injury, can cause fibrosis in some mice strains but do not cause HCC. Recently, elevated fructose consumption has been identified as an important contributor to NAFLD progression and mice maintained on a diet high in trans fats and high-fructose corn syrup along with sedentary lifestyle developed bridging fibrosis and six out of ten of them showed hepatocellular neoplasm after 1 year [Citation71]. The combination of HFD with CDAA (CDAA-HFD) results in obesity, NASH, fibrosis and progression to HCC in 25% of mice after 30 weeks [Citation72]. However, sensitivity to dietary models varies according to mouse strain and gender. C57BL/6 is significantly more prone to develop diet-induced hepatic inflammation and fibrosis compared with BALB/c mice [Citation73]. C57BL/6J mice are more insulin resistant compared with FVB/N strains [Citation74]. C57BL/6J male mice display steatohepatitis, whereas female only present steatosis suggesting a potential protecting role of estrogen [Citation75].
Association of toxic & diet models
Streptozotocin (STZ) is toxic to insulin producing pancreatic β-cells and induced Type 1 diabetes. The STAM model consists the administration of STZ immediately after birth following by HFD feeding at 4 weeks of age. This model leads to the occurrence of fibrosis after 9 weeks and HCC at 16 weeks [Citation76]. Even if this model is quick, contrary to human NASH, mice are lean and develop Type 1 diabetes. Moreover, we cannot rule out an independent carcinogenic effect of STZ as STZ is a DNA-damaging alkylating agent [Citation77].
Administration of diethylnitrosamine (DEN) in association with HFD or with HFD + CD lead to HCC occurrence within 20 weeks without underlying NASH features. However, these models provided the first demonstration that obesity and simple steatosis increase HCC by enhancing local production of TNF and IL-6 [Citation78]. Another possibility is the combination of weekly dose of intraperitoneal carbon tetrachloride (CCl4) with a WD, which leads to NASH features and HCC development within 24 weeks [Citation79].
Genetic models
Single mutations of appetite-regulating genes such as leptin-deficient (ob/ob mice) [Citation80], deficient leptin signaling (db/db mice) [Citation81], spontaneous mutations in the Alstrom syndrome 1 gene encoding for a protein localized to centrosomes and appetite-sensing neuronal cilia (foz/foz mice) [Citation73], PPARα-/- knockout mice [Citation82] and melanocortin receptor 4 knockout (Mc4r-/- mice) [Citation83] lead to overeating, obesity and insulin resistance. In these models, balanced diet leads to steatosis without inflammation nor NASH features nor HCC. Several specific murine knockout models exist to more specifically study the development of HCC. These models present HCC under normal diet but present limitations such as no obesity for phosphatase and tensin homolog (PTEN) [Citation84], acyl-coenzyme A oxidase (AOX) [Citation85], and methionine adenosyltransferase 1A (MAT1A) [Citation86] in global-deficient mice and in hepatic-deficient hepatocyte nuclear factor 4α (Hnf4α) [Citation87].
To mimic humans HCC occurring on NAFLD associated with metabolic features and NASH development, combination of diet and genetic mice seems more accurate. Feeding ob/ob and db/db mice with HFD or WD leads to more pronounced steatosis and inflammation but data about fibrosis differ according to the studies and no HCC are observed [Citation17]. Feeding Mc4r-/- mice and liver-specific Hnf4α-deficient mice with HFD is associated with HCC within 1 year [Citation83] and 36 weeks, respectively [Citation88]. HCC was also observed in foz/foz mice feed with a WD during more than 56 [Citation89] and 24 weeks after DEN injection [Citation90].
Recently, the DIAMOND mice for Diet-Induced Animal Model of NAFLD based on an isogenic strain of C57Bl6/J and S129S1/svlm mice fed an HFD with ad libitum glucose-fructose in drinking water were proposed. In this model, 90% of the mice developed HCC after 52 weeks [Citation91]. This HCC incidence was similar to another model based on feeding HFD to MUP-uPA transgenic mice, which express high amounts of urokinase plasminogen activator (uPA) specifically in hepatocyte during the first 6 weeks of life [Citation92].
Finally, few preclinical models are available to study HCC development in the context of NAFLD and, most of the time, required combined strategies (diet/toxic, genetic/diet, genetic/toxic). Further models are needed to replicate HCC occurrence in human NAFLD, in a reasonable time without using chemical carcinogens and taking into account the tumoral immune response in the era of immunotherapy.
Circadian dysfunction model
Chronic circadian disruption has been associated with induced leptin resistance in mice independent of diet choice. This was associated with NASH features including steatosis, fibrosis, inflammation and insulin resistance. Jet-lagged mice showed significantly reduced lifespan mostly due to cancer development such as HCC. This was associated with a global shift in liver metabolism to promote lipid synthesis and storage via accelerating cytoplasmic glycolysis as well as higher intracellular oxidative stress [Citation62].
Characteristics of background liver in HCC related to NAFLD
HCC mostly arise from the malignant transformation of preneoplastic/dysplastic nodules, consistent with a stepwise process of carcinogenesis. There is a clear association between severity of NAFLD and HCC development as reported in a Korean historical cohort study, enrolling more than 25,000 subjects who were evaliuated by noninvasive tests [Citation93]. Importantly, histological analysis of background cirrhotic liver in this population may not show the typical features of NAFLD, as steatosis and steatohepatitis may disappear with disease progression and fibrosis worsening, supporting the namely ‘burn-out cirrhosis.’ Accordingly, the contribution of NAFLD in HCC development, based on pathological examination, could be underestimated.
As previously stated, liver carcinogenesis related to NAFLD is somehow peculiar, as HCC can occur in absence of underlying cirrhosis (ranging from 23 to 65% according to the series) by contrast to other risk factors of chronic liver disease, in which HCC arise in more than 80% of cases in liver with cirrhosis. The histological analysis of nontumoral liver in HCC patients without cirrhosis usually shows steatosis (mostly mild to moderate) and some degree of steatohepatitis (grading 1 and 2 in >50% of cases). In terms of fibrosis, while some patients have no fibrosis at all, portal fibrosis with few septa is the rule [Citation94].
Although the molecular mechanisms involved in liver carcinogenesis related to NAFLD are not fully understood, some have been already shown to be involved in addition to genomic aberrations that accumulate along the course of the liver chronic disease. Among them, low-grade chronic inflammation state, associated with metabolic syndrome and especially obesity, plays a key role in the development of insulin resistance through the release of proinflammatory cytokines, mostly TNF-α and IL-6 via activation of IKK and JNK signaling pathways [Citation95]. Such inflammatory environment seems to occur early in the development of the disease, even before lipid accumulation. Accordingly, in mice models with nutrient overload, blocking IL-17A signaling reduces steatosis and liver injury, and prevents HCC [Citation96]. Chronic inflammation as well as hepatic metabolic reprogramming induces endoplasmic reticulum stress and oxidative stress in hepatocytes that result in DNA damages. In parallel, oxidative stress has been shown to promote pathological polyploidization of hepatocytes with increased number of polyploid cells, in experimental mice models of NAFLD and patients as well [Citation97].
Obesity is characterized by expansion of dysfunctional adipose tissue leading to a dysregulation of adipokines with increase and decrease in leptin and adiponectin, respectively. In addition to its role in development of insulin resistance, regulation of immune response, leptin exerts oncogenic effects by supporting cell growth and cell proliferation through the activation of molecular pathways, including JAK2/STAT3 and PI3K/Akt [Citation98]. Moreover, the low levels of adiponectin hamper its common anti-inflammatory properties and antagonizing effect on leptin. Contribution of additional adipokines has also been demonstrated. Among them, fatty-acid binding protein 4 (FABP4), an adipokine acting as a lipid chaperone protein mostly produced by adipocytes and macrophages, is known to play a role in insulin resistance [Citation99]. Circulating levels of FABP4 are increased in mice models of NAFLD, and also in patients with NAFLD, with a correlation with liver inflammation and fibrosis [Citation100]. More recently, the oncogenic effects of FABP4 have been reported in the context of HCC related to NAFLD using in vitro and in vivo models [Citation101]. Interestingly, a de novo FABP4 expression has been illustrated in peritumoral endothelial cells in human HCC suggesting the oncogenic role of endothelial cells in tumoral growth [Citation101].
Gut microbiota is another key player in the development of NAFLD and HCC, partly mediated by its role in the control of bile acid composition [Citation102]. In addition to the role of LPS from intestinal Gram-negative bacteria [Citation103], oncogenic effect of dysbiosis has been demonstrated through the increased levels of deoxycholic acid, a gut bacterial metabolite, able to cause DNA damage [Citation104]. Interestingly, deoxycholic acid has been shown to induce cellular senescence in HSCs, the main fibrocompetent liver cells, which, then, secrete proinflammatory and tumor-promoting factors within the liver, contributing to the HCC development in mice models [Citation104].
Dysregulated autophagy has been described in various liver diseases, including in NAFLD, with beneficial or deleterious effects according to the cell type. For instance, autophagy protects against fat accumulation and prevents liver injury in hepatocytes, exerts anti-inflammatory properties in macrophages, while autophagy has profibrogenic properties in HSCs [Citation105]. More recently, a defect in endothelial autophagy has been demonstrated in NASH, promoting liver inflammation, apoptosis and liver fibrosis [Citation106]. Increasing evidence has revealed the paradoxical roles of autophagy in cancer, in tumor initiation, progression and response to therapy, including in HCC [Citation107]. At early stages, defects in hepatocellular autophagy may act as a tumor-suppression mechanism while later on, autophagy may be detrimental, supporting tumor progression and resistance to therapies [Citation108].
Molecular drivers of HCC in NAFLD
Somatic genetic alterations identified in HCC are the consequences of the mutational processes operative in malignant hepatocytes [Citation109]. A median of 40–60 somatic genetic alterations are described in the coding sequence of each HCC genome. Several key pathways involved in liver carcinogenesis are recurrently mutated in HCC including mutations in driver genes such as TERT, TP53, CTNNB1, AXIN1, ARID1A, ARID2, RPS6KA3, FGF19 and VEGFA [Citation110,Citation111]. These genetic alterations are linked with transcriptomic subgroups (such as S1/S2/S3 or G1 to G6 molecular classification) related to specific risk factors and histological features [Citation109,Citation112]. Genomic analyses have linked the exposition to viral or environmental carcinogens with the type of substitutions at the nucleotide level. These mutational processes occurred early in liver carcinogenesis. HBV-related HCC are associated with a higher rate of mutations of TP53 and MLL4 and HBV is also responsible of insertional mutagenesis in the tumor genome targeting TERT, MLL4 and CCNE1 genes [Citation113]. In contrast, alcohol-related HCC are enriched in ARID1A, CTNNB1 and TERT promoter mutations [Citation110]. Genetic imprint of alcohol and tobacco have been observed in HCC with mutational signature characterized by a T > C mutations in ATN trinucleotides explaining the enrichment of mutations in CTNNB1 in alcohol-related HCC [Citation114]. Moreover, exposition to aflatoxin B1, a mycotoxin-contaminating culture in Africa and Asia, and aristolochic acid, a plant used in traditional medicine in Eastern countries, lead to pathognomonic mutational signatures identifiable in the HCC genome: C > A mutations in a GCC context and T > A transversions in a CTG trinucleotide context, respectively [Citation115,Citation116]. Few data are currently available about the genomic profile of human NAFLD-related HCC. Obesity is a well-known risk factor of development of inflammatory hepatocellular adenoma, a benign liver tumor characterized by an uncontrolled activation of JAK/STAT pathway induced by mutations in oncogenes such as IL6ST, STAT3, FRK, GNAS, JAK1 and ROS1 [Citation117,Citation118]. In contrast, data from large genomic data based on whole exome, whole genome and RNA sequencing from international consortium have not revealed a specific genomic pattern in NAFLD-related HCC [Citation110,Citation111,Citation119]. In a recent large genomic study of HCC, NASH-related HCC seems to be similar to alcohol-related HCC at the mutational and transcriptomic level [Citation112]. To note, this study was not properly designed to answer specifically this question and lacks of a large number of well-annotated NAFLD-related HCC. Overall, most of studies available on NASH-related HCC have been performed in preclinical models and analysis of a large cohort of human tumors is warranted to better decipher the pathogenesis of human NAFLD-related HCC.
Pathological characteristics of HCC related to NAFLD
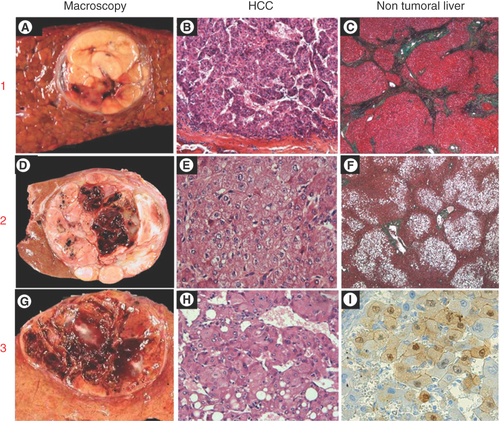
As already discussed, HCC related to NAFLD may occur in patients with cirrhosis or with no or mild liver damage in the background liver. When comparing HCC morphological characteristics according to the aspect of nontumoral liver (i.e., cirrhosis or not), differences have been reported mainly in terms of tumor size, with larger nodules observed in the absence of cirrhosis [Citation94]. However, and despite their larger size, HCC were more likely to be well differentiated with fewer signs of aggressiveness, such as microvascular invasion and satellite nodules [Citation94]. Noteworthy, in some cases occurring in absence of underlying cirrhosis, HCC may result from the malignant transformation of a pre-existing hepatocellular adenoma (observed in five out 31 patients [16%] with metabolic syndrome compared with none in 81 patients with HCV infection), as both aspects were recognized within the tumoral nodule [Citation94]. Such way of carcinogenesis is supported by the association of metabolic syndrome and development of hepatocellular adenoma [Citation120]: in this surgical series of transformed adenomas, metabolic syndrome fulfilling the WHO definition was observed in six out of 12 patients. It is well known than obesity is a risk factor of inflammatory HCA development. Moreover, steatosis on the nontumor liver was identified in 40% of inflammatory HCA [Citation117]. It is also known than inflammatory HCA could give rise to HCC mainly in conjunction with activation of the Wnt/B-catenin pathway [Citation121]. Nevertheless, the real prevalence of malignant transformation of HCA in patients with HCC developed on NAFLD remains unknown. In this context, inflammatory and the newly recognized sonic Hedgehog subtypes are the most frequent observed [Citation122]. Whereas no specific molecular pathways have been identified to explain such distinctive patterns of HCC development, a comparative genomic hybridization of HCC related to metabolic syndrome or HCV infection identified several genomic alterations more frequent in HCC related to metabolic syndrome [Citation123]. More interestingly, among them, in the group of HCC related to metabolic syndrome, 6p21.1 amplification leading to cullin 7 overexpression, was significantly associated with the presence of advanced fibrosis in the background liver (67% in advanced fibrosis [including stages 3 and 4] vs 25% in nonadvanced fibrosis [from F0 to F2], p < 0.01) [Citation123]. The different morphological patterns of HCC associated with metabolic syndrome/NAFLD are recapitulated in .
1. HCC arising in a patient with cirrhosis (A) well-limited encapsulated nodule of 3 cm within a cirrhotic liver; (B) Moderately differentiated HCC (hematein & eosin staining); (C) Nontumoral liver showing cirrhotic nodules (trichrome staining). 2. HCC arising in a patient without advanced chronic liver disease (D) well-limited nodule of 6 cm; (E) Well-differentiated HCC (hematein and eosin staining); (F) Nontumoral liver showing normal architecture with steatosis (trichrome staining). 3. HCC arising in a pre-existing hepatocellular adenoma (G) well-limited nodule of 4 cm showing hemorrhagic areas in a background normal liver, (H) Well-differentiated HCC (hematein & eosin staining); (I) β-catenin immunostaining showing nuclear positivity of tumoral hepatocytes from the hepatocellular adenoma component.
HCC: Hepatocellular carcinoma; NAFLD: Non-alcoholic fatty liver disease.
Morphologically, HCC define a wide spectrum of hepatocellular proliferations with recognition of at least eight subtypes (WHO classification of tumours, Digestive System Tumours, 5th edition, IARC 2019). Although all subtypes could be observed (, the variant called ‘steatohepatitic HCC’ appears to be more often reported in patients with NAFLD. Interestingly, this variant, initially described in HCV-transplanted patients, is characterized by morphological hallmarks recapitulating NASH picture, including steatosis, ballooning malignant hepatocytes, Mallory-Denk bodies within tumor cells, inflammatory infiltrates and fibrosis with a pericellular ‘chicken-wire’ or trabecular patterns () [Citation124]. Then, the steatohepatitic-hepatocellular carcinoma (SH-HCC) variant was reported in 35.5% of HCV liver explants, among them two-thirds of patients displayed at least one risk factor for metabolic syndrome, with NASH features in 63.6% of cases. In a follow-up study including liver resections and transplantations, SH-HCC accounted for 13.5% of overall HCC, arising more frequently in the context of either chronic alcohol consumption or NAFLD compared with other chronic liver diseases (35.7 vs 1.3%, p < 0.0001) [Citation125]. Again, steatosis and steatohepatitis were much more frequent in the background liver of patients with SH-HCC [Citation125]. In a series from India, Jain et al. identified the SH variant of HCC in 18.8% of explanted livers, with approximately half of them related to either NAFLD or HCV hepatitis [Citation126]. In a retrospective Japanese surgical cohort of HCC, SH-HCC accounted for 31.4% of overall HCC, also showing association with signs of metabolic syndrome and NAFLD in the underlying non tumoral liver [Citation127]. Such differences in proportion of SH-HCC variant may result from the distinct morphological criteria used for their diagnosis. Indeed, no consensus definition has been recognized so far, and, according to the studies, SH-HCC may be referred to HCC showing SH pattern in more than 5% to more than 50% of tumor area. In addition, the number of elementary features (steatosis, ballooned cells, Mallory-Denk bodies, inflammation and fibrosis) required for the diagnosis may vary [Citation125–127].
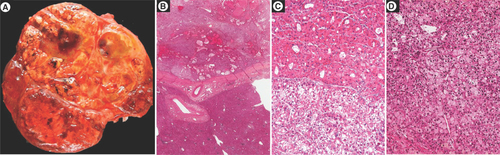
(A) Macroscopic view showing a large heterogeneous tumor nodule with firm brown and soft yellowish areas. Histological analysis showing (B) at low magnification an encapsulated tumor arising in a normal liver, (C & D) different microscopic patterns (architectural and cytological) are observed throughout the tumor.
NAFLD: Non-alcoholic fatty liver disease.
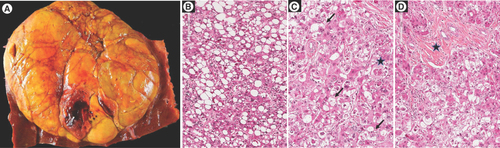
(A) Macroscopic view showing a large, well-limited, unencpasulated yellowish tumor nodule with few hemorragic areas; H&E staining showing (B) steatosis with large droplets with tumoral hepatocytes, (C) presence of ballooned tumoral hepatocytes (arrow) and Mallory-Denk bodies within ballooned cells (black star) and (D) fibrous stoma (black star).
H&E: Hematoxylin and eosin.
The SH variant of HCC is macroscopically nodular, well-limited and more yellowish (due to steatosis) compared with other subtypes [Citation125]. In one study, SH-HCC tended to be smaller and better differentiated independently of the presence of cirrhosis in the background liver [Citation127]. Immunophenotypically, most SH-HCC display common markers of HCC including Glypican-3, Heat Shock Protein-70 (HSP-70) and glutamine synthetase (GS) [Citation124,Citation126]. In addition, tumor-ballooned hepatocytes are negative for Cytokeratin 8/18, except for the Mallory-Denk bodies, which are also labeled by ubiquitin [Citation125]. In a larger series, including 197 HCC with 70 SH-HCC, lower incidences of GS overexpression and β-catenin nuclear positivity were observed in the SH-HCC variant [Citation128]. These results were confirmed by the lower frequency of CTNNB1 mutations in the group of SH-HCC. Interestingly, all SH-HCCs were diffusely stained with sonic Hedgehog ligand, while a minority of them express progenitor markers including SALL4, EpCAM and CK19 [Citation128]. Lastly, they did not observe at the protein level any significant alteration of the AKT/mTOR signaling pathway [Citation128]. Molecular characterization of the fibrous compartment, a distinctive feature of SH-HCC, has been performed, demonstrated higher number of cancer-associated fibrosblast (α-smooth muscle actin positive cells) with greater proportion of CAF expressing markers of senescence (p21), DNA damage (γ-H2AX) markers as well as IL-6 [Citation129]. These results are in line with previous experimental data showing that HSCs may acquire a senescence-associated secretory phenotype and contribute to HCC development in obese mice [Citation104].
High-throughput sequencing techniques were able to describe the mutational landscape of HCC [Citation130]. Among the different molecular classifications available, the G1–G6 classification identified two main tumor groups associated with clinical and genetic features, characterized by either chromosomal instability (from G1 to G3) or chromosomal stability (from G4 to G6) [Citation131]. Accordingly, thanks to a comprehensive pathomolecular analysis, the SH-HCC was assigned to the G4 transcriptomic subgroup characterized by a lack of Wnt/β-catenin pathway activation and low GS expression [Citation132], supporting the immunophenotypical data obtained by Ando et al. [Citation128]. While no significant changes in gene involved in lipid metabolism were observed, activation of the IL-6/AKT/STAT pathway was frequent in this subgroup, which is consistent with the involvement of this pathway in the transition from NAFL to NASH [Citation132]. Although the strong association between SH-HCC and NAFLD, a number of SH-HCC may be observed in absence of a context of metabolic syndrome and NAFLD, suggesting that the SH pattern of HCC may be unrelated to the clinical condition and rather result from tumor specific pathways and genetic alterations. To address this issue, Yeh et al. described a set of 12 cases of SH-HCC without any associated features of MS or NAFLD and performed a genomic microarray analysis to search for specific chromosomal alterations [Citation133]. In addition to previously reported recurrent abnormalities in HCC (loss of 8p and gains of 1q, 7 and 8q), a new deletion (9q12–q31.1) was observed in this SH-HCC subset, especially in cases without cirrhosis in the background [Citation133].
Whether SH-HCC may present a better or worse prognosis compared with conventional HCC remains difficult to conclude, as available data are derived from resected or transplanted patients. Nevertheless, almost of all them did not show any statistical difference in terms of overall survival or disease-free survival [Citation125,Citation127]. Such clinical behavior is supported by the less aggressive histological phenotype with a lack of satellite nodules and microvascular invasion of SH-HCC seem to display [Citation132].
Conclusion
HCC developed in NAFLD patients presents specific penotypic as well as constitutional and functional genetic features. Its pathophysiology is mostly unknown and has largely benefited from the continuous study of dedicated animal models. Translational research both at the population and liver sample levels are key to unravel specific mechanisms impliaced in this particular carcinogenic process.
Future perspective
attempts to provide an integrative approach of translational research in the field of NAFLD-related HCC. The incorporation of genetic traits into complex GRS will allow the selection of individuals who should be included in surveillance programs among the general population. Such approach will also refine screening modalities in patients bearing the highest risk, particularly in case of extensive fibrosis, and in whom implementation of costly procedures such as MRI or circulating biomarkers might be cost-effective. Deciphering the biological pathways implicated in NAFLD-related hepatocarcinogenesis will foster the development of dedicated primary and secondary prevention. Finally, improving pathological and molecular classification of tumors should help physicians to adapt therapeutic management through complex strategies aimed at decreasing recurrence/progression rates and improving survival. Overall, all these combined efforts should pave the way for personalized management and will impact the management of patients throughout the entire spectrum of NAFLD.
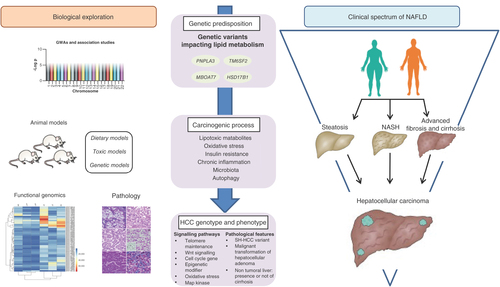
NAFLD-related hepatocellular carcinoma (HCC) has been the focus of increasing scientific interest in view of the growing burden of obesity worldwide.
Genome-wide association studies have highlighted several genetic traits modulating lipid turnover as liver carcinogenesis predisposing factors.
Numerous animal models allowed deciphering specific oncogenic process.
Pathological examination revealed different subtypes linked to HCC molecular classification and associated with differences in outcome.
Although dedicated studies at the functional genomic level are still scarce, some genetic defects may be similar to that observed in alcohol-related HCC.
Translating these findings into clinical practice to tailor precision medicine is the next challenge.
Author contributions
P Nahon, M Allaire, J-C Nault and V Paradis participated in all stages of manuscript production, design, figures, tables, writing and review of final version.
Financial & competing interests disclosure
P Nahon has received honoraria or grants from Abbvie, Astra-Zeneca, Bayer, Bristol-Myers Squibb, EISAI, Gilead, Ipsen and Roche. He consults for Bristol-Myers Squibb and Roche. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- YounossiZ, AnsteeQM, MariettiMet al.Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15(1), 11–20 (2018).
- EstesC, RazaviH, LoombaR, YounossiZ, SanyalAJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67(1), 123–133 (2018).
- BaffyG, BruntEM, CaldwellSH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J. Hepatol. 56(6), 1384–1391 (2012).
- KleinS, DufourJF. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Hepat. Oncol. 4(3), 83–98 (2017).
- WhiteDL, KanwalF, El-SeragHB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 10(12), 1342–1359, e1342 (2012).
- KanwalF, KramerJR, MapakshiSet al.Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 155(6), 1828–1837, e1822 (2018).
- SanyalA, PoklepovicA, MoyneurE, BarghoutV. Population-based risk factors and resource utilization for HCC: US perspective. Curr. Med. Res. Opin. 26(9), 2183–2191 (2010).
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J. Hepatol. 69(1), 182–236 (2018).
- HeimbachJK, KulikLM, FinnRSet al.AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 67(1), 358–380 (2018).
- KawamuraY, AraseY, IkedaKet al.Large-scale long-term follow-up study of Japanese patients with non-alcoholic fatty liver disease for the onset of hepatocellular carcinoma. Am. J. Gastroenterol. 107(2), 253–261 (2012).
- LoombaR, LimJK, PattonH, El-SeragHB. AGA clinical practice update on screening and surveillance for hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: expert review. Gastroenterology 158(6), 1822–1830 (2020).
- SingalAG, LamperticoP, NahonP. Epidemiology and surveillance for hepatocellular carcinoma: new trends. J. Hepatol. 72(2), 250–261 (2020).
- KimHL, AnJ, ParkJA, ParkSH, LimYS, LeeEK. Magnetic resonance imaging is cost-effective for hepatocellular carcinoma surveillance in high risk patients with cirrhosis. Hepatology 69(4),1599–1613 (2019).
- KimSY, AnJ, LimYSet al.MRI with liver-specific contrast for surveillance of patients with cirrhosis at high risk of hepatocellular carcinoma. JAMA Oncol. 3(4), 456–463 (2017).
- EslamM, ValentiL, RomeoS. Genetics and epigenetics of NAFLD and NASH: clinical impact. J. Hepatol. 68(2), 268–279 (2018).
- SchwimmerJB, CeledonMA, LavineJEet al.Heritability of nonalcoholic fatty liver disease. Gastroenterology 136(5), 1585–1592 (2009).
- AnsteeQM, ReevesHL, KotsilitiE, GovaereO, HeikenwalderM. From NASH to HCC: current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 16(7), 411–428 (2019).
- RomeoS, KozlitinaJ, XingCet al.Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40(12), 1461–1465 (2008).
- KozlitinaJ, SmagrisE, StenderSet al.Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 46(4), 352–356 (2014).
- MancinaRM, DongiovanniP, PettaSet al.The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150(5), 1219–1230, e1216 (2016).
- Abul-HusnNS, ChengX, LiAHet al.A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 378(12), 1096–1106 (2018).
- DongiovanniP, RomeoS, ValentiL. Genetic factors in the pathogenesis of nonalcoholic fatty liver and steatohepatitis. Biomed. Res. Int. 2015, 460190 (2015).
- Vespasiani-GentilucciU, GalloP, Dell’untoC, VolpentestaM, Antonelli-IncalziR, PicardiA. Promoting genetics in non-alcoholic fatty liver disease: combined risk score through polymorphisms and clinical variables. World J. Gastroenterol. 24(43), 4835–4845 (2018).
- TrepoE, NahonP, BontempiGet al.Association between the PNPLA3 (rs738409 C > G) variant and hepatocellular carcinoma: evidence from a meta-analysis of individual participant data. Hepatology 59(6), 2170–2177 (2014).
- HuangY, CohenJC, HobbsHH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 286(43), 37085–37093 (2011).
- PirazziC, AdielsM, BurzaMAet al.Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 57(6), 1276–1282 (2012).
- YuanX, WaterworthD, PerryJRet al.Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am. J. Hum. Genet. 83(4), 520–528 (2008).
- KolleritsB, CoassinS, KiechlSet al.A common variant in the adiponutrin gene influences liver enzyme values. J. Med. Genet. 47(2), 116–119 (2010).
- ChambersJC, ZhangW, SehmiJet al.Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet. 43(11), 1131–1138 (2011).
- ValentiL, Al-SerriA, DalyAKet al.Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 51(4), 1209–1217 (2010).
- SpeliotesEK, ButlerJL, PalmerCD, VoightBF, HirschhornJN. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 52(3), 904–912 (2010).
- RotmanY, KohC, ZmudaJM, KleinerDE, LiangTJ. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 52(3), 894–903 (2010).
- HolmenOL, ZhangH, FanYet al.Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat. Genet. 46(4), 345–351 (2014).
- MaY, BelyaevaOV, BrownPMet al.17-Beta hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology 69(4), 1504–1519 (2019).
- MancinaRM, DongiovanniP, PettaSet al.The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150(5), 1219.e6–1230.e6 (2016).
- LiuYL, PatmanGL, LeathartJBet al.Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 61(1), 75–81 (2014).
- DonatiB, DongiovanniP, RomeoSet al.MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 7(1), 4492 (2017).
- FalletiE, CussighA, CmetS, FabrisC, ToniuttoP. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig. Liver Dis. 48(1), 69–75 (2016).
- YangJ, TrepoE, NahonPet al.A 17-beta-hydroxysteroid dehydrogenase 13 variant protects from hepatocellular carcinoma development in alcoholic liver disease. Hepatology 70(1), 231–240 (2019).
- LiL, CheL, TharpKMet al.Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 63(6), 1900–1913 (2016).
- NaultJC, NahonP. Genetic predisposition to hepatocellular carcinoma in alcoholic cirrhosis: the NCAN-PNPLA3-lipid connection?J. Hepatol. 61(5), 971–972 (2014).
- NahonP, Zucman-RossiJ. Single nucleotide polymorphisms and risk of hepatocellular carcinoma in cirrhosis. J. Hepatol. 57(3), 663–674 (2012).
- NahonP, NaultJC. Constitutional and functional genetics of human alcohol-related hepatocellular carcinoma. Liver Int. 37(11), 1591–1601 (2017).
- NahonP, SuttonA, ZiolM, Zucman-RossiJ, TrinchetJC, Ganne-CarrieN. Genetic risk markers for hepatocellular carcinoma in patients with alcoholic liver disease. Hepat. Oncol. 2(1), 63–78 (2015).
- Ganne-CarrieN, NahonP. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 70(2), 284–293 (2019).
- GuyotE, SuttonA, RufatPet al.PNPLA3 rs738409, hepatocellular carcinoma occurrence and risk model prediction in patients with cirrhosis. J. Hepatol. 58(2), 312–318 (2013).
- YangJ, TrepoE, NahonPet al.PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int. J. Cancer 144(3), 533–544 (2019).
- BurzaMA, PirazziC, MaglioCet al.PNPLA3 I148M (rs738409) genetic variant is associated with hepatocellular carcinoma in obese individuals. Dig. Liver Dis. 44(12), 1037–1041 (2012).
- GrimaudoS, PipitoneRM, PennisiGet al.Association between PNPLA3 rs738409 C > G variant and liver-related outcomes in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 18(4), 935–944e933 (2020).
- Gellert-KristensenH, RichardsonTG, DaveySmith G, NordestgaardBG, Tybjaerg-HansenA, StenderS. Combined effect of PNPLA3, TM6SF2, and HSD17B13 variants on risk of cirrhosis and hepatocellular carcinoma in the general population. Hepatology 72(3), 845–856 (2020).
- SugrueLP, DesikanRS. What are polygenic scores and why are they important?JAMA 321(18), 1820–1821 (2019).
- MarraF, LotersztajnS. Pathophysiology of NASH: perspectives for a targeted treatment. Curr. Pharm. Des. 19(29), 5250–5269 (2013).
- MotaM, BaniniBA, CazanaveSC, SanyalAJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 65(8), 1049–1061 (2016).
- BoursierJ, MuellerO, BarretMet al.The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63(3), 764–775 (2016).
- MeekTH, MortonGJ. The role of leptin in diabetes: metabolic effects. Diabetologia 59(5), 928–932 (2016).
- MallatA, LotersztajnS. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am. J. Physiol. Cell Physiol. 305(8), C789–799 (2013).
- Svegliati-BaroniG, RidolfiF, DiSario Aet al.Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology 29(6), 1743–1751 (1999).
- PradereJP, TroegerJS, DapitoDH, MencinAA, SchwabeRF. Toll-like receptor 4 and hepatic fibrogenesis. Semin. Liver Dis. 30(3), 232–244 (2010).
- MarraF, Svegliati-BaroniG. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 68(2), 280–295 (2018).
- KunneC, AccoA, DuijstSet al.FXR-dependent reduction of hepatic steatosis in a bile salt deficient mouse model. Biochim. Biophys. Acta 1842(5), 739–746 (2014).
- McbrideRL, FeringaER, SmithBE. The fate of prelabeled Clarke’s column neurons after axotomy. Exp. Neurol. 102(2), 236–243 (1988).
- KettnerNM, VoicuH, FinegoldMJet al.Circadian homeostasis of liver metabolism suppresses hepatocarcinogenesis. Cancer Cell 30(6), 909–924 (2016).
- TovarV, AlsinetC, VillanuevaAet al.IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J. Hepatol. 52(4), 550–559 (2010).
- ChettouhH, LequoyM, FartouxL, VigourouxC, Desbois-MouthonC. Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int. 35(10), 2203–2217 (2015).
- MichelottiGA, MachadoMV, DiehlAM. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 10(11), 656–665 (2013).
- AllaireM, NaultJC. Type 2 diabetes-associated hepatocellular carcinoma: a molecular profile. Clin. Liver Dis. (Hoboken) 8(2), 53–58 (2016).
- HaczeyniF, YehMM, IoannouGNet al.Mouse models of non-alcoholic steatohepatitis: a reflection on recent literature. J. Gastroenterol. Hepatol. 33(7), 1312–1320 (2018).
- MachadoMV, MichelottiGA, XieGet al.Correction: mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 10(6), e0132315 (2015).
- IbrahimSH, HirsovaP, MalhiH, GoresGJ. Animal models of nonalcoholic steatohepatitis: eat, delete, and inflame. Dig. Dis. Sci. 61(5), 1325–1336 (2016).
- MiuraK, KodamaY, InokuchiSet al.Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139(1), 323–334, e327 (2010).
- DowmanJK, HopkinsLJ, ReynoldsGMet al.Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high-fat/fructose diet and sedentary lifestyle. Am. J. Pathol. 184(5), 1550–1561 (2014).
- WolfMJ, AdiliA, PiotrowitzKet al.Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26(4), 549–564 (2014).
- FarrellGC, MridhaAR, YehMMet al.Strain dependence of diet-induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype. Liver Int. 34(7), 1084–1093 (2014).
- HaluzikM, ColomboC, GavrilovaOet al.Genetic background (C57BL/6J versus FVB/N) strongly influences the severity of diabetes and insulin resistance in ob/ob mice. Endocrinology 145(7), 3258–3264 (2004).
- GanzM, CsakT, SzaboG. High fat diet feeding results in gender specific steatohepatitis and inflammasome activation. World J. Gastroenterol. 20(26), 8525–8534 (2014).
- FujiiM, ShibazakiY, WakamatsuKet al.A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 46(3), 141–152 (2013).
- RidolfiR, AmaducciL, DerniS, FabbriL, InnocentiMP, VignutelliP. Chemotherapy with 5-fluorouracil and streptozotocin in carcinoid tumors of gastrointestinal origin: experiences with 13 patients. J. Chemother. 3(5), 328–331 (1991).
- KishidaN, MatsudaS, ItanoOet al.Development of a novel mouse model of hepatocellular carcinoma with nonalcoholic steatohepatitis using a high-fat, choline-deficient diet and intraperitoneal injection of diethylnitrosamine. BMC Gastroenterol. 16(1), 61 (2016).
- TsuchidaT, LeeYA, FujiwaraNet al.Corrigendum to ‘A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer’. [ J. Hepatol.69, 385–395 (2018)]. J. Hepatol. 69(4), 988 (2018).
- LindstromP. The physiology of obese-hyperglycemic mice [ob/ob mice]. Sci. World J. 7, 666–685 (2007).
- WangB, ChandrasekeraPC, PippinJJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr. Diabetes Rev. 10(2), 131–145 (2014).
- ZhangN, ChuES, ZhangJet al.Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-kappaB signaling pathway. Oncotarget 5(18), 8330–8340 (2014).
- ItohM, SuganamiT, NakagawaNet al.Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am. J. Pathol. 179(5), 2454–2463 (2011).
- HorieY, SuzukiA, KataokaEet al.Hepatocyte-specific PTEN deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest. 113(12), 1774–1783 (2004).
- FanCY, PanJ, ChuRet al.Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J. Biol. Chem. 271(40), 24698–24710 (1996).
- LuSC, MatoJM. Role of methionine adenosyltransferase and S-adenosylmethionine in alcohol-associated liver cancer. Alcohol 35(3), 227–234 (2005).
- BonzoJA, FerryCH, MatsubaraT, KimJH, GonzalezFJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J. Biol. Chem. 287(10), 7345–7356 (2012).
- FekryB, Ribas-LatreA, BaumgartnerCet al.HNF4alpha-deficient fatty liver provides a permissive environment for sex-independent hepatocellular carcinoma. Cancer Res. 79(22), 5860–5873 (2019).
- Van RooyenDM, LarterCZ, HaighWGet al.Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 141(4), 1393–1403; e1391–e1395 (2011).
- ArfiantiE, LarterCZ, LeeSet al.Obesity and diabetes accelerate hepatocarcinogenesis via hepatocyte proliferation independent of NF-kappaB or Akt/mTORC1. J. Clin. Transl. Res. 2(1), 26–37 (2016).
- AsgharpourA, CazanaveSC, PacanaTet al.A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 65(3), 579–588 (2016).
- NakagawaH, UmemuraA, TaniguchiKet al.ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26(3), 331–343 (2014).
- KimG, JangSY, NamCM, KangES. Statin use and the risk of hepatocellular carcinoma in patients at high risk: a nationwide nested case-control study. J. Hepatol. 68(3), 476–484 (2018).
- ParadisV, ZalinskiS, ChelbiEet al.Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology 49(3), 851–859 (2009).
- HirosumiJ, TuncmanG, ChangLet al.A central role for JNK in obesity and insulin resistance. Nature 420(6913), 333–336 (2002).
- GomesAL, TeijeiroA, BurenSet al.Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30(1), 161–175 (2016).
- GentricG, MailletV, ParadisVet al.Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Invest. 125(3), 981–992 (2015).
- VillanuevaA, ChiangDY, NewellPet al.Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 135(6), 1972–1983, e1971–e1911 (2008).
- HotamisligilGS, PeraldiP, BudavariA, EllisR, WhiteMF, SpiegelmanBM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271(5249), 665–668 (1996).
- KohJH, ShinYG, NamSM, LeeMY, ChungCH, ShinJY. Serum adipocyte fatty acid-binding protein levels are associated with nonalcoholic fatty liver disease in type 2 diabetic patients. Diabetes Care 32(1), 147–152 (2009).
- LaouiremS, SannierA, NorkowskiEet al.Endothelial fatty liver binding protein 4: a new targetable mediator in hepatocellular carcinoma related to metabolic syndrome. Oncogene 38(16), 3033–3046 (2019).
- JansenPL. Endogenous bile acids as carcinogens. J. Hepatol. 47(3), 434–435 (2007).
- DapitoDH, MencinA, GwakGYet al.Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21(4), 504–516 (2012).
- YoshimotoS, LooTM, AtarashiKet al.Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499(7456), 97–101 (2013).
- AllaireM, RautouPE, CodognoP, LotersztajnS. Autophagy in liver diseases: time for translation?J. Hepatol. 70(5), 985–998 (2019).
- HammouteneA, BiquardL, LasselinJet al.A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J. Hepatol. 72(3), 528–538 (2020).
- RybsteinMD, Bravo-SanPedro JM, KroemerG, GalluzziL. The autophagic network and cancer. Nat. Cell Biol. 20(3), 243–251 (2018).
- TakamuraA, KomatsuM, HaraTet al.Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25(8), 795–800 (2011).
- DhanasekaranR, NaultJC, RobertsLR, Zucman-RossiJ. Genomic medicine and implications for hepatocellular carcinoma prevention and therapy. Gastroenterology 156(2), 492–509 (2019).
- SchulzeK, ImbeaudS, LetouzeEet al.Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 47(5), 505–511 (2015).
- TotokiY, TatsunoK, CovingtonKRet al.Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 46(12), 1267–1273 (2014).
- NaultJC, MartinY, CarusoSet al.Clinical impact of genomic diversity from early to advanced hepatocellular carcinoma. Hepatology 71(1), 164–182 (2020).
- SungWK, ZhengH, LiSet al.Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 44(7), 765–769 (2012).
- LetouzeE, ShindeJ, RenaultVet al.Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat Commun 8(1), 1315 (2017).
- NaultJC, LetouzeE. Mutational processes in hepatocellular carcinoma: the story of aristolochic acid. Semin. Liver Dis. 39(3), 334–340 (2019).
- BressacB, KewM, WandsJ, OzturkM. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 350(6317), 429–431 (1991).
- NaultJC, CouchyG, BalabaudCet al.Molecular classification of hepatocellular adenoma associates with risk factors, bleeding, and malignant transformation. Gastroenterology 152(4), 880–894, e886 (2017).
- BayardQ, CarusoS, CouchyGet al.Recurrent chromosomal rearrangements of ROS1, FRK and IL6 activating JAK/STAT pathway in inflammatory hepatocellular adenomas. Gut 69(9), 1667–1676 (2020).
- Cancer Genome Atlas Research Network. Electronic Address WBE, Cancer Genome Atlas Research N. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169(7), 1327–1341, e1323 (2017).
- FargesO, FerreiraN, DokmakS, BelghitiJ, BedossaP, ParadisV. Changing trends in malignant transformation of hepatocellular adenoma. Gut 60(1), 85–89 (2011).
- PilatiC, LetouzeE, NaultJCet al.Genomic profiling of hepatocellular adenomas reveals recurrent FRK-activating mutations and the mechanisms of malignant transformation. Cancer Cell 25(4), 428–441 (2014).
- ParadisV, ChampaultA, RonotMet al.Telangiectatic adenoma: an entity associated with increased body mass index and inflammation. Hepatology 46(1), 140–146 (2007).
- ParadisV, AlbuquerqueM, MebarkiMet al.Cullin7: a new gene involved in liver carcinogenesis related to metabolic syndrome. Gut 62(6), 911–919 (2013).
- SalomaoM, YuWM, BrownRSJr, EmondJC, LefkowitchJH. Steatohepatitic hepatocellular carcinoma (SH-HCC): a distinctive histological variant of HCC in hepatitis C virus-related cirrhosis with associated NAFLD/NASH. Am. J. Surg. Pathol. 34(11), 1630–1636 (2010).
- SalomaoM, RemottiH, VaughanR, SiegelAB, LefkowitchJH, MoreiraRK. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum. Pathol. 43(5), 737–746 (2012).
- JainD, NayakNC, KumaranV, SaigalS. Steatohepatitic hepatocellular carcinoma, a morphologic indicator of associated metabolic risk factors: a study from India. Arch. Pathol. Lab. Med. 137(7), 961–966 (2013).
- ShibaharaJ, AndoS, SakamotoY, KokudoN, FukayamaM. Hepatocellular carcinoma with steatohepatitic features: a clinicopathological study of Japanese patients. Histopathology 64(7), 951–962 (2014).
- AndoS, ShibaharaJ, HayashiA, FukayamaM. beta-catenin alteration is rare in hepatocellular carcinoma with steatohepatitic features: immunohistochemical and mutational study. Virchows Arch. 467(5), 535–542 (2015).
- LeeJ, LeeS, ZhangH, HillMA, ZhangC, ParkY. Interaction of IL-6 and TNF-alpha contributes to endothelial dysfunction in type 2 diabetic mouse hearts. PLoS ONE 12(11), e0187189 (2017).
- Zucman-RossiJ, VillanuevaA, NaultJC, LlovetJM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 149(5), 1226–1239, e1224 (2015).
- BoyaultS, RickmanDS, DeReynies Aet al.Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 45(1), 42–52 (2007).
- CalderaroJ, CouchyG, ImbeaudSet al.Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 67(4), 727–738 (2017).
- YehMM, LiuY, TorbensonM. Steatohepatitic variant of hepatocellular carcinoma in the absence of metabolic syndrome or background steatosis: a clinical, pathological, and genetic study. Hum. Pathol. 46(11), 1769–1775 (2015).