
Abstract
Aim: To explore the roles of lncRNA MALAT1 and SHOC2 in breast cancer, and the potential connections to chemotherapy resistance in breast cancer. Materials & methods: Paclitaxel-resistant breast cancer cells were induced by gradually increasing intermittent doses. Bioinformatic analyses were performed to predict the regulated miRNAs of MALAT1. Results: High expressions of MALAT1 and SHOC2 contribute to paclitaxel resistance in breast cancer cells. MALAT1 sponges miR-497-5p enhance SHOC2 expression in paclitaxel-resistant breast cancer cells and contribute to paclitaxel resistance in breast cancer cells. Conclusion: Patients with high expression of MALAT1 and SHOC2 have a poorer response to paclitaxel. Upregulation of miR-497-5p could improve the treatment response to paclitaxel in patients with breast cancer by inhibiting MALAT1 and SHOC2.
Keywords::
The World Health Organization’s International Agency for Research on Cancer released the latest global cancer burden data at the end of 2020. There were 2.26 million new cases of breast cancer (BC) worldwide, which surpassed lung cancer, making BC the world’s most prevalent cancer in the female population [Citation1]. Currently, the mechanisms of BC pathogenesis are still unclear, but it is related to factors attributed to estrogen secretion, environment and heredity [Citation2]. Chemotherapy is one of the important treatment methods for cancer. However, drug resistance seriously affects the effect of chemotherapy. Epigenetic abnormalities and specific cell membrane transporters (by changing drug transport and pumping drugs out of tumor cells) have been reported to be related to the induction of drug resistance [Citation3].
The abnormal activation of the Ras–Raf–ERK pathway is the key step to initiating tumorigenesis. The activation process of the ERK pathway originates from extracellular stimulation signals binding GTP to Ras, then Raf is recruited to GTP–Ras and activated. After Raf activation, MAPK (MEK1/2) is activated, and finally, MEK1/2 phosphorylation activates ERK1/2 [Citation4,Citation5]. However, in the wide range of biological reactions controlled by the ERK pathway, how the specificity of the signal is regulated is not very clear. Studies have shown that scaffold proteins can promote the interaction and function of core signaling factors of the ERK pathway and play an indispensable role in controlling ERK1/2 activity [Citation6,Citation7]. SHOC2 is a member of the scaffold protein family and can positively regulate the Ras pathway and promote the occurrence and development of tumors by activating the Ras–Raf–ERK pathway in a variety of tumor cells [Citation8]. SHOC2 also could enhance the malignant potential of tumor cells, promote tumorigenesis and induce drug resistance by regulating microfilament microtubule remodeling, promoting abnormal cell proliferation and inhibiting apoptosis [Citation9]. Geng et al. examined 120 formalin-fixed paraffin-embedded BC specimens and found that overexpression of SHOC2 may be associated with a poor prognosis for BC patients [Citation10].
MALAT1 is a new transcript discovered when predicting the metastasis and survival of early non-small-cell lung cancer in 2003. It was named MALAT1 at the beginning of discovery and is located on human chromosome 11q13.1; the main sequence has more than 8000 nucleotides [Citation11]. By analyzing the MALAT1 sequence, Ji et al. found that the transcript lacked an ORF of sufficient length; the longest ORF in the whole sequence could only encode a peptide of 52 or 55 amino acids, and MALAT1 had no protein or peptide formation during translation in vitro [Citation11]. Therefore, the new transcript MALAT1 was confirmed to be one of the noncoding RNAs. Further studies found that MALAT1 can play its molecular role by regulating selective splicing, regulating gene transcription and acting as a miRNA sponge [Citation12,Citation13], and is closely related to tumor metastasis, clinical stage, recurrence and prognosis [Citation14–16]. MALAT1 plays a carcinogenic role in BC [Citation17]; however, the mechanism of its action on paclitaxel resistance in BC cells remains unclear. A previous study indicated that miR-497 is involved in the suppression of metastasis and invasion of BC in vitro and in vivo [Citation18]. In a domestic study in China, Ma et al. found that miR-497-5p inhibits the proliferation and induces apoptosis of paclitaxel-resistant BC (PR-BC) cells by targeting PD-L1 [Citation19].
This study aims to explore the roles of MALAT1 and SHOC2 in BC and the potential connections to chemotherapy resistance. MALAT1 and SHOC2 were detected in PR-BC cell lines MCF-7-PR and MDA-MB-231-PR, and the cells’ sensitivity to paclitaxel was evaluated by observing MALAT1 silencing, miR-497-5p overexpression and SHOC2 rescue experiments, to reveal their mutually targeted mechanism of action and provide a new research direction for BC treatment.
Materials & methods
Regents & cell lines
Fetal bovine serum (#10099141), RPMI-1640 medium (#11875119) and Dulbecco’s modified Eagle medium (DMEM, #30030) were purchased from Gibco (OK, USA). Paclitaxel (#Sigma-T7191) was purchased from Sigma-Aldrich (MO, USA). Thiazolyl blue (MTT, #ST316) and Trizol™ reagent (#R0016) were purchased from Beyotime Biotechnology (Shanghai, China). Transwell® cell culture inserts (#3428) were purchased from Corning Life Sciences (NY, USA). The primary antibodies for SHOC2, β-actin (#PA5-96278 and #MA5-11869; 1:1000 dilution), ERK1/2 (#61-7400, 1:1000) and pERK1/2 (#44-680G, 1:1000) and the second goat anti-rabbit IgG (#32460, 1: 500) were purchased from Thermo Fisher (MA, USA). The si-MALAT1, miR-497-5p negative control (NC) for mimic, miR-497-5p mimic and overexpression sequences of SHOC2 (SHOC2 OE) and MALAT1 (MALAT1 OE) were designed and purchased from GenePharma Biotech (Shanghai, China). Power SYBR™ Green RNA-to-Ct ™1Step kit (#4391178) and Lipofectamine™ 2000 transfection reagent (#11668027), pGL3 luciferase reporter vectors and Luciferase Assay System (#E1751, #E1741, #E1500) were all purchased from Thermo Fisher. qPCR primers were designed by Sangon Biotech Co., Ltd (Shanghai, China) and were performed on a 7500 Real-Time PCR System (Applied Biosystems, CA, USA).
BC cell lines MCF-7 and MDA-MB-231 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). MCF-10A and SKBR3 were purchased from American Type Culture Collection (VA, USA). MCF-7, MDA-MB-231 and SKBR3 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 0.1 mg/ml streptomycin. Human mammary epithelial cell line MCF-10A was maintained in Dulbecco’s modified Eagle medium. All cells were cultured at 37°C in a constant temperature incubator with 5% CO2. When the cell confluence reached 70–90%, 0.55% trypsin was used for digestion and subculture.
Tissue samples & Gene Expression Omnibus data collection
A total of 40 pathologically confirmed (preoperative puncture pathological examination or postoperative lesion tissue examination) primary BC and adjacent normal tissues were collected for analysis. These patients all received paclitaxel monotherapy, which included paclitaxel injection, liposome paclitaxel, docetaxel and albumin-bound paclitaxel. The demographic data of included samples are listed in . The experiment was reviewed by the ethics committee, and patients signed informed consent forms and agreed to allow the use of samples for scientific research. All samples were collected before neoadjuvant chemotherapy after the operation and preserved in our hospital.
Table 1. Demographic data of included patients (n = 40).
To validate the expression of SHOC2 in BC tissues, the related microarray data were retrieved from the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/gds/) with the following filtering criteria: ‘homo sapiens’ and ‘breast cancer’ and ‘expression’ and ‘chemosensitivity’. GSE22513 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE22513+) was finally adopted. Expression profiling data from 19 pretreatment breast needle biopsy samples of BC patients were analyzed. In this data set, all patients received three cycles of paclitaxel followed by concurrent paclitaxel/radiation. The differentially expressed genes in patients who achieved pathological complete response (pCR) and non-pCR patients were compared.
Treatment response assessment
The treatment response was evaluated according to Response Evaluation Criteria In Solid Tumors 1.1 [Citation20]. Patients who achieved complete remission, partial remission or stable disease were classified as sensitive to paclitaxel treatment, while patients with progressive disease were classified as resistant to paclitaxel treatment.
Establishment of paclitaxel-resistant cell lines
PR-BC cells were induced by gradually increasing intermittent doses. Briefly, 1 nmol/l paclitaxel was added to logarithmic growth phase BC cells for 48 h, then the medium was replaced by taxol-free culture medium. Subculture of steady-growth cells was continued, and the concentration of paclitaxel gradually increased to 5, 10, 20, 40, 80, 160, 320, 640 and 1280 nmol/l, respectively. Finally, the paclitaxel-resistant cells that could be stably passaged were selected and cultured in a medium supplemented with paclitaxel to maintain the paclitaxel resistance.
Cell transfection
The digested cells were inoculated in a six-well culture plate to a fusion extent of 70–90%. Cells were transfected with Lipofectamine 2000 according to the manufacturer’s instructions. The plasmids containing the designed sequence were diluted by Opti-MEM™ medium (Thermo Fisher Scientific [CO, USA]). After incubation at room temperature for 5 min, plasmids containing si-MALAT1, miR-497-5p NC, miR-497-5p mimic and SHOC2 OE were added to the cells (concentration of 1 × 106 cells/ml) for transfection and cultured for 4 h following the manufacturer’s instructions [Citation21].
RNA extraction & qPCR
Trizol reagent was added to tissue homogenate or centrifuged cells to extract total RNA. The 260-nm absorbance value (A260) was measured by a Nanodrop™ spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific), and the concentration of RNA was determined by the formula (single-stranded RNA concentration = A260 × 40 μg/ ml). A total of 100 ng RNA was used as the template for qPCR and was performed in the Applied Biosystems 7500 system using the Power SYBR Green RNA-to-Ct 1-Step Kit. The total reaction system was 20 μl, including Power SYBR Green RT-PCR Mix (2×) 10 μl, 200 nmol of the upstream and downstream primers and 0.16 μl of RT enzyme mix (125×). Reaction conditions were: holding at 48°C 30 min, 95°C for 10 min, 95°C for 15 s, 60°C for 1 min followed by cycling (40 cycles) of 95°C for 15 s, 60°C for 15 s, 95°C for 15 s. U6 was used as an internal reference for miR-497-5p. GAPDH was used as the internal reference for the expression of MALAT1 and SHOC2. The quantitative results were calculated by the 2-ΔΔ Ct method. The sequences used are listed in .
Table 2. Sequences for primer, mimic and siRNAs.
Cell proliferation assay
For the cell proliferation assay, 200 μl of cells were inoculated on 96-well plates to a final concentration of 2 × 103 cells/well, with four multiple wells in each group. After culture for 24 h, different concentrations of paclitaxel (final concentrations of 0.1, 1, 3, 6, 12 and 24 μM) were added. After being cultured for another 72 h, 10 μl MTT was added to each well and cultured at 37°C for 4 h. Finally, 50 μl of dimethyl sulfoxide was added to terminate the reaction. The absorbance value at 490 nm was detected by the microplate reader 10 min later. The proliferation of cells in each well was evaluated as the relative cell viability of cells only supplemented with medium (NC).
Cell invasion assay
The number of transmembrane penetrating cells was detected in 24-well plates with 8-μm Transwell inserts. For this, 300 μl of serum-free medium containing extracellular matrix was added to the upper Transwell chamber, then a total of 100 μl serum-free RPMI-1640 culture medium (containing 2 × 104 cells) was added in the upper chamber and 600 μl RPMI-1640 medium containing 10% fetal bovine serum was added in the lower chamber. After culture for 24 h, cells that remained in the upper chamber of the inserts were removed with cotton swabs. Cells that had migrated to the lower chamber of the inserts were fixed with 4% paraformaldehyde for 30 min and stained with 0.5% crystal violet for 20 min. The cells were observed and counted under a light microscope.
Bioinformatics analysis & luciferase reporter assay
The potential binding site of lnc MALAT1, miR-497-5p and SHOC2 was analyzed by the ENCORI online prediction website [Citation22]. To verify the potential binding site, cells were inoculated into 96-well plates cultured for 6 h and transfected with wild or mutant pGL3-MALAT1/SHOC2 vector (200 ng, containing wild-type or mutant sequence of the miR-497-5p predicted binding site) and miR-497-5p mimics or NC into MDA-MB-231 by Lipofectamine 2000. After culturing for 48 h, cells were washed with phosphate-buffered saline and the fluorescence intensity was detected by the luciferase reporter analysis system with 100 μl luciferase assay reagent loaded in the luminometer tubes according to the manufacturer’s protocol. The final expression was calculated as the relative value of invalid sequence reading values.
Western blotting analysis
The total protein was extracted by the radioimmunoprecipitation assay lysis method, and the protein concentration was measured by the bicinchoninic acid method. A total of 30 μg protein in each sample was loaded in 15% SDS-PAGE, and the protein was transferred to the polyvinylidene difluoride membrane. After being closed with 5% skim milk powder, proteins were incubated overnight with related primary antibodies, and cultured with secondary antibodies, then developed by electrochemiluminescence. The image was captured by Image J software (National Institutes of Health, MD, USA). The results were shown as the optical density ratio between the target strip and the internal reference strip [Citation22].
Statistical analysis
SPSS v. 22.0 software (IBM Corp., NY, USA) was used for statistical analysis. The continuous variables were expressed by mean ± standard deviation. The t-test was used to compare the two groups, and one-way analysis of variance was used to compare multiple groups (the Student–Newman–Keuls method was used for comparison between two groups). A p < 0.05 was considered statistically significant.
Results
MALAT1 contributes to paclitaxel resistance in BC cells
We first explored the roles of MALAT1 and SHOC2 on paclitaxel resistance in BC cells. Cells with paclitaxel resistance were constructed by gradually increasing paclitaxel concentration. Among the four cell lines, MCF-7 and MDA-MB-231 grew relatively well under the action of paclitaxel; these drug-resistant cell lines were named MDA-MB-231-PR and MCF-7-PR. Further analysis demonstrated that the expression of MALAT1 in the two drug-resistant cell lines was upregulated to varying degrees compared with the same cell lines before paclitaxel treatment (A & B). The analysis of resected samples before paclitaxel treatment revealed that the expression of SHOC2 increased in BC tissues (p < 0.0001) compared with the adjacent tissues at the transcriptional level (C). The paclitaxel treatment response also showed that the expression of SHOC2 was also significantly increased in patients with treatment resistance (D). Analysis in the GSE22513 data set also confirmed that SHOC2 was higher in patients did who not achieve pCR, compared with patients who achieved pCR (E). Further analysis showed that the expression of SHOC2 in BC tissue was proportional to MALAT1 expression (p < 0.001; F).
(A) The expression levels of MALAT1 were measured in human mammary epithelial cell line MCF-10A and breast cancer cell lines MCF-7, SKBR3 and MDA-MB-231. (B) The expression of MALAT1 in MCF-7-PR and MDA-MB-231-PR cells was upregulated compared with cells without paclitaxel treatment. (C) The expression of SHOC2 increased in breast cancer tissues compared with adjacent normal tissues. (D) Expression levels of SHOC2 were higher in PR patients compared with paclitaxel-sensitive patients. (E) Expression levels of SHOC2 in the GSE22513 data set. The expression was higher in patients did who not achieve a pathological complete response compared with those who achieved a pathological complete response. (F) The expression of SHOC2 in breast cancer tissue was proportional to MALAT1 expression in collected tissues.
**p < 0.01.
PR: Paclitaxel-resistant.
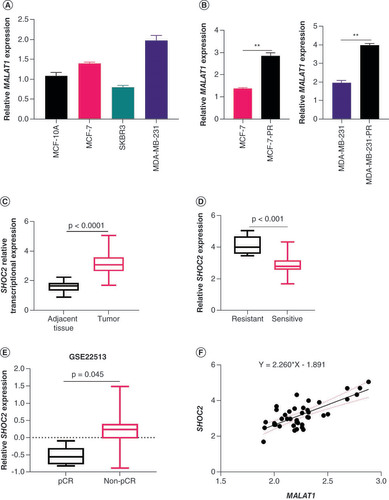
Knockdown of MALAT1 inhibited survival & migration of PR-BC cells
To further explore the roles of MALAT1 in PR-BC cells, a si- MALAT1 plasmid with the highest transfection efficiency in the pre-experiment and control plasmid NC- MALAT1 were transfected into MDA-MB-231-PR and MCF-7-PR cells. Analysis by qPCR showed that the expression of MALAT1 in the cells transfected with the si- MALAT1 plasmid was significantly downregulated (A). The IC50 of the si- MALAT1-transfected cells was lower than that of the NC- MALAT1-transfected cells under the same concentration of paclitaxel, and the survival rate of the si- MALAT1-transfected group was significantly lower than that of the NC- MALAT1 group (B & C). Transwell assay demonstrated that migration of MDA-MB-231-PR and MCF-7-PR cells also decreased with knockdown of MALAT1 (D & E).
(A) Relative expression of MALAT1 in MCF-7-PR and MDA-MB-231-PR cells after MALAT1 was interfered with si- MALAT1. (B & C) Cell viabilities measured by methyl thiazolyl tetrazolium assay. The survival rate of the si- MALAT1-transfected group was significantly lower than that of the negative control MALAT1 group in (B) MDA-MB-231-PR and (C) MCF-7-PR cells. (D & E) Interfering with MALAT1 expression decreased the invasion ability of MDA-MB-231-PR and MCF-7-PR cells.
**p < 0.01.
PR: Paclitaxel-resistant.
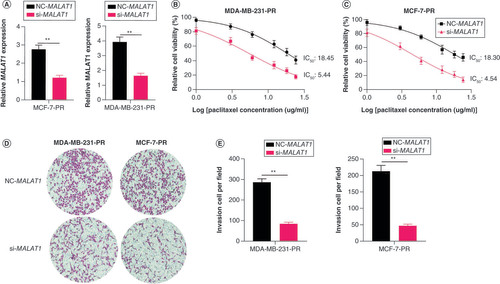
MALAT1 downregulated miR-497-5p in BC
Bioinformatics analysis demonstrated that MALAT1 does not directly regulate SHOC2, and miR-497-5p, a reported tumor suppressor gene, showed a potential binding site with MALAT1 (A). The qPCR results demonstrated that the expression of miR-497-5p was significantly decreased in PR-BC cells but increased after blocking of MALAT1 (p < 0.005; B–D). Then the luciferase construct containing the wild-type or mutated binding sequence of MALAT1 was cotransfected with the miR-497-5p mimic or miR-497-5p-NC into MDA-MB-231 cells. The results demonstrated that the luciferase activity of the wild-type MALAT1 sequence was suppressed by the miR-497-5p mimic but that the mutant MALAT1 sequence was not affected (E). The expressions of miR-497-5p were also analyzed in BC tissues (F & G) and the results showed that lower miR-497-5p expression was related to paclitaxel resistance in BC tissues.
(A) The potential binding site of miR-497-5p with MALAT1. (B) The relative expression of miR-497-5p in breast cancer cell lines MCF-7, MDA-MB-231, MCF-7-PR and MDA-MB-231-PR, measured by qPCR. The PR cells have lower miR-497-5p expression. (C) Relative miR-497-5p expression levels in MCF-7 and MDA-MB-231 cells after MALAT1 was interfered with si- MALAT1. (D) Relative miR-497-5p expression levels in MCF-7-PR and MDA-MB-231-PR cells after MALAT1 was interfered with si- MALAT1. (E) Comparison of the luciferase activity after MALAT1 was cotransfected with the miR-497-5p mimic or negative control miR (miR-497-5p NC) into cells. The luciferase activity of the wild-type MALAT1 sequence was suppressed by the miR-497-5p mimic but the mutant MALAT1 sequence was unaffected. (F) The expression levels of miR-497-5p in breast cancer tissue and adjacent normal tissues. (G) Expression of miR-497-5p was higher in breast cancer tissue than adjacent normal tissues.
**p < 0.01.
PR: Paclitaxel-resistant.
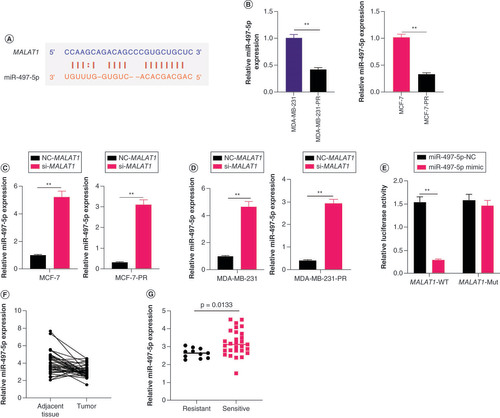
miR-497-5p decreased paclitaxel resistance of PR-BC cells by downregulating SHOC2 expression
The potential target mRNA of miR-497-5p was screened via the online bioinformatic website starBase2 (A) [Citation22]. Luciferase reporter assays indicated direct binding between miR-497-5p and SHOC2 in MDA-MB-231-PR or MCF-7-PR cells (B). We observed a negative relationship between miR-497-5p and SHOC2 expression levels in BC tissues (C). To verify the regulatory effect of miR-497-5p on SHOC2, cell viability was observed after SHOC2 was overexpressed in MDA-MB-231-PR and MCF-7-PR cells with and without miR-497-5p; the results demonstrated that overexpression of SHOC2 restored paclitaxel resistance of MCF-7-PR and MDA-MB-231-PR cells by upregulation of miR-497-5p (D & E).
(A) The potential target site for miR-497-5p in SHOC2 as predicted by starBase2. (B) The binding effect of miR-497-5p with SHOC2 verified by luciferase reporter assays. (C) The negative relationship between miR-497-5p and SHOC2 expression levels in breast cancer tissues. (D & E) Overexpression of SHOC2 restored the paclitaxel resistance of (D) MDA-MB-231-PR and (E) MCF-7-PR cells caused by upregulation of miR-497-5p (significance levels are compared with miR-497-5p mimic).
*p < 0.05; **p < 0.01.
PR: Paclitaxel-resistant.
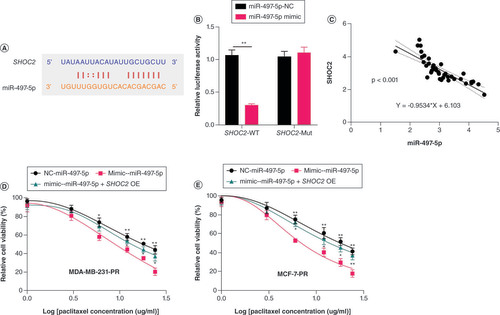
MALAT1 increased paclitaxel resistance of BC cells dependent on enhancing the SHOC2 axis
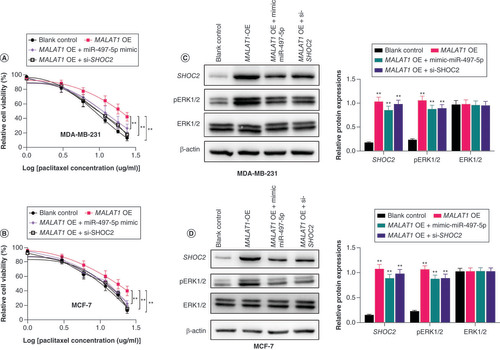
The inhibitory effects of miR-497-5p on SHOC2 in MDA-MB-231 and MCF-7 cells were confirmed (Supplementary Figure 1). We next measured the effect of MALAT1 on BC cells through upregulation of MALAT1. The results showed that upregulation of MALAT1 in MDA-MB-231 and MCF-7 cells led to increased paclitaxel resistance in both cell lines (A & B). Further upregulation of miR-497-5p almost restored the sensitivity of MDA-MB-231 and MCF-7 cells to paclitaxel (A & B). In addition, knockdown of SHOC2 partially abolished the paclitaxel resistance in both cell lines that were increased by upregulation of MALAT1 (A & B). The western blot analysis also demonstrated that upregulation of MALAT1 increased protein expression of SHOC2 and its subsequent downstream activator protein pERK1/2, while these upregulations were decreased by miR-497-5p mimic and knockdown of SHOC2 in BC cells (C & D). Taken together, these results demonstrated that MALAT1 contributed to the paclitaxel resistance of BC cells via sponging miR-497-5p to increase SHOC2 expression.
(A & B) Upregulation of MALAT1 in MDA-MB-231 and MCF-7 cells led to increased paclitaxel resistance in both cell lines. Significance levels are relative to MALAT1 overexpression in the same paclitaxel dose. (C) Representative western blot image and column diagram of relative expression of SHOC2, ERK1/2 and pERK1/2 in MDA-MB-231 cells. (D) Representative western blot image and column diagram of relative expression of SHOC2, ERK1/2 and pERK1/2 in MCF-7-PR cells. Significance is relative to the blank control.
*p < 0.05; **p < 0.01.
PR: Paclitaxel-resistant.
Discussion
BC is the most common malignancy in women worldwide and is also the leading cause of cancer-related death in women. It is estimated that there were more than 2 million new cases in 2018, which accounts for about one-quarter of the total number of cancers in women [Citation23]. The occurrence of BC is related to abnormal gene expression and reproductive factors, higher testosterone levels and the consumption of high-fat food, among other factors [Citation24]. The treatment methods include local surgery, radiotherapy, systemic chemotherapy, endocrine therapy and molecular-targeted therapy [Citation25]. Despite the progress of the treatment, a large number of patients still suffer from drug resistance, recurrence and metastasis [Citation26]. For patients with metastatic BC, the overall 5-year survival rate is less than 30% [Citation27]. Chemotherapy is one of the important treatment methods for BC. However, the emergence of drug resistance in the process of chemotherapy invalidates the treatment.
Clarifying the mechanism of BC at the molecular level could lead to improvements or adjustments of the treatment regimen, making the treatment plan more effective and ultimately improving the prognosis of patients. Whole-genome transcription sequencing analysis has shown that up to 80% of the human genome is transcribed into RNA, while only <2% of human genomic DNA is composed of protein-coding genes [Citation28]. Research has also found that noncoding RNA plays a regulatory role in physiological and pathological conditions. lncRNA and miRNA are genomic transcripts with >200 and 18–15 nucleotides, respectively; most of them have no protein-coding function. They participate in transcription, posttranscription, epigenetic gene regulation and play an important role in normal development and tumorigenesis [Citation29,Citation30]. MALAT1 is one of the highly abundant and highly conserved lncRNAs [Citation31]. Its 3′ end forms a unique triple helix structure to maintain stability and it is widely distributed in various tissues and organs of the human body [Citation32]. Current reports indicate that the roles of MALAT1 might be inconsistent in the progression of malignant BC. Kim et al. reported that in a mouse model of BC, an inactivated MALAT1 gene contributes to lung metastasis, while exogenous upregulation of MALAT1 suppresses the lung metastasis [Citation33]. Our analysis showed that MALAT1 expression increased in BC, and its expression was positively correlated with SHOC2, which is consistent with Wang’s report [Citation34]. The inconsistent role of MALAT1 in BC metastasis deserves further study; perhaps it acts as a ‘double-edged sword’ – that is, it can inhibit the progress of BC within a ‘safe’ threshold range, whereas if it is overexpressed, it will activate downstream oncogenes to promote tumor invasion. Moreover, the consistency between the expression pattern/amount of MALAT1 in model animals and humans may need further verification. In conclusion, the ‘excessive’ expression of MALAT1 in the breast may require further quantification and investigation to clarify its function.
The role of MALAT1 in chemoresistance has also been reported [Citation35,Citation36], but an association with paclitaxel resistance in BC is rarely reported. Our study showed that silencing MALAT1 could enhance the sensitivity to paclitaxel in paclitaxel-resistant cells. Furthermore, knockdown of MALAT1 could inhibit cell motility and inhibit tumor metastasis in cancer mouse models [Citation37]. This suggests that MALAT1 antisense oligonucleotides may be an effective method for the treatment of metastatic BC. However, due to the huge information network inside the body, currently, the strategy of using MALAT1 as an antimetastatic targeted therapy for BC still needs to be re-evaluated in combination with the actual situation of patients.
miRNA plays an important role not only in tumors but also in the production of tumor resistance to chemoradiotherapy. miR-497 is a highly conserved miRNA encoded by the first intron of the miR497hg gene on human chromosome 17p13.1. It is an important member of the miR-15/16/195/424/497 family and shares the same 3′ UTR seed sequence. miR-497 exists in almost all human organs and tissues, including the brain, chest, lungs, kidneys, stomach, liver and blood. It regulates the expression of related factors of a variety of signal pathways, promotes apoptosis, inhibits cell proliferation and metastasis and participates in a variety of physiological and pathological processes [Citation38]. Our study shows that miR-497-5p is a key miRNA linking MALAT1 and SHOC2 and works as a tumor suppressor gene in BC. In addition, MALAT1 sponges miR-497-5p to enhance SHOC2 expression in PR-BC cells. In a non-small-cell lung cancer analysis, Hou et al. indicated that miR-497-5p is sponged by KRT17P3 and acts as a tumor suppressor gene in non-small-cell lung cancer [Citation39]. We also demonstrate that miR-497-5p targets SHOC2 and alleviates paclitaxel resistance of BC cells.
In the 1990s researchers found a variety of scaffold proteins related to the Ras–ERK pathway in gene screening of Drosophilamelanogaster and nematodes, including kinase suppressor of Ras, MEK partner 1 and SHOC2 [Citation40,Citation41]. SHOC2 – also known as the suppressor of Ras-8, with a molecular weight of 65 kDa – is widely expressed in organisms, suggesting that it plays a key role in the Ras pathway. Signaling through the ERK1/2 pathway (the downstream kinase proteins of the Ras pathway) is fine-tuned by scaffold proteins. Reports have indicated that phosphorylation of ERK1/2 kinases promotes paclitaxel resistance [Citation42] and that SHOC2 is a critical positive modulator of ERK1/2 activity [Citation43]. This shows that the SHOC2 scaffold protein provides an active platform for the activation of the Ras–ERK signaling pathway. It locates signal molecules at specific sites in cells, provides a microenvironment for their contact with high-concentration substrates, regulates the intensity of pathway signals through positive and negative feedback signals, and protects the activity of activated signal molecules. Rodriguez et al. found that silencing SHOC2 expression can still block the activation of the ERK1/2 pathway even if Ras and RAF oncogenic mutations exist in tumor cells [Citation44]. In malignant melanoma, SHOC2 mediates the acquired resistance of tumor cells to the Raf inhibitor vemurafenib by continuously activating NRAS [Citation45]. SHOC2 also promotes the expression of glycoproteins (such as LGALS3BP) to control the effect of the ERK1/2 pathway on cell movement and adhesion [Citation43]. The high expression of these proteins in tumor tissues is associated with a poor prognosis [Citation46,Citation47]. In our data, we observed that lncRNA MALAT1 is upregulated in PR-BC cells and that overexpressed SHOC2 indicates a poor therapy response to paclitaxel in patients with BC. We speculate that the decreased response is caused by increased expression of SHOC2.
Conclusion
In summary, our research demonstrates that patients with high expression of MALAT1 and SHOC2 have a poorer response to paclitaxel. The lncRNA MALAT1 regulates SHOC2 expression and promotes the progression of BC by sponging miR-497-5p. In addition, lncRNA MALAT1 can regulate the resistance of BC cells to paclitaxel, and the mechanism may be related to the upregulation of SHOC2 expression by targeting miR-497-5p. The limitation of this study lies in the lack of validation of the Ras–ERK pathway, which is mainly regulated by SHOC2; this will be further addressed in future work. Our study provides important theoretical significance and clinical value for the targeted therapy against SHOC2.
Most of the standard treatment methods for breast cancer (BC) include chemotherapy. However, the poor response to chemotherapy of some patients, as well as emerging drug resistance, affects the efficacy of treatment.
Paclitaxel is a first-line chemotherapy agent which is widely used in BC, but drug resistance restricts its long-term usage in clinical therapy.
Recently, researchers found that epigenetic mechanisms contribute to drug resistance; many lncRNA–miRNA–mRNAs axes have been identified and confirmed to play important roles in tumor progression, including drug resistance in BC treatment.
In this study we analyzed expression levels of MALAT1 and SHOC2 in BC tissues through qPCR.
MALAT1 and SHOC2 expression levels increased in paclitaxel-resistant tumor tissues and cell lines.
Bioinformatic analyses and luciferase assay were performed to predict the miRNA interaction targets of MALAT1.
We demonstrated that lncRNA MALAT1 upregulates SHOC2 expression and promotes paclitaxel resistance in BC by sponging miR-497-5p.
The upregulation of miR-497-5p could improve the treatment response to paclitaxel.
In summary, the current study suggests that lncRNA MALAT1 might be a potential gene therapy target to increase paclitaxel sensitivity.
Author contributions
Q Li conceived and designed the study. C Shi performed the experiment and acquired analyzed data. S Ren and X Zhao were responsible for bioinformatic analysis and conducted the statistical analysis. C Shi drafted the manuscript.
Ethical conduct of research
The clinical sample used in the current study was obtained from the Second Hospital of Hebei Medical University. All patients signed informed consent for the analysis of the samples. The ethics committee of the Second Hospital of Hebei Medical University approved the protocols of the current study.
Supplemental Figure 1
Download TIFF Image (361.7 KB)Supplementary data
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Data sharing statement
The data used can be obtained from the corresponding author on reasonable request.
References
- Ferlay J , ColombetM, SoerjomataramIet al. Cancer statistics for the year 2020: an overview. Int. J. Cancer doi:10.1002/ijc.33588 (2021) ( Epub ahead of print).
- Liu HM , MaLL, LiCet al. The molecular mechanism of chronic stress affecting the occurrence and development of breast cancer and potential drug therapy. Transl. Oncol.15(1), 101281 (2022).
- Zhang T , ZhouH, WangKet al. Role, molecular mechanism and the potential target of breast cancer stem cells in breast cancer development. Biomed. Pharmacother.147, 112616 (2022).
- Guo YJ , PanWW, LiuSB, ShenZF, XuY, HuLL. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med.19(3), 1997–2007 (2020).
- Lavoie H , GagnonJ, TherrienM. ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol.21(10), 607–632 (2020).
- Stuart DD , SellersWR. Targeting RAF–MEK–ERK kinase–scaffold interactions in cancer. Nat. Med.19(5), 538–540 (2013).
- Frodyma D , NeilsenB, Costanzo-GarveyD, FisherK, LewisR. Coordinating ERK signaling via the molecular scaffold Kinase Suppressor of Ras. F1000Res6, 1621 (2017).
- Jang H , StevensP, GaoT, GalperinE. The leucine-rich repeat signaling scaffolds Shoc2 and Erbin: cellular mechanism and role in disease. FEBS J288(3), 721–739 (2021).
- Jones GG , DelRio IB, SariSet al. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat. Commun.10(1), 2532 (2019).
- Geng W , DongK, PuQ, LvY, GaoH. SHOC2 is associated with the survival of breast cancer cells and has prognostic value for patients with breast cancer. Mol. Med. Rep.21(2), 867–875 (2020).
- Ji P , DiederichsS, WangWet al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene22(39), 8031–8041 (2003).
- Qiao FH , TuM, LiuHY. Role of MALAT1 in gynecological cancers: pathologic and therapeutic aspects. Oncol. Lett.21(4), 333 (2021).
- Yoshimoto R , MayedaA, YoshidaM, NakagawaS. MALAT1 long non-coding RNA in cancer. Biochim. Biophys. Acta1859(1), 192–199 (2016).
- Arun G , SpectorDL. MALAT1 long non-coding RNA and breast cancer. RNA Biol.16(6), 860–863 (2019).
- Huang XJ , XiaY, HeGFet al. MALAT1 promotes angiogenesis of breast cancer. Oncol. Rep.40(5), 2683–2689 (2018).
- Wang Z , WangX, ZhangTet al. lncRNA MALAT1 promotes gastric cancer progression via inhibiting autophagic flux and inducing fibroblast activation. Cell Death Dis.12(4), 368 (2021).
- Jadaliha M , ZongX, MalakarPet al. Functional and prognostic significance of long non-coding RNA MALAT1 as a metastasis driver in ER negative lymph node negative breast cancer. Oncotarget7(26), 40418–40436 (2016).
- Liu J , ZhouY, ShiZet al. microRNA-497 modulates breast cancer cell proliferation, invasion, and survival by targeting SMAD7. DNA Cell Biol.35(9), 521–529 (2016).
- Ma Y , YanJ, QiH, MaL. Effects of miR-497-5p on proliferation and apoptosis of paclitaxel resistant breast cancer cells. Chin. J. Clin. Pharmacol.37(20), 2763–2765,2769 (2021).
- Eisenhauer EA , TherasseP, BogaertsJet al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer45(2), 228–247 (2009).
- Invitrogen . Lipofectamine® product information sheet. http://tools.thermofisher.com/content/sfs/manuals/Lipofectamine_2000_Reag_protocol.pdf
- Li JH , LiuS, ZhouH, QULH, YangJH.:starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data.Nucleic Acids Res42(Database issue), D92–97 (2014)
- Bray F , FerlayJ, SoerjomataramI, SiegelRL, TorreLA, JemalA. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.68(6), 394–424 (2018).
- Ataollahi MR , SharifiJ, PaknahadMR, PaknahadA. Breast cancer and associated factors: a review. J. Med. Life8(Spec Iss 4), 6–11 (2015).
- Lukasiewicz S , CzeczelewskiM, FormaA, BajJ, SitarzR, StanislawekA. Breast cancer – epidemiology, risk factors, classification, prognostic markers, and current treatment strategies – an updated review. Cancers (Basel)13(17), 4287 (2021).
- Gote V , NookalaAR, BollaPK, PalD. Drug resistance in metastatic breast cancer: tumor targeted nanomedicine to the rescue. Int. J. Mol. Sci.22(9), 4673 (2021).
- Othman A , WinogradzkiM, LeeL, TandonM, BlankA, PratapJ. Bone metastatic breast cancer: advances in cell signaling and autophagy related mechanisms. Cancers (Basel)13(17), 4310 (2021).
- ENCODE Project Consortium , BirneyE, StamatoyannopoulosJAet al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature447(7146), 799–816 (2007).
- Kazimierczyk M , KasprowiczMK, KasprzykME, WrzesinskiJ. Human long noncoding RNA interactome: detection, characterization and function. Int. J. Mol. Sci.21(3), 1027 (2020).
- Tsai MC , SpitaleRC, ChangHY. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res.71(1), 3–7 (2011).
- Fu S , WangY, LiH, ChenL, LiuQ. Regulatory networks of lncRNA MALAT-1 in cancer. Cancer Manag. Res.12, 10181–10198 (2020).
- Zhang X , HamblinMH, YinKJ. The long noncoding RNA MALAT1: its physiological and pathophysiological functions. RNA Biol.14(12), 1705–1714 (2017).
- Kim J , PiaoHL, KimBJet al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet.50(12), 1705–1715 (2018).
- Wang N , CaoS, WangX, ZhangL, YuanH, MaX. lncRNA MALAT1/miR26a/26b/ST8SIA4 axis mediates cell invasion and migration in breast cancer cell lines. Oncol Rep46(2), 181 (2021).
- Ren S , LiuY, XuWet al. Long noncoding RNA MALAT-1 is a new potential therapeutic target for castration resistant prostate cancer. J. Urol.190(6), 2278–2287 (2013).
- Liu C , HanX, LiBet al. MALAT-1 is associated with the doxorubicin resistance in U-2OS osteosarcoma cells. Cancer Manag. Res.13, 6879–6889 (2021).
- Chen D , XuT, ChangHHet al. The role of MALAT1 in cancer. J. Cancer Sci. Clin. Ther.3, 5–27 (2019).
- Yang G , XiongG, CaoZet al. miR-497 expression, function and clinical application in cancer. Oncotarget7(34), 55900–55911 (2016).
- Hou Z , WangY, XiaN, LvT, YuanX, SongY. Pseudogene KRT17P3 drives cisplatin resistance of human NSCLC cells by modulating miR-497-5p/mTOR. Cancer Sci112(1), 275–286 (2021).
- Wassarman DA , TherrienM, RubinGM . The Ras signaling pathway in Drosophila. Curr Opin Genet Dev5(1), 44–50 (2016).
- Lavoie H , TherrienM.Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol16(5), 281–298 (2015).
- Huang C , ZhangX, JiangL, ZhangL, XiangM, RenH. FoxM1 induced paclitaxel resistance via activation of the FoxM1/PHB1/RAF–MEK–ERK pathway and enhancement of the ABCA2 transporter. Mol. Ther. Oncolytics14, 196–212 (2019).
- Jeoung M , JangER, LiuJet al. Shoc2-tranduced ERK1/2 motility signals – novel insights from functional genomics. Cell. Signal.28(5), 448–459 (2016).
- Rodriguez-Viciana P , Oses-PrietoJ, BurlingameA, FriedM, McCormickF. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol. Cell22(2), 217–230 (2006).
- Kaplan FM , KugelCH3rd, DadpeyN, ShaoY, AbelEV, AplinAE. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J. Biol. Chem.287(50), 41797–41807 (2012).
- Zhao S , QiuZX, ZhangL, LiWM.Prognostic values of ERK1/2 and p-ERK1/2 expressions for poor survival in non-small cell lung cancer. Tumour Biol36(6), 4143–4150 (2015).
- Stampolidis P , UllrichA, IacobelliS. LGALS3BP, lectin galactoside-binding soluble 3 binding protein, promotes oncogenic cellular events impeded by antibody intervention. Oncogene.34(1), 39–52 (2015).