Abstract
Despite worldwide efforts biomarker discovery by plasma proteomics was not successful so far. Several reasons for this failure are obvious. Mainly, proteome diversity is remarkable between different individuals and is caused by genetic, environmental and life style parameters. To recognize disease related proteins that could serve as potential biomarkers is only feasible by investigating a non realizable large number of patients. Furthermore, plasma proteomics comprises enormous technical hurdles for quantitative analysis. High reproducibility of blood sampling in clinical routine is hard to achieve. Quantitative proteome analysis has to struggle with the complexity of millions of protein species comprising typical plasma proteins, cellular leakage proteins and antibodies and concentration differences of more than 1011 between high and low abundant proteins. Therefore, no successful quantitative and comprehensive plasma proteome analysis is reported so far.
A novel proteomics strategy is proposed for biomarker discovery in plasma. Instead of comparing the plasma proteome of different individuals it is recommended to analyze the proteomes of different time points of a single individual during the development of a disease. This strategy is realized by the use of plasma of the Bavarian Red Cross Blood Bank, were three million samples are stored under standardized conditions. To achieve reliable data the isotope coded protein labelling proteomics technology was used.
Key Words::
Introduction
So far, only very few biomarkers have been discovered using proteomics techniques. Cellular proteomics, analyzing the protein pattern of relevant tissues from diseased patients compared with healthy control persons, is state of the art. This strategy usually requires invasive sample collection and enrichment of the diseased material by e.g. microdissection. After quite sophisticated quantitative analyses using DIGE or mass spectrometry based techniques numerous biomarker candidates usually can be found. However, almost all of them do not survive the validation phase. In contrast, proteomics starting from blood plasma is noninvasive and easily applicable and thus much more desirable to discover prognostic and diagnostic markers. Unfortunately, despite incredible academic and industrial efforts, plasma proteomics has not delivered so far. The reasons are rather obvious.
i) Sample acquisition is critical and reproducible plasma samples can only be obtained by precise standardized operating procedures of blood collection, hardly achievable in routine clinical work.
ii) The enormous complexity of several millions of molecular distinct protein species and the wide dynamic range of more than 10 orders of magnitude between high and low abundant proteins confronts biomarker discovery with almost intractable technical challenges. All protein fractionation techniques necessary for the reduction of complexity are connected with unpredictable protein loss and unpredictable recovery. Especially low abundant proteins, suspected to contain the majority of potentially relevant biomarkers, are currently out of reach for the common proteomics analysis methods. For all these reasons, today very few reports on quantitative plasma proteomics are published.
iii) Usually many differences in the proteome pattern of different individuals can be detected. However, due to their high number, the correlation of any of these changes with a disease state is difficult. The patient numbers required for solid statistics can hardly be investigated due to cost and time constrains.
Several strategic considerations have to be made to overcome the causes of failure of plasma proteomics studies.
i) Standardization of sample collection is mandatory. Nevertheless, in clinical routine reproducible sampling of plasma will remain difficult. Consequently, only rather robust markers can be detected.
ii) Doubtlessly, the proteomics techniques for biomarker discovery have to be quantitative. Label free techniques have been quite successfully applied in low complex systems and most of the published plasma proteomics projects have used label free techniques. However, these techniques are probably not really suited for plasma proteomics. Since the reduction of complexity has to involve multidimensional fractionation steps, unpredictable loss of distinct proteins can occur. Unfortunately, these losses depend on the composition of the sample, which in the case of plasma can be quite different with different blood donors (e.g. lipids, carbohydrates, etc.). A further disadvantage is that label free proteomics techniques have to be performed separately for each sample. Better suited for plasma proteomics are techniques based on isotopic labelling where up to four samples can be multiplexed at a time. The quantitative ratios of all the proteins in different samples are fixed at an early stage and quantitative results remain valid even after multidimensional fractionation steps.
iii) The type of proteomics technique applied plays a major role on the accuracy and the validity of the results. Entirely peptide based (bottom up) strategies, where the proteome is cleaved into peptides as a first step, carries especially for plasma a certain risk. After enzymatic cleavage a certain peptide may be released from different protein species. Thus, the quantitative analysis of a peptide may not reflect the amount of one distinct protein but rather the amount of all protein species and isoforms containing this peptide. In plasma it is well documented that many proteins appear in tens of different forms (genetic isoforms, glycosylation heterogeneity, phosphorylated forms, degradation products, etc). All these individual protein species can only be quantified correctly if all the existing forms are known, which is almost impossible. Therefore, proteomic strategies, where the analyzed peptides are undoubtedly linked to a single distinct protein species, produce in principle much more meaningful results. This can be achieved by protein based (top-down) proteomics strategies.
iv) The individual heterogeneity is probably the major cause for the inefficacy of proteomics in biomarker discovery. The protein patterns of two individuals are much more different than the protein patterns of one single individual in diseased and healthy state. To discriminate between individual heterogeneity, caused by genetic and environmental factors and disease related protein changes is extremely challenging. This can only be overcome by either a large number of individuals analyzed, which is prohibitively costly, or by a kind of personalized proteomics approach, where healthy and diseased states of one single individual are compared. Here at least genetic variability can be neglected.
An example of a feasibility study
A biomarker discovery study on colon cancer was carried out to evaluate plasma proteomics in the light of the arguments given. Following the idea of an individualized proteomics strategy, samples from individual blood donors were chosen as starting material. The Blutspendedienst of the Bavarian Red Cross collects hundreds of thousands blood samples a year. For legal reasons, a portion of these samples has to be stored in a blood bank for several years. After informed consent of the blood donor, these samples may then become accessible for research. 16 blood donors were selected, who have donated blood repeatedly and who were diagnosed with colon cancer with similar staging and grading. Additionally, a blood sample after successful therapy was obtained.
As quantitative proteomics technique the protein based isotope coded protein label (ICPL) strategy was chosen [Citation1]. With the ICPL technique up to four samples can be compared in one single experiment. Dedicated software, ICPLQuant [Citation2] covers the whole ICPL workflow shown in .
Figure 1. Workflow of ICPL-based proteomics of human blood plasma.
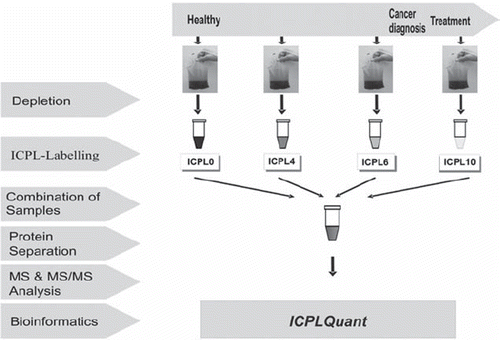
The 20 most abundant proteins account for about 97% of the protein content of blood plasma. Any analysis performed directly on whole plasma mainly results in data derived from these major proteins and will thereby obscure information on low abundance proteins. Therefore, the 20 most abundant proteins were extracted by immunoaffinity chromatography (IGMA). The bound fraction containing mainly albumin and immunoglobulin is eluted and can be further used e.g. to investigate the antibody repertoire of the patient [Citation3]. In our opinion the depletion of the high abundant proteins is an indispensible fractionation step for two reasons. The dynamic range of the plasma proteome is reduced by at least 2–3 orders of magnitude and the isotopic labelling of large protein quantities is rather expensive. At least 100 μL of plasma (corresponding to about 7 mg protein) has to be used to get access to low abundant proteins. Only then cellular leakage proteins which are in the mg/L interval can be detected with state of the art mass spectrometry instrumentation. After depletion, less than 1 mg protein material from a 100 μL plasma proteome sample remains. This is a reasonable amount for labeling and further sample workup. However, the depletion step is done without any isotopic control and the large amount of protein material removed may unintentionally eliminate certain proteins from the plasma sample by specific or unspecific binding. Therefore, great care has to be taken to reproducibly perform the depletion under standard operating procedures.
Following ICPL labelling, four samples derived from 4 different blood donation time points (as indicated in ) were combined and fractionated by preparative SDS-gels. After cutting the gels into 20 slices, the different molecular size fractions were cleaved enzymatically and subjected to mass spectrometry for quantification of the peptides. The ICPLQuant software, specially designed to follow the ICPL workflow, recognizes and quantifies peptide multiplets and stores the data (e.g. mass, retention time, quantitative, identifications) in a database. Only peptides exhibiting the expected concentration change pattern correlated to the progression of the disease were further investigated for identification by MSMS. Quantitative peptide patterns were recorded for all 16 patients. However, bioinformatic analysis revealed no single protein consistently regulated in all patients.
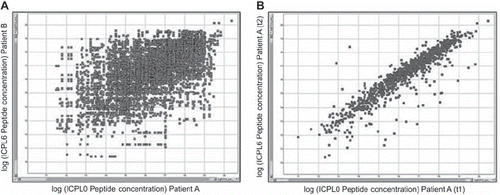
The results obtained in this feasibility study showed the high selectivity of the approach but suggested that the analysis depth of the proteomics study was not sufficient to reveal a new biomarker. However, an important result supports the applied strategy. When comparing the protein abundances in different patients, only very few proteins are present in similar concentrations. In contrast, comparing the protein pattern of different time points within one patient the majority of the proteins remain almost unchanged (). Only few proteins changed in concentrations even if the time points of sample collection were more than two years apart.
Figure 2. Comparison of peptide amounts in two different patients (A) and two different time points of the same patient (B).
Currently, these samples are reanalyzed using a similar proteomics workflow but with much higher fractionation of the depleted plasma. This is obtained by depletion, ICPL labeling, multiplexing and extensive fractionation by preparative isoelectric focusing (OffGel, Agilent). After this, reversed phase fractionation of each of the isoelectric focusing fractionation steps is performed on the peptide level. However, since the number of fractions now become quite large (i.e. several thousand fractions per patient), ESI-LC analyses are too time consuming. Automated sample preparation for rapid MALDI-MS analyses is being developed.
Discussion and conclusion
The targeted mass spectrometry techniques to monitor protein changes in many samples are at a mature stage and expected to enter routine clinical diagnosis and therapy monitoring rather soon. However, the targets of interest have to be well known and extensively characterized on the molecular level. Quantitative proteomics for biomarker discovery still is in its infancy. Today, probably the most successful approach is to quantitatively compare the proteomes of diseased tissues with their healthy counterparts. Proteins found to differ significantly in amount in the two proteomic states have then to be extensively validated for specificity and sensitivity by other techniques. Some of these putative biomarkers are disseminated into the circulation and can be traced directly in blood by targeted proteomics techniques with a high detectability like SRM or immuno-assays. The major obstacle in finding new biomarkers is the genetic polymorphism of different individuals. Therefore, personalized proteomics techniques in combination with accurate quantification and sophisticated bioinformatic methods may improve biomarker discovery in the future.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Schmidt A, Kellermann J, Lottspeich F. A novel strategy for quantitative proteomics using isotope-coded protein labels. Proteomics 2005;5:4–15.
- Brunner A, Keidel E, Dosch D, Kellermann J, Lottspeich F. ICPL Quant – a software for non-isobaric isotopic labeling proteomics. Proteomics 2010;10:315–23.
- Seliger B, Kellner R. Design of proteome-based studies in combination with serology for the identification of biomarkers and novel targets. Proteomics 2002;2:1641–51.