
Abstract
Aim: To evaluate pharmacokinetic interactions of atogepant with sumatriptan, an open-label, randomized, crossover study was conducted. Patients & methods: Thirty healthy adults received atogepant 60 mg, sumatriptan 100 mg, or coadministered drugs. Primary end point was geometric mean ratios (GMRs) and 90% CIs of interventions for area under the plasma concentration–time curve from time 0 to t (AUC0-t) or infinity (AUC0-∞) and peak plasma concentration (Cmax). Results: Atogepant GMRs for AUC0-t and AUC0-∞ versus with sumatriptan were within 90% CI 0.80–1.25, indicating no interaction; atogepant Cmax was reduced by 22% (GMR: 0.78; 90% CI: 0.69–0.89) with sumatriptan. Sumatriptan GMRs for AUC0-t, AUC0-∞ and Cmax versus with atogepant were within 90% CI 0.80–1.25. Conclusion: Atogepant with sumatriptan had no clinically relevant pharmacokinetic interactions.
Lay abstract
We evaluated the possibility of interactions between atogepant, a new drug for the prevention of migraine, and sumatriptan, a commonly used drug to treat active migraines. A group of 30 healthy adults received atogepant alone, sumatriptan alone or the two drugs taken together, and we measured how the body absorbed, distributed and got rid of the two drugs when given together compared with each drug given alone. There was no indication of any clinically important interactions between atogepant and sumatriptan.
Migraine is a neurologic disease characterized by recurrent attacks of severe headache pain, which are commonly accompanied by nausea, phonophobia and photophobia [Citation1]. The high global prevalence of migraine (~1.3 billion individuals worldwide in 2017) [Citation2] and its incapacitating symptoms [Citation3] contribute to a high global and individual burden of disease [Citation4,Citation5].
Acute treatment options for migraine attacks include nonprescription and prescription medications [Citation6,Citation7]. Triptans – serotonin receptor agonists – are a frequently used prescription acute treatment option for migraine attacks [Citation7]. Sumatriptan is the most commonly used triptan for acute treatment of migraine [Citation8]. Preventive medications for migraine have historically included prescription medications that are not migraine-specific (beta-blockers, antiepileptics, calcium channel blockers, antidepressants) [Citation6]. Recently, calcitonin gene-related peptide (CGRP) and its receptors have been implicated in migraine pathophysiology and are now targets for acute and preventive treatment of migraine attacks [Citation9–12].
Atogepant is a potent, selective, competitive, oral small-molecule CGRP receptor antagonist that has recently received US FDA approval for the preventive treatment of episodic migraine in adults [Citation13]. In a 12-week, randomized, placebo-controlled, multidose, Phase IIb/III trial, atogepant had a favorable safety profile and demonstrated efficacy at all doses compared with placebo [Citation14]. It is likely that atogepant will be used concomitantly with acute treatments for migraine attacks, including triptans in select individuals [Citation15]. Specifically, those with severe or frequent migraine attacks, or poor response to acute treatments alone, may require combination therapy to reduce attack symptoms as well as the frequency of attacks [Citation16]. Considering that both preventive and acute treatments can be administered in certain populations, there is a potential for drug–drug interactions (DDIs) with combination therapies [Citation17]. Investigating DDIs and the pharmacokinetic (PK) parameters of coadministered medications while taking into account their mechanisms of action can better inform prescribers and minimize any potential safety issues. Furthermore, individual patient characteristics should be considered when choosing the safest combination therapy option [Citation16–19].
Although atogepant and sumatriptan are metabolized by different isozymes in the liver (CYP3A4 for atogepant and MAO-A for sumatriptan), there is potential to increase sumatriptan exposure because atogepant is an inhibitor of the hepatic organic cation transporter OCT1, which mediates hepatic uptake of sumatriptan [Citation20]. Therefore the primary objective of this study was to evaluate the potential for PK interactions between atogepant and sumatriptan in healthy adult participants. The secondary objectives were to evaluate safety and tolerability profiles of atogepant and sumatriptan when administered alone and when coadministered in healthy participants.
Patients & methods
Study design
This open-label, randomized, three-way crossover, Phase I trial was designed to assess the potential of a PK interaction between atogepant and sumatriptan in healthy adult participants. The study was conducted at a single site in the USA (Miami, FL). Eligible participants were randomly assigned based on a randomization code generated by the sponsor to one of six intervention sequences to receive single oral 60 mg atogepant dose (1 × 60 mg tablet; Allergan, Clonshaugh, Ireland), single oral 100 mg sumatriptan dose (1 × 100 mg tablet; Imitrex tablets, GlaxoSmithKline, NC, USA) or 60 mg atogepant coadministered with 100 mg sumatriptan under fasted conditions after a screening period of up to 21 days (Supplementary Table 1). Interventions were given on days 1, 8 and 15 with a 7-day washout between interventions. The duration of each participant’s participation in the study (post-screening) was up to 45 days, including a 30-day safety follow-up visit following the last dose of study treatment (day 15).
This study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation E6 guideline for Good Clinical Practice. The study protocol was approved by IntegReview IRB (TX, USA). All participants provided written informed consent prior to initiation of study-specific procedures. The authors had full access to all data from the trial.
Participants
Participants were healthy adults aged 18–45 years who were nonsmokers (never smoked, or had not smoked or used nicotine products within the previous 2 years) with a BMI of ≥18 and ≤30 kg/m2 and sitting pulse rate between ≥45 and ≤100 beats per minute. Men and women of childbearing potential agreed to use an effective method of contraception. Women with a negative result from a serum pregnancy test at screening and a negative result from a serum or urine pregnancy test on day -1 were included. Exclusion criteria included a known hypersensitivity to CGRP receptor antagonists or sumatriptan; sitting systolic blood pressure ≥140 or ≤90 mmHg; sitting diastolic blood pressure ≥90 or ≤50 mmHg; abnormal ECG results considered clinically relevant by the investigator; or QT prolongation (QTcF ≥450 ms for men; ≥470 ms for women) at screening; and any clinical condition or previous surgery that may have affected the absorption, distribution, biotransformation or excretion of atogepant or sumatriptan.
Participants were prohibited from taking any medications (including over-the-counter medications) for at least 14 days or five half-lives (whichever was longer) before the first study dose and during the course of the study, or hormonal drug products and St John’s wort within 30 days before study intervention administration. These medications, as well as vitamin preparations, herbal medicines and dietary or herbal supplements, were prohibited until the completion of the follow-up visit. Additionally, participants could not consume or ingest foods or beverages that could affect drug-metabolizing enzymes and transporters, including foods containing quinine, poppy seeds, grapefruit, vegetables from the mustard green family or charbroiled meats within 14 days before dosing through the end of dosing. Participants were required to abstain from alcohol from 72 h before study intervention and throughout the study. The ingestion of caffeine- or xanthine-containing products was prohibited for 48 h before study intervention administration and until after PK sampling (day 17).
Study procedures
Participants were admitted to the study center on days -1, 7 and 14, and stayed the nights of days -1, 1, 2, 7, 8, 9, 14, 15 and 16. Study interventions were administered under fasting conditions (10-h overnight fast) with 240 ml of water on days 1, 8 and 15. Additional water was restricted 1 h before and after study intervention administration. Participants continued to fast and remain seated upright and awake for 4 h after dosing. Participants were released from the Clinical Research Unit on the mornings of days 3, 10 and 17. Meals were provided at appropriate times each day.
PK assessments
Blood samples for the determination of plasma atogepant and sumatriptan concentrations were collected on days 1, 8 and 15 at 0 (predose), 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36 and 48 h after each dose. Collected blood for PK analysis was centrifuged at 2500 × g for 10 min at 4°C within 30 min of collection. Plasma samples were flash-frozen and stored at -70°C or colder. Drug concentrations in plasma samples were analyzed by Altasciences (Laval, Quebec, Canada) using validated liquid chromatography with tandem mass spectrometry detection methods. The lower limit of quantitation was 1.00 ng/ml for both atogepant and sumatriptan.
Safety assessments
Safety and tolerability were monitored by physical examinations, vital signs, clinical laboratory tests and 12-lead ECGs. Participants were monitored for adverse events (AEs), including treatment-emergent AEs (TEAEs) and serious AEs (SAEs), throughout the study. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 21.1.
Statistical analysis
A sample size of 28 participants was estimated to provide at least 90% power to show that the 90% CIs for the geometric mean ratios (GMRs) for mean PK parameters of peak plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) of atogepant with and without coadministration of sumatriptan would be within 80–125%. This estimate was based on the assumptions that the within-participant coefficient of variation was 25% for atogepant Cmax and AUC, and the true ratio of test (drug in combination) to reference (drug alone) geometric means was 1. This sample size was also estimated to be sufficient to ensure at least 90% power for the comparison of sumatriptan PK parameters, based on the assumption that the within-participant coefficients of variation for sumatriptan Cmax and AUC were 23 and 17%, respectively.
All standard noncompartmental PK parameters were calculated using WinNonlin Phoenix version 8.0 (Certara, NJ, USA). Concentrations below the limit of quantification were set to zero for the analyses. PK parameters that were derived from plasma concentrations included Cmax, AUC from time 0 to time t (AUC0-t) and from time 0 to infinity (AUC0-∞), time to Cmax (Tmax), apparent terminal elimination half-life (t1/2), apparent total body clearance of drug from plasma after extravascular administration (CL/F) and apparent volume of distribution during the terminal phase after extravascular administration (VZ/F). The PK parameters t1/2, AUC0-∞, CL/F and Vz/F were not calculated if R2 was <0.80 for the regression analysis on the terminal linear phase of semilogarithmic plots of individual atogepant concentration–time data in the elimination phase.
A linear mixed-effects model was used to compare log-transformed PK parameters (Cmax, AUC0-t, AUC0-∞) of atogepant and sumatriptan when coadministered versus when each study intervention was administered alone. The intervention, period and sequence were fixed effects in this model, while participant within sequence was a random effect. The 90% CIs to evaluate DDIs were two-sided for the GMRs between the test intervention (coadministration of atogepant with sumatriptan) and reference intervention (atogepant or sumatriptan alone). In keeping with FDA guidance for DDI studies, no significant effect between the test and reference interventions was concluded if the 90% CIs were within 80–125% [Citation21].
All participants who received at least one study dose were included in the safety analyses. The atogepant and sumatriptan PK populations comprised all participants who had evaluable PK parameters for atogepant and sumatriptan, respectively.
Results
Participants
This study was conducted between 9 April 2019 and 30 May 2019. A total of 30 participants were enrolled and randomized to an intervention sequence; of these, 27 (90.0%) received all doses and completed the study. Three participants were withdrawn from the study by the investigators after period 1 due to positive urine drug screens on day 7 (). Of the participants who were withdrawn, one received single-dose atogepant and two received single-dose sumatriptan. The mean age of the safety population was 32.9 years (range: 19–44), 56.7% of participants were male, and most (70.0%) were white (). The safety population included 30 participants, and the PK populations included 28, 29 and 27 participants for atogepant, sumatriptan and coadministration, respectively.
PK: Pharmacokinetics.
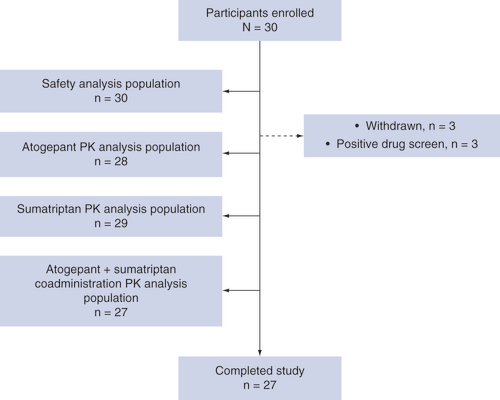
Table 1. Demographics and baseline characteristics (safety population).
Atogepant & sumatriptan pharmacokinetics
The mean plasma concentrations of atogepant following the administration of atogepant alone or in combination with sumatriptan are shown in . Coadministration of atogepant and sumatriptan delayed the median atogepant Tmax by 1.5 h but had minimal impact on the mean apparent terminal t1/2 compared with administration of atogepant alone ().
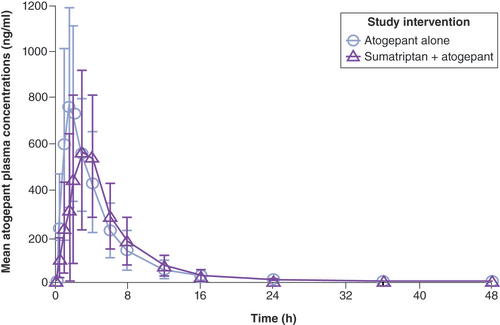
Table 2. Pharmacokinetic parameters of atogepant following single-dose oral administration of atogepant alone or when coadministered with sumatriptan.
The mean plasma concentrations of sumatriptan following the administration of sumatriptan alone or in combination with atogepant are shown in . For sumatriptan, the median Tmax and mean apparent terminal t1/2 were similar when coadministered with atogepant or administered alone ().
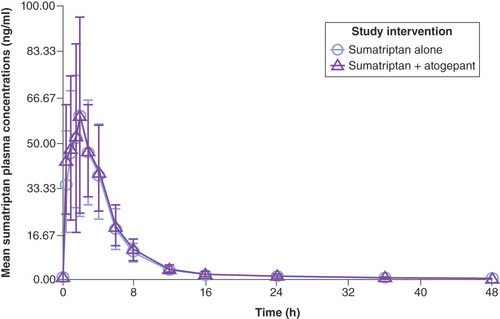
Table 3. Pharmacokinetic parameters of sumatriptan following single-dose administration of sumatriptan alone or when coadministered with atogepant.
The GMRs and their 90% CIs for the comparison of Cmax and AUC parameters of atogepant and sumatriptan coadministered versus atogepant administered alone are summarized in . The 90% CIs for the GMRs for atogepant coadministered with sumatriptan versus administered alone were within the range 0.80–1.25 for AUC0-t and AUC0-∞; however, the Cmax of atogepant was reduced by 22% (GMR: 0.78; 90% CI: 0.69–0.89). The GMR 90% CI for Cmax was outside the range 0.85–1.25, suggesting a statistically significant DDI.
Table 4. Statistical analysis of pharmacokinetic parameters: comparison of atogepant administered alone and when coadministered with sumatriptan and comparison of sumatriptan administered alone and when coadministered with atogepant.
The GMRs and 90% CIs for the comparison of Cmax and AUC parameters of sumatriptan and atogepant coadministered versus sumatriptan administered alone are summarized in . The 90% CIs were within the range 0.80–1.25 for AUC0-t, AUC0-∞ and Cmax for sumatriptan when coadministered with atogepant versus administration alone, indicating no DDI.
Safety
In total, four participants (13%) reported a TEAE. All TEAEs were mild in severity, and no TEAEs were reported when atogepant was administered alone. Two participants reported somnolence after taking sumatriptan alone, and two reported events of nausea and headache after taking atogepant coadministered with sumatriptan. No SAEs or deaths occurred during the study, and no participants discontinued because of a TEAE. Mean changes from baseline in clinical laboratory parameters and vital signs were small, not considered clinically meaningful, and displayed no clinically relevant trends or patterns. No participant met the criteria for a potential Hy’s Law case (alanine aminotransferase or aspartate aminotransferase greater than or equal to three-times the upper limit of normal [ULN], total bilirubin greater than or equal to two times the ULN, and alkaline phosphatase less than two times the ULN) or had concurrent elevations of liver laboratory parameters during the intervention period [Citation22]. Physical examinations, vital sign evaluations and ECG assessments revealed no clinically relevant findings.
Discussion
The results of this DDI study which was designed to evaluate potential interactions between atogepant, a gepant for preventive treatment of migraine, with sumatriptan, a commonly used medication for acute treatment of migraine attacks, suggest no clinically relevant PK interaction. When coadministered with sumatriptan, AUC0-t and AUC0-∞ for atogepant were similar, while Cmax was lower. The reduction of 22% in the mean atogepant Cmax was accompanied by a delay of 1.5 h in median Tmax. Administration of sumatriptan to healthy individuals 30 min prior to a test meal has been shown to delay gastric emptying for 4 h after eating [Citation23]. The reduction in Cmax and delay in Tmax observed in the present study could be attributed to the effects of sumatriptan on gastric emptying when atogepant was coadministered with sumatriptan. Changes in atogepant Cmax when coadministered with sumatriptan are expected to have minimal clinical relevance because the overall systemic exposure of atogepant (AUC) was within the prespecified no-effect boundaries of 80–125%. Atogepant has been found to be efficacious with doses ranging from 10 mg once daily to 60 mg twice daily, and safety has been demonstrated up to a single supratherapeutic dose of 300 mg [Citation14]. The results for atogepant Cmax and AUC in this study are consistent with other atogepant studies. From a study of single-dose atogepant 60 mg in healthy adults, the typical Cmax was approximately 589–788 ng/ml, with an AUC approximately 2757–3628 ng·h/ml [Citation24,Citation25]. Studies of multiple-dose atogepant have reported a mean Cmax of 1165 ng/ml with once-daily 60 mg administration for 17 days [Citation26], and 1865 ng/ml following once-daily 170 mg administration for 28 days [Citation27].
The Cmax and AUC values of sumatriptan increased dose-proportionally and linearly in studies in healthy adults [Citation28]. Sumatriptan Tmax showed a trend upward with increasing dose [Citation28]. In the present study, PK parameters for sumatriptan were similar to values reported for sumatriptan 100 mg in other studies, and relatively consistent with values reported for sumatriptan administered orally at other doses [Citation28–34]. Sumatriptan Cmax and AUC values were unchanged when sumatriptan was coadministered with atogepant.
Coadministration of atogepant with sumatriptan was well tolerated in this study, with no SAEs, deaths or premature discontinuations due to a TEAE. Overall, the incidence of TEAEs was low, and all were mild in severity. No participants reported a TEAE following administration with atogepant. Although these results are for a single dose of atogepant, results from the Phase III trial of atogepant in participants with migraine, with doses up to 60 mg once daily administered for 12 weeks, suggest that atogepant is safe and generally well tolerated [Citation35]. In the Phase III trial, the most commonly reported AEs were constipation (7–8% across doses vs 0.5% with placebo) and nausea (4–6% across doses vs 2% with placebo); none were considered serious. Together, these studies suggest that atogepant is well tolerated and safe when administered alone or when coadministered with a triptan for the acute treatment of migraine attacks.
A strength of the study was the use of a crossover design, which allowed participants to serve as their own controls. The study also benefited from the short half-lives of the medications (~11 h for atogepant and 2.5 h for sumatriptan), which allowed for a relatively short (7-day) washout between intervention administrations. This feature of the study design may have contributed to the large number of participants (90%) who completed the study.
The study had several limitations. The study was powered to detect statistical differences in PK parameters but was not powered to thoroughly assess the safety of atogepant when coadministered with sumatriptan. The study enrolled healthy adults, whereas atogepant is designed for the preventive treatment of migraine, and the results of this study may not be generalizable to the target patient population (i.e., those with chronic migraine). Additionally, migraine is associated with significant comorbidities, including vascular, neurologic, psychiatric and nonmigraine pain conditions [Citation36] which were not represented in this Phase I trial of healthy adult participants.
Conclusion
Atogepant AUC was unchanged and Cmax was reduced by 22% when coadministered with sumatriptan; this change in Cmax is expected to have minimal clinical relevance. The PK profile of sumatriptan was unchanged when coadministered with atogepant. Coadministration of atogepant and sumatriptan is safe and well tolerated, with no safety concerns identified.
Atogepant is a potent, selective, competitive, oral, small-molecule calcitonin gene-related peptide receptor antagonist recently approved for preventive treatment of episodic migraine in adults; sumatriptan is the most commonly used triptan for acute treatment of migraine.
The present open-label, randomized, three-way crossover trial was designed to assess the potential pharmacokinetic interactions between atogepant and sumatriptan in healthy adult participants.
When coadministered with sumatriptan, area under the plasma concentration–time curve from time 0 to time t (AUC0-t) and to infinity (AUC0-∞) for atogepant were similar, while peak plasma concentration (Cmax) was lower. The slight decrease in atogepant Cmax when coadministered with sumatriptan is expected to have minimal clinical relevance because the overall systemic exposure of atogepant (AUC) was within the prespecified no-effect boundaries of 80–125%.
Sumatriptan Cmax and AUC values were unchanged when sumatriptan was coadministered with atogepant.
Coadministration of atogepant with sumatriptan was well tolerated.
Author contributions
Study concept and design: R Boinpally, A Jakate, A Periclou. Acquisition of data: A Jakate, M Butler. Analysis and interpretation of data: all authors. Revising the manuscript for intellectual content: all authors. Final approval of the completed manuscript: all authors.
Ethical conduct of research
This study was conducted in accordance with the principles of the Declaration of Helsinki and the ICH E6 guideline for Good Clinical Practice. The study protocol was approved by IntegReview IRB (TX, USA). All participants provided written informed consent prior to initiation of study-specific procedures.
Supplemental document 1
Download MS Word (20.8 KB)Acknowledgments
The authors thank the study participants, as well as the site/clinical research unit personnel, including clinical and data management staff and the biostatisticians.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2217/pmt-2021-0073
Financial & competing interest disclosure
This study was sponsored by Allergan (prior to its acquisition by AbbVie). R Boinpally is an employee of AbbVie and may hold AbbVie stock. A Jakate, M Butler and A Periclou were employees of AbbVie at the time of the study and may hold AbbVie stock. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing and editorial assistance were provided to the authors by L Feder of Peloton Advantage, an OPEN Health company (Parsippany, NJ, USA) and were funded by AbbVie.
Data availability
The authors certify that this manuscript reports original clinical trial data. AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Additional information
Funding
References
- Pietrobon D , MoskowitzMA. Pathophysiology of migraine. Annu. Rev. Physiol.75, 365–391 (2013).
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet392(10159), 1789–1858 (2018).
- Lipton RB , BigalME , DiamondM , FreitagF , ReedML , StewartWF. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology68(5), 343–349 (2007).
- GBD 2016 Headache Collaborators . Global, regional, and national burden of migraine and tension-type headache, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol.17(11), 954–976 (2018).
- Lipton RB , ManackAdams A , BuseDC , FanningKM , ReedML. A Comparison of the Chronic Migraine Epidemiology and Outcomes (CaMEO) study and American Migraine Prevalence and Prevention (AMPP) study: demographics and headache-related disability. Headache56(8), 1280–1289 (2016).
- Lew C, Punnapuzha S. Migraine medications. StatPearls Publishing LLC, FL, USA (2020).
- Becker WJ . Acute migraine treatment in adults. Headache55(6), 778–793 (2015).
- Ferrari MD , RoonKI , LiptonRB , GoadsbyPJ. Oral triptans (serotonin 5-HT1B/1D agonists) in acute migraine treatment: a meta-analysis of 53 trials. Lancet358(9294), 1668–1675 (2001).
- Iyengar S , OssipovMH , JohnsonKW. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain158(4), 543–559 (2017).
- Edvinsson L . CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br. J. Clin. Pharmacol.80(2), 193–199 (2015).
- Charles A , Pozo-RosichP. Targeting calcitonin gene-related peptide: a new era in migraine therapy. Lancet394(10210), 1765–1774 (2019).
- Dodick DW . CGRP ligand and receptor monoclonal antibodies for migraine prevention: evidence review and clinical implications. Cephalalgia39(3), 445–458 (2019).
- Source: AbbVie . FDA approves QULIPTA (atogepant), the first and only oral CGRP receptor antagonist specifically developed for the preventive treatment of migraine (2021). https://news.abbvie.com/news/press-releases/fda-approves-qulipta-atogepant-first-and-only-oral-cgrp-receptor-antagonist-specifically-developed-for-preventive-treatment-migraine.htm
- Goadsby PJ , DodickDW , AilaniJet al. Safety, tolerability, and efficacy of orally administered atogepant for the prevention of episodic migraine in adults: a double-blind, randomised Phase 2b/3 trial. Lancet Neurol.19(9), 727–737 (2020).
- Martelletti P . Combination therapy in migraine: asset or issue?Expert Rev. Neurother.20(10), 995–996 (2020).
- Borro M , GuglielmettiM , SimmacoM , MartellettiP , GentileG. The future of pharmacogenetics in the treatment of migraine. Pharmacogenomics20(16), 1159–1173 (2019).
- Lionetto L , BorroM , CurtoMet al. Choosing the safest acute therapy during chronic migraine prophylactic treatment: pharmacokinetic and pharmacodynamic considerations. Expert. Opin. Drug. Metab. Toxicol.12(4), 399–406 (2016).
- Martelletti P , CipollaF , CapiM , CurtoM , LionettoL. Atogepant. Calcitonin gene-related peptide (CGRP) receptor antagonist, preventive treatment of migraine. Drugs Future45(5), 285 (2020).
- Martelletti P , LucianiM , SpuntarelliV , BentivegnaE. Deprescribing in migraine. Expert Opin. Drug Saf.20(6), 623–625 (2021).
- Matthaei J , KuronD , FaltracoFet al. OCT1 mediates hepatic uptake of sumatriptan and loss-of-function OCT1 polymorphisms affect sumatriptan pharmacokinetics. Clin. Pharmacol. Ther.99(6), 633–641 (2016).
- US Food & Drug Administration . Clinical drug interaction studies – study design, data analysis, and clinical implications guidance for industry. (2017).
- US Food & Drug Administration . Guidance for industry drug-induced liver injury: premarketing clinical evaluation (2009). www.fda.gov/regulatory-information/search-fda-guidance-documents/drug-induced-liver-injury-premarketing-clinical-evaluation
- Sakamoto Y , SekinoY , YamadaEet al. Effect of sumatriptan on gastric emptying: a crossover study using the BreathID system. World J. Gastroenterol.18(26), 3415–3419 (2012).
- Boinpally R , JakateA , ButlerM , BorbridgeL , PericlouA. Single-dose pharmacokinetics and safety of atogepant in adults with hepatic impairment: results from an open-label, phase I trial. Clin. Pharmacol. Drug Development doi:10.1002/cpdd.916 (2021).
- Boinpally R , SpaventaJ , ChenK , ButlerM. Evaluation of the pharmacokinetic interaction and safety of atogepant co-administered with acetaminophen or naproxen in healthy participants: a randomized trial. Clin. Drug Investig.41(6), 557–567 (2021).
- Ankrom W , XuJ , ValleeMHet al. Atogepant has no clinically relevant effects on the pharmacokinetics of an ethinyl estradiol/levonorgestrel oral contraceptive in healthy female participants. J. Clin. Pharmacol.60(9), 1157–1165 (2020).
- Min KC , KraftWK , BondiskeyPet al. Atogepant is not associated with clinically meaningful alanine aminotransferase elevations in healthly adults. Clin. Transl. Sci.14(2), 599–605 (2021).
- Scott AK . Sumatriptan clinical pharmacokinetics. Clin. Pharmacokinet.27(5), 337–344 (1994).
- Imitrex [package insert]. GlaxoSmithKline, NC, USA (2017).
- Scott AK , WalleyT , BreckenridgeAM , LaceyLF , FowlerPA. Lack of an interaction between propranolol and sumatriptan. Br. J. Clin. Pharmacol.32(5), 581–584 (1991).
- Van Hecken AM , DepréM , DeSchepper PJ , FowlerPA , LaceyLF , DurhamJM. Lack of effect of flunarizine on the pharmacokinetics and pharmacodynamics of sumatriptan in healthy volunteers. Br. J. Clin. Pharmacol.34(1), 82–84 (1992).
- Duquesnoy C , MametJP , SumnerD , FuseauE. Comparative clinical pharmacokinetics of single doses of sumatriptan following subcutaneous, oral, rectal and intranasal administration. Eur. J. Pharm. Sci.6(2), 99–104 (1998).
- Moore KH , LeesePT , McNealSet al. The pharmacokinetics of sumatriptan when administered with clarithromycin in healthy volunteers. Clin. Ther.24(4), 583–594 (2002).
- Moore KH , McNealS , BrittoMR , ByeC , SaleM , RichardsonMS. The pharmacokinetics of sumatriptan when administered with norethindrone 1 mg/ethinyl estradiol 0.035 mg in healthy volunteers. Clin. Ther.24(11), 1887–1901 (2002).
- Ailani J , LiptonRB , GoadsbyPJet al. Atogepant for the preventive treatment of migraine.. N. Engl. J. Med.385(8), 695–706 (2021).
- Burch RC , BuseDC , LiptonRB. Migraine: epidemiology, burden, and comorbidity. Neurol. Clin.37(4), 631–649 (2019).