Abstract
Purpose: In previous reports, laboratory-made lysolecithin-containing thermosensitive liposome encapsulating doxorubicin (LTSL-DOX) showed potent anticancer effects in FaDu human squamous cell carcinoma. To further study the spectrum of LTSL-DOX activity, the efficacy of its commercial formulation was re-examined in FaDu and compared in HCT116, PC3, SKOV-3 and 4T07 cancer cell lines. Factors that may influence differences in HT-LTSL-DOX efficacy were also examined.
Methods: Anticancer effect was measured using standard growth delay methods. We measured doubling time and clonogenic survival after doxorubicin exposure in vitro, and interstitial pH and drug concentrations in vivo.
Results: In all five tumour types, HT-LTSL-DOX increased median tumour growth time compared with untreated controls (p < 0.0006) and HT alone (p < 0.01), and compared with LTSL-DOX alone in FaDu, PC-3 and HCT-116 (p < 0.0006). HT-LTSL-DOX yielded significantly higher drug concentrations than LTSL-DOX (p < 0.0001). FaDu was most sensitive (p < 0.0014) to doxorubicin (IC50 = 90 nM) in vitro, compared to the other cell lines (IC50 = 129–168 nM). Of the parameters tested for correlation with efficacy, only the correlation of in vitro doubling time and in vivo median growth time was significant (Pearson r = 0.98, p = 0.0035). Slower-growing SKOV-3 and PC-3 had the greatest numbers of complete regressions and longest tumour growth delays, which are clinically important parameters.
Conclusions: These results strongly suggest that variations in anti-tumour effect of HT-LTSL-DOX are primarily related to in vitro doubling time. In the clinic, the rate of tumour progression must be considered in design of treatment regimens involving HT-LTSL-DOX.
Introduction
Liposomally encapsulated drugs have been a subject of considerable attention in experimental and clinical oncology for more than 20 years Citation[1]. Several methods have been investigated for enhancing release of drugs from liposomes, including development of pH Citation[2], lipase Citation[3] and thermally sensitive formulations Citation[4–6]. The work described in this paper addresses liposomes that incorporate lysolipid to facilitate rapid content release upon reaching mild hyperthermia (40°–42°C). This low temperature-sensitive liposome (LTSL) formulation offers great promise and is currently in clinical trials Citation[7], Citation[8]. In this paper we investigate the extent to which hyperthermia plus LTSL can influence the growth rate of tumours. While many variables may influence tumour growth rate after LTSL plus hyperthermia in the five tumour types tested, the most important of the variables studied here is the intrinsic in vitro doubling time of each tumour type.
Hyperthermia (HT) has been shown to improve drug accumulation through increased perfusion and vascular permeability. This allows HT to augment liposome accumulation Citation[9] and serve as a mechanism for drug release Citation[10]. Thus, HT can enhance the activity of three fundamentally different types of liposomes that have been used for drug delivery to tumour tissue: non-thermosensitive liposome (NTSL), traditional thermosensitive liposome (TTSL), and most recently, LTSL. HT can enhance delivery of liposomes to tumour tissue as a result of hyperthermia-mediated increases in endothelial gap sizes Citation[11]. Whereas use of HT with non-thermally sensitive formulations yielded some enhancement in anti-tumour effect Citation[10], it has been hypothesised that further enhancement in anti-tumour effect could be achieved if HT could be used to trigger local drug release from liposomes.
After introduction of thermally sensitive liposomes by Yatvin et al. in 1978 Citation[6], significant improvements in thermally sensitive formulation development came with the use of lysolecithin by Needham et al. Citation[7], Citation[12] and with Lindner et al., who recently reported on a different formulation that behaves in a similar manner Citation[4], Citation[5]. With inclusion of 10 mol% of lysolecithin, a single-chain phospholipid that facilitates the opening of pores in the lipid bilayer, rapid drug release occurs as the bilayer solid-to-liquid phase transition is approached around 41°C Citation[12]. Rapid release of contents from the original formulation of LTSL is triggered at temperatures above 39°C, with ∼50% being released in <20 s at 40°C Citation[12]. The formulation of LTSL that was later commercialized as ThermoDox® releases 80–100% of encapsulated drug in 20–40 s at 41.3°C, with minimal (∼15%) release at 39°C Citation[13]. This rapid release quality, which can achieve drug release in the blood stream, provides a stark contrast to the non-thermally sensitive Doxil™ formulation, which exhibits peak tumour drug concentrations up to 48 h after treatment and slow release over days to weeks Citation[14].
The rapid release of doxorubicin from LTSL, when administered during HT (HT-LTSL-DOX), provides tumour drug levels five times higher than non-thermally sensitive formulations and 30-fold greater concentrations than free drug Citation[15]. Such fast, intravascular drug release may be responsible for vascular targeting effects after HT-LTSL-DOX treatment of FaDu human squamous cell carcinoma xenografts Citation[16] and 4T07 murine mammary carcinoma Citation[17]. Ability to control the location of drug release with heat makes HT-LTSL-DOX effective at maintaining stable drug encapsulation in circulation and achieving rapid release at the heated tumour site.
The LTSL-DOX formulation has been used previously in the FaDu tumour line Citation[15] and in canine soft tissue sarcomas in a phase I trial Citation[18]. Two phase I trials have been conducted using LTSL-DOX in humans for liver tumours in conjunction with radiofrequency ablation Citation[8], Citation[19], Citation[20]. The drug is now in a Phase III trial in this patient population Citation[21]. It is also currently being tested in a phase I trial in patients with chest wall recurrences of breast cancer Citation[22].
The purpose of the work reported here was to extend the original preclinical observations made in FaDu Citation[7], Citation[15] to a broader group of human xenografts and a murine tumour line using the commercial LTSL formulation (Thermodox®). Therefore, we conducted a new set of experiments to compare FaDu to other tumour lines (SKOV3, PC3, HCT116, 4T07). These lines varied in their in vivo response to LTSL-DOX with HT in standard growth delay studies, although all showed enhanced effectiveness when compared with LTSL without HT.
To examine possible causes of this variation, factors potentially affecting drug performance were compared between the tumour lines and examined for correlations with anti-tumour effect. These factors included tumour doxorubicin concentration in vivo, in vitro cellular doubling time, drug sensitivity of the tumour lines in vitro and tumour pH. The main point of this research has been to examine whether there are definable mechanisms that may explain the variation in what otherwise is a very effective treatment across the board. Identifying such mechanisms may allow for a predictor of treatment efficacy and offer guidance to clinical study design and ultimate treatment protocols.
Materials and methods
Liposomes and doxorubicin
Monostearoylphosphatidylcholine (MSPC), dipalmitoylphosphatidylcholine (DPPC) and distearoylphosphatidylethanolamine-PEG 200 (DSPE-PEG-2000) were purchased from Avanti Polar Lipids (Alabaster, AL). The LTSL was composed of DPPC, MSPC and DSPE-PEG-2000 in the molar ratio of 90:10:4 and had the mean diameter of 100 nm. Doxorubicin (DOX) was purchased from Sigma (St. Louis, MO). Encapsulation of DOX into LTSL was carried out using a pH gradient-driven loading protocol Citation[23], Citation[24] yielding a final lipid concentration of 10 mg/mL and a 0.05:1 drug:lipid weight ratio Citation[25], Citation[26].
Cell lines and culture
All five tumour cell lines were obtained from ATCC. FaDu human squamous cell carcinoma was cultured in Eagle's minimal essential medium with Earle's BSS with 2 mM L-glutamine (EMEM), 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 1.5 g/L sodium bicarbonate and 10% fetal bovine serum (FBS). HCT116 human colon carcinoma and SKOV-3 human ovarian adenocarcinoma cells were cultured in 90% McCoy's 5a medium, supplemented with 10% FBS. PC-3 human prostate adenocarcinoma was cultured in 9:1 Ham's F12K medium:FBS by volume. 4T07 mouse mammary carcinoma cells were cultured in 9:1 DMEM:FBS. Tumour cells were grown in the above culture media prior to in vivo implantation. In the clonogenic evaluation of sensitivity to DOX, cells were cultured as mentioned above with the exception of HCT116 and FaDu being cultured in DMEM supplemented with 10% FBS. All cell lines were cultured with 1% antibiotic-antimycotic. However, in vitro doubling time evaluation of all cell lines was performed in 9:1 DMEM:FBS (see Cell doubling time below).
Animals and tumours
Six-week-old female homozygous NCr athymic nude and BALB/c mice (20 ± 3 g) were purchased from Taconic (Germantown, NY). Tumours were established by injections of 1 × 106 tumour cells (50 µL single cell suspension) subcutaneously in the left leg. Animals whose normal organ drug concentration was evaluated were injected with FaDu tumour cells on both hind legs (see Doxorubicin concentrations in tumour and normal tissue). Tumours were allowed to grow to 5–7 mm in diameter before being stratified according to tumour volume and randomised into the following four groups (9–10 animals/group): control (no treatment), hyperthermia (HT) alone, LTSL-DOX alone, and HT-LTSL-DOX.
Animals were weighed, and tumour volumes were measured three times per week (volume = length × width2× π/6). Animals were followed until the tumours reached five times their initial tumour volume or 60 days, whichever occurred first. According to Institutional Animal Care & Use Committee (IACUC) guidelines, if mouse body weight loss was ≥15% of the initial body weight, animals were to be euthanised in compliance with IACUC guidelines. No animals were sacrificed early because of weight loss in this study, however. All animal procedures were approved by the institutional IACUC.
Chemotherapy and hyperthermia treatment
All animals were anaesthetised with an intraperitoneal injection of pentobarbital (80 mg/kg) prior to treatment. LTSL-DOX was administered as a single intravascular injection via the tail vein (50–100 µL) at a dose of 6 mg DOX/kg, with the exception of animals carrying 4T07 tumours, where 7 mg DOX/kg were used. These doses were chosen, based on a preliminary LD50 study, in which mice were exposed to a range of doses; 6–7 mg/kg was established as the maximum tolerated dose. Doses >7 mg/kg resulted in greater than 15% body weight loss (data not shown), which is considered moribund by the IACUC. Local hyperthermia was applied immediately after injection of LTSL for the HT-LTSL-DOX group and immediately after the onset of anaesthesia for the HT group. To obtain 34°C and 42°C tumour temperatures for control and heated groups, a water bath set at 35°C and 43°C was used as reported previously Citation[15]. A fan was used to keep the rectal temperature of the mice at 36°–37°C.
Median growth time, relative median growth time, growth delay, response fraction, tumour regression and body weight
Median growth time (MGT), measured in days, was the median number of days it takes for each tumour of one treatment group to reach the endpoint of five times its original volume, or 60 days after treatment. In order to isolate the effects of HT, relative MGT was obtained by normalising MGT of the treated group to the MGT of a non-heated control group. Growth delay (GD) was defined as MGT of treated group minus MGT of a control group at the same temperature, with the exception of the HT group, where GD is defined as MGT of the HT group minus MGT of unheated control group. Response fraction (RF) was the ratio of animals with tumours less than five times the original tumour volume to the total number of all animals in a treatment group of one tumour type, at the end of the experiment. Complete tumour regression was defined as absence of palpable tumours 60 days after treatment. Changes in body weight were quantified as maximum percentage loss of body weight after treatment.
Doxorubicin concentrations in tumour and normal tissue
To compare drug delivered to the tumour by LTSL with and without HT we measured total doxorubicin concentration ([DOX]) in 8–10 tumours of each cell line, grown on the left leg of nude mice. HT-LTSL-DOX and LTSL-DOX treatments were performed as described above. After one hour of heating, the tumours were surgically excised, snap frozen and stored at −80°C. Total (bound and unbound) tumour [DOX] concentrations were measured by high performance liquid chromatography (HPLC) as previously published Citation[27]. Concentrations obtained from two halves of each tumour were averaged to obtain tumour [DOX] for each animal. The reported average [DOX] was obtained by averaging all tumour [DOX] values from all animals of a given tumour type (n = 8–10 for all tumour types).
Liver, kidney, spleen, and lung [DOX] were determined concurrently with tumour [DOX] in 11 additional mice with FaDu tumours on both legs. The tumour on the left leg received HT while the other tumour was kept at room temperature for 1 hour after LTSL-DOX injection. Doxorubicin concentrations in the organs and both tumours were analysed as above, though tumour [DOX] from these additional animals was not used in comparisons between tumour types. Following surgical excision, snap freezing and storage at −80°C, each organ/tumour was homogenised and processed for HPLC analysis as a separate sample (n = 11 for each organ, heated tumour and unheated tumour).
Cell doubling time
Exponentially growing cells of each of the five cell lines were inoculated at 500–2000 cells/well in 96-well microtitre plates, in 150 µL of 9:1 DMEM:FBS. Because the MTS assay that we used to measure doubling time depends on the rate of cellular metabolism, one medium that enabled growth of all five cell lines was chosen. A set of separate experiments was conducted using different growth media (data not shown) and the doubling times of all cell lines agreed with those reported here using the MTS assay. At 24, 36, 48, 60, 72, and 84 h after inoculation, MTS assay was performed with the addition of 20 µL of the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI) to each of the four wells containing cells as well as two control wells (medium only). Absorbance at 490 nm was measured 6 h after MTS reagent addition. In three separate experiments the doubling times were calculated using a linear regression fitted to log(absorbance–background) normalised to the first time point, vs. time. Error was calculated using error of the slope and y-intercept, and while the error was not evenly centred at the mean doubling time, the average of the positive and negative error bounds is reported in the text for clarity. Three independent experiments were conducted, with 4 wells being analysed at each time point.
In vitro DOX sensitivity
Clonogenic survival after 24 h exposure to DOX was assessed without adjuvant hyperthermia treatment (HT; 42°C for 1 h). Three independent experiments were performed, each containing six cultures at each [DOX], with three wells at each cell density, for each tumour line. Before drug treatment, single cell suspensions were plated overnight. The cell density was varied according to different cell lines and DOX concentrations, from 150 to 15,000 in order to yield >30 colonies/plate after treatment. The cells were treated with 20 to 500 nM of DOX while incubated for 24 h. Subsequently the cells were washed with PBS and the medium was changed to stop drug exposure.
After 8–14 days of culture, cells were washed with PBS, fixed (10% methanol, 10% glacial acetic acid, and 80% water), and stained with crystal violet (0.4% solution in ethanol and water). Colonies were counted in each dish. Plating efficiency and survival fractions were calculated for each cell line at different DOX concentrations. Wells that were disrupted by washing and those containing colonies at high, uncountable densities were excluded from analysis, resulting in N = 3–6 in each independent experiment. The drug concentration that resulted in 50% reduction of colony number relative to untreated plates (IC50) was calculated from the fit of Equation 1 to the survival fraction vs. log([DOX]) Citation[28], using GraphPad Prism (version 5.00 for Windows, GraphPad Software, San Diego, CA).
In the equation, SF is the surviving fraction; SFmin and SFmax are the minimum and the maximum values SF attains, IC50 is [DOX] that causes a 50% decrease in SF, C is log[DOX], and γ is the unitless Hill slope factor. For fitting, SFmin was constrained between 0 and 1, SFmax was set to 1, and average survival fraction results from the 6 wells in each experiment, at each concentration, were used as independent points (n = 3 at each [DOX]), with the fitting routine accounting for the number of independent experiments and standard error of the mean SF from all experiments.
Tumour pH
Tumour interstitial pH was determined using combination interstitial needle electrodes (Microelectrode, Londonderry, NH; Agulian, Hamden, CT), following previously published protocols Citation[29]. Prior to each experiment, and upon its completion, the electrodes were calibrated using buffered solutions of pH 6.0, 7.0, 7.4 and 8.0. All tumour measurements were corrected using the average of the two calibration curves. During the experiment, multiple points were measured at 0.5–1 cm intervals along the probe track. Measurements from two tracks in each tumour were averaged to arrive at average tumour pH. Five tumours of each tumour type were sampled (n = 5).
Statistical analysis
All of the statistical analysis was performed with the GraphPad Prism computer program (GraphPad, San Diego, CA). Analysis of the difference in Kaplan-Meier survival curves was performed using the Mantel-Haenszel log-rank test, with survival being defined as having a tumour that is less than five times its original volume. The p-values reported with MGT were obtained using this method. Differences in median values of maximum per cent loss of body weight were compared using the Kruskal-Wallis test followed by Dunn's post test. In most instances in this work the magnitude of values and not just their relative rank could be of importance, and therefore the Pearson correlation statistic was used for correlation analysis. Tukey's multiple comparison test was employed as a post test after one-way ANOVA to verify significance between means (DT and tumour [DOX]). Curves fitted to the cytotoxicity assay data and the resulting IC50 values were compared using the F test for difference in fitted parameters. The Bonferroni method was used to adjust the p-values for pairwise comparisons of survival curves. Results were considered significant with p < 0.05, and two-tailed p-values were obtained in all cases. Pairwise comparisons were only considered when ANOVA, Kruskal-Wallis or the Mantel-Haenszel log-rank test showed significant differences between all tested groups.
Results
Tumour growth delay
To assess the therapeutic effects of the different treatments on tumour growth inhibition, we treated murine (4T07) and human xenograft (FaDu, HCT116, PC3, SKOV3) tumours in mice with saline (control), HT alone, LTSL-DOX alone, or HT-LTSL-DOX, and measured tumour growth delay following treatment (). Replicate experiments were performed for all of the cell lines except FaDu and SKOV-3 (). The growth delay experiment in was used for analysis and/or calculations, whereas data from other experiments were similar for each tumour type in replicate experiments ().
Figure 1. Treatment groups: Control ![]()
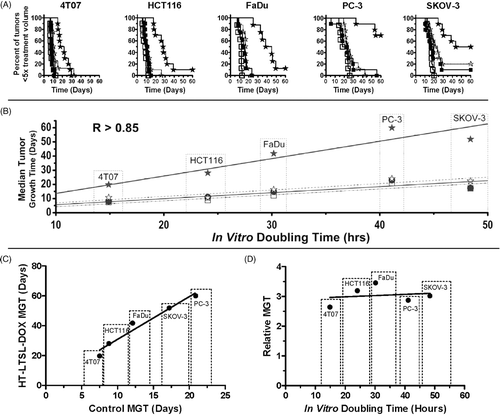
Table I. Summary of all growth delay experiments with 4T07, HCT116, FaDu, SKOV-3 and PC-3. A single dose of 6 mg/kg of LTSL-DOX i.v. and 1-h long HT treatment at 42°C were used in all experiments, with the exception of 4T07, where 7 mg/kg was used. The table lists results of 1–3 independent experiments with each tumour type. Data from the experiment in and are shown in bold. Median growth time (MGT) is the median number of days required to reach five times initial tumour volume, or 60 days after treatment. Growth delay (GD) is defined as (MGT of treatment group) minus (MGT of control group at the same temperature), with the exception of the HT group, where GD is defined as the difference in MGT between HT and the unheated control groups. Response fraction (RF) is the ratio of tumour regressions (complete and incomplete) to the number of animals in all experiments of that tumour type and treatment, where an incomplete regression is a reduction >30% of starting tumour volume. The number of complete regressions, if they are present, is listed after RF (for example, for the HT group and SKOV-3, 2/10, 1 means two out of ten responses, with 1 complete regression). n = 8–10 animals for all treatment groups of each tumour, in all experiments. ![]() indicate a significant difference with p < 0.05 and p < 0.01, respectively, from MGT of a control group treated at the same temperature (Mantel-Haenszel log-rank test), and therefore no Bonferroni correction was performed here. p > 0.05 is denoted by X.
indicate a significant difference with p < 0.05 and p < 0.01, respectively, from MGT of a control group treated at the same temperature (Mantel-Haenszel log-rank test), and therefore no Bonferroni correction was performed here. p > 0.05 is denoted by X.
Comparisons between treatments
For all five tumour types, HT-LTSL-DOX treatment resulted in a significant increase in median growth time compared with saline controls (p < 0.0006) and HT alone (p < 0.01) when assessed by the Bonferroni-adjusted Mantel-Haenszel log-rank test. HT-LTSL-DOX treatment significantly increased MGT compared with LTSL-DOX alone for FaDu, HCT116 and PC-3 (p < 0.0006), but not for 4T07 (p = 0.79) and SKOV-3 (p = 0.13). LTSL-DOX without HT significantly prolonged MGT compared with saline controls for FaDu (p = 0.005), but not for any other tumour types (0.06 < p < 0.47). When compared to HT alone, LTSL-DOX without HT did not improve MGT in any of the tumour types (0.05 < p < 1.0).
Comparisons between tumour types
MGT of PC3 with or without any treatment was longer than that of 4T07–the most rapidly growing tumour (). For treated groups, PC-3 also had the highest percentage of complete regressions, whereas there were no complete regressions in the 4T07 tumour line (). Despite the higher dose of drug given to animals bearing 4T07 tumours (7 mg/kg), tumour growth was impeded less than for all other tumour types.
The MGT of HT-LTSL-DOX linearly correlated with the control MGT (, Pearson r = 0.98, p = 0.0035). This correlation and the response fraction data from demonstrate that the tumour types that display the greatest number of complete tumour regressions, namely SKOV-3 and PC-3 are the slowest growing. Normalisation of MGT to the MGT of unheated controls demonstrates that FaDu and HCT-116 exhibit approximately 35% more growth inhibition following HT-LTSL-DOX than 4T07 ().
Total [DOX] in tissues
Comparisons between treatments
For all five tumour lines, the average total drug concentration in tumours treated with HT-LTSL-DOX was significantly higher than that of the LTSL-DOX without HT group (One-way ANOVA, p < 0.001, followed by pairwise Tukey, p < 0.0001; ).
Figure 2. Mean total doxorubicin concentration one hour after LTSL-DOX and HT-LTSL-DOX treatments in five tumour lines and some normal organs. [DOX] in tumours was assessed in animals with only one tumour, whereas [DOX] in normal tissues was assessed in animals with two tumours, one of which was heated. Brackets with ![]()
![Figure 2. Mean total doxorubicin concentration one hour after LTSL-DOX and HT-LTSL-DOX treatments in five tumour lines and some normal organs. [DOX] in tumours was assessed in animals with only one tumour, whereas [DOX] in normal tissues was assessed in animals with two tumours, one of which was heated. Brackets with Display full size and * indicate pairwise comparisons where differences were significant at p < 0.0001 and p < 0.05, respectively. 4T07 with HT was also significantly less than all other tumour types, as well as liver, spleen and kidney (p < 0.05). Tukey's multiple comparison test was used for pairwise comparisons, after one-way ANOVA confirmed significant differences across all groups (p < 0.001). n = 8–11.](/cms/asset/49c603e8-058d-4dcb-aaa3-708ffd5660ea/ihyt_a_479450_f0002_b.gif)
Comparisons between tumour types
The degree of hyperthermic enhancement of drug delivery was variable between tumour types (). As illustrated in , HT-LTSL-DOX induced a 15.4-fold increase in [DOX] over non-heated LTSL-DOX in PC-3 tumours and an 11.7-fold increase in [DOX] in HCT-116. In contrast, HT-LTSL-DOX resulted in a 6-fold higher tumour [DOX] in FaDu and SKOV-3 and 4.7-fold enhancement in 4T07 compared with LTSL-DOX without HT.
Drug concentration in normal tissues compared to tumour
Doxorubicin concentrations in the liver, spleen, kidney, lung and the tumour were measured in 11 additional FaDu-bearing animals. All of these measurements of [DOX] were done in animals that had one of two leg tumours receiving HT (). Drug levels in heated tumours of animals with two tumours were not significantly different from tumour [DOX] in animals that only had one heated FaDu tumour (p = 0.12, unpaired t-test). Tumour [DOX] in the unheated tumour of animals with two tumours was also not significantly different from tumour [DOX] in animals with one tumour that was not heated (p = 0.18, unpaired t-test). The [DOX] in organs of these animals should be similar to animals with one tumour treated with HT-LTSL-DOX.
DOX concentrations in liver (21 ± 4 ng/mg tissue), spleen (15 ± 3 ng/mg tissue), kidney (19 ± 3 ng/mg tissue) and all heated tumours were higher than the 6 ± 1 ng/mg tissue [DOX] found in lung (Tukey, p < 0.01). After HT-LTSL-DOX treatment, only PC-3 and HCT116, but not the other tumours had significantly greater [DOX] than liver, spleen, kidney and lung (Tukey, p < 0.05).
Toxicity
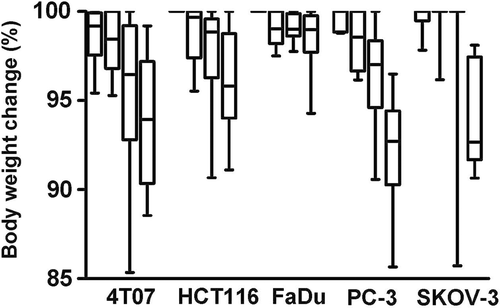
The treatment regimens described resulted in mild toxicity, as assessed by body weight loss (). The HT-LTSL-DOX group showed the greatest toxicity, with a median maximum body weight loss of 7.4% for all tumour types. Body weight recovered for all animals in all groups <20 days after treatment. For all tumour types, weight loss was significantly different between treatment groups (Kruskal-Wallis, p < 0.013), though only the difference between control and HT-LTSL-DOX was consistently significant for all tumour types (p < 0.05, Dunn). The higher dose used to treat 4T07 (7 mg/kg vs. 6 mg/kg for others), did not result in significantly greater body weight loss, except for the comparison of HT-LTSL-DOX between 4T07 with FaDu (p < 0.05, Dunn). No animals in this study met the body weight loss criteria for early euthanasia (body weight dropping 15% or more of the initial value).
Figure 3. Maximum decrease in body weight over a period of 60 days after treatment. For each tumour type, control, HT, LTSL-DOX and HT-LTSL-DOX are shown, in that order. Boxes show the upper and lower quartiles and the bar in the box shows the median value. The whiskers demonstrate the range of the data. Body weight recovered for all animals in all groups <20 days after treatment. For all tumour types weight loss was significantly different between treatment groups (Kruskal-Wallis, p < 0.013). The difference between control and HT-LTSL-DOX maximum decrease in body weight was consistently significant for all tumour types (p < 0.05, Dunn).
Cell doubling time
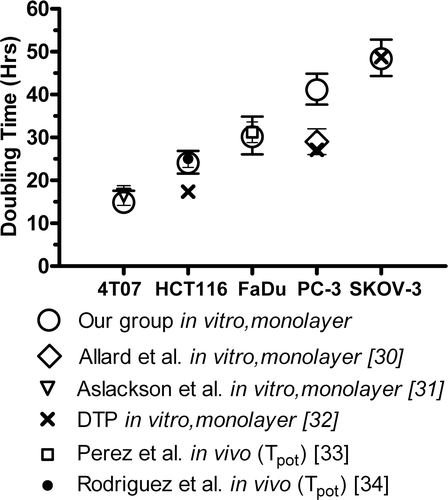
We evaluated in vitro doubling times of the five tumour lines (). 4T07 had the shortest DT (14 ± 2 h), followed by HCT116 (24 ± 3 h), FaDu (30 ± 4 h), PC-3 (41 ± 4 h), and SKOV-3 (48 ± 4 h). One-way ANOVA showed that the differences in doubling time among these cell lines were significant (p < 0.0001), with all pairwise Tukey tests showing significant differences (p < 0.01). The results of this study generally agree with previously published doubling times measured in vitro and potential doubling times, measured in vivo by flow cytometry after BrdUrd labelling Citation[30–34] ().
Figure 4. Doubling times for each cell line, compared with previously published results. The doubling times were measured in the same growth medium using the MTS assay in three replicate experiments. With the exception of PC-3, the results from our group generally agree with previously published doubling times measured in vitro and potential doubling times measured following in vivo using BrdUrd labelling and flow cytometry.
In vitro DOX sensitivity
In vitro sensitivity of the five tumour lines to doxorubicin was tested using the clonogenic assay (). Following 24 h exposure to DOX and subsequent incubation in DOX-free medium, FaDu was most sensitive to doxorubicin (IC50 = 90 ± 6 nM), followed by SKOV3 (IC50 = 128 ± 28 nM), PC-3 (IC50 = 151 ± 14 nM), HCT116 (IC50 = 165 ± 22 nM), and 4T07 (IC50 = 167 ± 16 nM). The standard error of IC50 was not centred on the mean values () because it was obtained from the fitted parameter log(IC50), and therefore it is averaged in the text for clarity. The IC50 of FaDu was significantly smaller than all others (p < 0.0014, F test). SKOV-3, HCT116, PC-3 and 4T07 had IC50 values that were statistically indistinguishable from each other, with the exception of a statistically significant difference between SKOV-3 and 4T07 (p = 0.02, F test) in pairwise comparisons.
Figure 5. IC50 values after 24 h exposure to DOX without HT. IC50 values that result from fitting Equation 1 to surviving fraction vs. log[DOX]. R2 was greater than 0.62 for all fitted curves. Comparison of the fitted parameters using the F test shows that the IC50 values in this dataset are statistically different overall (p < 0.0001). The IC50 of FaDu was significantly smaller than all others (p < 0.0014, F test). In pairwise comparisons, SKOV-3, HCT116, PC-3 and 4T07 had IC50 values that were statistically indistinguishable from each other, with the exception of a 1.3-fold significant difference between SKOV-3 and 4T07 (p = 0.02, F test).
![Figure 5. IC50 values after 24 h exposure to DOX without HT. IC50 values that result from fitting Equation 1 to surviving fraction vs. log[DOX]. R2 was greater than 0.62 for all fitted curves. Comparison of the fitted parameters using the F test shows that the IC50 values in this dataset are statistically different overall (p < 0.0001). The IC50 of FaDu was significantly smaller than all others (p < 0.0014, F test). In pairwise comparisons, SKOV-3, HCT116, PC-3 and 4T07 had IC50 values that were statistically indistinguishable from each other, with the exception of a 1.3-fold significant difference between SKOV-3 and 4T07 (p = 0.02, F test).](/cms/asset/4bcc560e-0ff5-4be8-b3c3-601b1fc238c8/ihyt_a_479450_f0005_b.gif)
Tumour pH
Measurements of pH were performed in five mice of each tumour line. Average interstitial pH values for all tumour lines ranged 6.8–7.0, with no significant differences between the tumour types (data not shown). Since no differences in tumour pH were found, pH was omitted from multi-factor comparisons between tumour types.
Multi-factor comparisons between tumour types
In this section we compare in vivo tumour growth delay data with parameters we hypothesised might influence the anti-tumour effect of this treatment.
Tumour [DOX]
Concentration of DOX in the tumour correlated with greater anti-tumour effect, when treatment groups of the same tumour type are compared (). This is evidenced by a significant correlation between [DOX] and MGT for any one tumour type (Pearson r > 0.77, p < 0.005). However, different tumour types responded differently to tumour [DOX]; relative MGT of HT-LTSL-DOX did not correlate with tumour [DOX] when compared across different tumour types (Pearson r = 0.36, p = 0.6). For instance, the MGT of HCT116 increases 3.2 times with an 11-fold increase in tumour [DOX], whereas the MGT of SKOV-3 increases 3-fold with only a 6.5-fold increase in tumour [DOX].
Table II. Summary of key comparative parameters for each tumour type. For all five tumour lines, tumour [DOX], in vitro DT, in vitro IC50, median growth time and relative median growth time are shown. The in vivo parameters are shown only for the HT-LTSL-DOX treatment group. For in vitro DT and IC50, means and average standard error are shown.
In vitro doubling time
In vitro doubling times correlated linearly with MGT in each of the treatment groups (Pearson r > 0.85, p < 0.02), as shown in . In vitro doubling times also linearly correlated with GD for the HT-LTSL-DOX treatment groups across tumour types (Pearson r = 0.9, p = 0.014). In addition, the longest in vitro doubling times were most common in tumours (SKOV3 and PC-3) where response fraction was highest (, and ). However, no statistically significant correlation (Pearson r = 0.18, p > 0.77) was observed between in vitro cell doubling time and relative MGT ().
In vitro DOX sensitivity
There was no significant correlation between in vitro cellular DOX sensitivity measured as IC50 and MGT (Pearson r =–0.4, p = 0.5). Furthermore, PC-3, which was not among the most sensitive to DOX in vitro, displayed greatest growth delay, longest MGT and the highest number of complete regressions in vivo (, ). 4T07 was among the least sensitive in vitro as well as in vivo. Hyperthermia alone did not have an effect on clonogenic survival at these temperatures (data not shown), though in vivo, SKOV-3 had a complete regression in the HT group but none in the non-heated control group (, ).
Discussion
We designed this study to test the possibility of a correlation of relative therapeutic efficacy of HT-assisted LTSL delivery of doxorubicin with factors that could influence the efficacy of this treatment, such as tumour drug concentration, in vitro tumour cell doubling time, in vitro sensitivity of these cells to doxorubicin and in vivo tumour pH. We gathered data on these parameters in several tumour models (4T07, HCT116, FaDu, SKOV-3, and PC-3). Ultimately, our goal was to define parameters that might predict efficacy of hyperthermia-assisted LTSL treatment. For example, one might expect that a higher intratumoural drug concentration might correlate with longer median growth times; or faster growth times following treatment might correlate with faster doubling times of the untreated tumour. To discuss this issue it is important to define treatment efficacy.
Evaluation of efficacy
In order to assess the antitumour effects of HT-LTSL-DOX, it is important to draw a distinction between the various measures of treatment efficacy. In this work we mostly discuss median growth times, relative MGT and the number of complete regressions, though we also provide other measures of efficacy, such as the response fraction and growth delay (). These measures must be interpreted carefully: for example, our data shows that relative MGT following treatment is not significantly different between tumour types (), although HT-LTSL-DOX consistently exhibited better anti-tumour effect for all tumours studied. Since the relative MGT is similar across all tumour types, we could conclude that none of the other parameters had an important influence on treatment outcome. However, the real question for any given patient is the absolute amount of prolongation of anti-tumour effect achieved, not the relative improvement over hyperthermia alone. For example, in a clinical scenario, a significant benefit due to administration of HT plus LTSL-DOX would clearly be derived from increasing the probability of local control or survival of a patient with a slow growing tumour from months to years. Conversely, a similar relative increase in anti-tumour effects in a patient with an extremely aggressive and lethal tumour may only add a few weeks to months to the patient's life. This paradox is clearly demonstrated in our pre-clinical data, with the slow growing prostate xenograft PC-3 having many complete regressions () while displaying similar relative MGT () to the much more aggressive 4T07 mammary carcinoma. In addition, the numbers of complete and incomplete tumour regressions are extremely important in the clinic, as they provide an early assessment of improvement of quality of life and perhaps durability of local control. In our data, the slowest growing tumours with longest MGT and longest GD also had the highest number of tumour regressions. Therefore, the absolute measurements of efficacy, such as MGT and GD are more clinically relevant than relative MGT in the context of HT-LTSL-DOX treatment. We will thus refer to MGT and GD when discussing treatment efficacy. It is certainly possible in the clinic that multiple treatments with HT-LTSL-DOX in a more rapidly growing tumour could lead to greater anti-tumour effect than a single treatment, so we do not want to imply that more rapidly growing tumours would not benefit from this therapy. Knowledge of growth rates may influence the frequency or absolute number of treatments in this context.
Choice of tumour lines
The five tumour lines investigated here vary in their rate of growth and vascularity and are derived from different tissues. Efficacy of HT-LTSL-DOX has only been evaluated in the FaDu tumour line until now, and in order to see if further development of this treatment method is necessary it was important to evaluate its efficacy in a number of substantially differing tumour lines. All of the lines represent tumour types that could ultimately be heated; indeed human hyperthermia clinical trials have previously been conducted on all tumour types represented in this cohort Citation[35–37].
Tumour [DOX] and treatment efficacy
Kong et al. previously demonstrated a direct correlation of tumour [DOX] and growth time for FaDu Citation[15]. In another study Chen et al. demonstrated that the efficacy of HT-LTSL-DOX treatment of 4T07 tumours, in terms of microvascular damage in tumours, can be significantly enhanced when interstitial [DOX] is increased via drug release in the blood stream and an increase in microvascular permeability Citation[17].
Tumour drug concentrations have been shown to correlate with treatment efficacy in other reports Citation[15], Citation[38], Citation[39], and our data confirm this correlation within each tumour type. When compared across tumour types, however, MGT and RF do not show any relationship to tumour drug levels (Tables I and II). It is important to note that tumour [DOX] was measured only 1 h after treatment, while growth delay was monitored over a much longer period of time, and that we did not separate intravascular [DOX] from interstitial or intracellular [DOX]. It is likely that majority of DOX was released within the vascular and subendothelial spaces and then diffused into the surrounding interstitial regions. As a result, tumour [DOX] measured in this study was the average of both concentrations, weighted by the fraction of the vascular and extravascular volumes. In addition, organ [DOX] was evaluated only in mice that were subjected to the combination of hyperthermia and LTSL-DOX, thus the data cannot be used to determine whether organ accumulation of DOX is due to accumulation of free or liposome-encapsulated DOX. We are currently investigating this particular question in a study that also includes measurement of heart [DOX], which is of interest due to doxorubicin-associated cardiomyopathy Citation[40].
Our findings indicate that drug deposition in tumour tissue is highly variable among the five tumour types under investigation (). This variability can exist due to several possible differences between tumour types, including differences in perfusion and permeability as well as factors related to intracellular drug transport. For instance, vascular permeability is variable across different tumour types Citation[41], Citation[42]. It has recently been reported that differences in vascular permeability between FaDu and 4T07 are correlated with the anti-tumour effect of HT-LTSL-DOX Citation[17]. HT increases tumour blood flow Citation[43] and permeability of blood vessel walls, and these effects could differ across the tumour lines examined herein Citation[10], Citation[44]. We did not investigate these parameters, and they could affect HT-LTSL-DOX efficacy.
In vitro doubling time and treatment efficacy
It has been previously shown that DOX efficacy in vitro correlates with untreated in vitro cell doubling time Citation[45–48]. Our results expand upon this knowledge to show that MGT in vivo is also correlated with in vitro DT for the HT-LTSL-DOX treatment for all tumours (). This correlation may be used to evaluate the likelihood of certain slower growing tumours to be controlled by HT-LTSL-DOX in a clinical treatment protocol. Thus, clinically obtainable measurements of potential doubling time and proliferation may be useful prognostic indicators for this treatment (See Future directions below).
In vitro sensitivity to DOX and treatment efficacy
Our data demonstrate significant differences in the sensitivity between the five tumour lines to doxorubicin in vitro (). However, the tumour that is most sensitive to DOX in vitro, FaDu (IC50 = 90 nM DOX), is not affected by HT-LTSL-DOX as much as PC-3 (IC50 = 150 nM DOX), when MGT is used as an assessment of treatment efficacy (magnitude of MGT in ).
The mechanism of cell kill by doxorubicin is not entirely known Citation[49], but there are cellular mechanisms for variation in drug sensitivity, such as up-regulation of p-glycoprotein and variation in levels of topoisomerase II, the main target of doxorubicin toxicity Citation[50]. We are now examining this question in more detail, but it is likely to be a complex answer, because three of the cell types tested in this series do not express p-glycoprotein Citation[51–53].
The relationship between in vitro drug sensitivity and in vivo treatment efficacy is further complicated by several other variables, such as exposure time and cellular uptake kinetics in vitro Citation[54], and differential vascular permeability and perfusion in vivo Citation[17], Citation[41], Citation[42]. Since the accumulation and wash-out rates of DOX are unknown for the five tumour types investigated here, the duration and severity of exposure to DOX is uncertain in vivo. We have noticed that 4T07 was more resistant to DOX than FaDu when the cells were treated in vitro for 24 h in the current study. However, the difference in resistance was minimal when the same cells were treated for only 1 h Citation[17]. Thus, testing in vitro sensitivity to DOX may still be helpful, if the exposure time is varied or matched to the kinetics of drug delivery and retention. Such data are now being acquired in separate experiments.
Tumour pH and treatment efficacy
The hypothesis that pH may impact treatment efficacy was based on the fact that the extracellular pH of tumours is known to vary widely Citation[55]. Low pH can increase the hydrophilic component of DOXH+ because it is a weak base. This increase in the hydrophilic charged component would lead to a concomitant decrease in the fraction of membrane soluble and permeable hydrophobic DOX and lower its uptake by cells. However, we did not observe significant differences in interstitial pH between the tumour lines, while they differed greatly in their response to LTSL-DOX treatment. Thus, our data do not show a dependence of treatment response on pre-treatment pH. However, our results do not discount the possibility that pH could play a major role in modulating tumour response to doxorubicin in other tumour types that exhibit larger variations in extracellular pH. Wike-Hooley et al., reported on a survey of extracellular pH measurements performed in a large series of human patients Citation[56]. In a cohort of 105 tumours studied, the median value was approximately 7.2, but individual measurements ranged from 6.4 to 7.7. Prescott et al. examined extracellular pH in a series of 40 dogs with soft tissue sarcomas Citation[57]. A similar range of pH values was obtained, and low extracellular pH has been shown to inhibit doxorubicin cytotoxicity Citation[58], Citation[59].
Toxicity
The weight loss in the HT-LTSL-DOX group was slightly but consistently higher than in the other treatment groups. This suggests that there may be enhanced systemic toxicity with this treatment. In a previously reported phase I trial in canine tumours, the maximally tolerated dose of this drug was not substantially different from that achieved with free drug, however Citation[18]. The difference in maximum body weight reduction between 4T07 and FaDu tumour bearing mice treated with HT-LTSL-DOX is likely due to tumour burden, since 12 days after treatment, the body weight in the control group increased on average by 16 ± 2% for FaDu and by only 2 ± 0.4% for the faster growing 4T07.
Comparison to previous HT-LTSL-DOX studies with these tumour lines
Previous studies at 5 mg/kg reported much higher numbers of complete regressions (up to 11/11) for FaDu Citation[15]. Despite the slightly higher drug dose we employed (6–7 mg/kg vs. 5 mg/kg), we were not able to achieve equivalent anti-tumour effect in the FaDu tumour. There are two likely reasons for this: the liposomal formulation has been modified since the original studies with FaDu were performed, i.e. the original studies in 2000 (Needham et al. Citation[7] and Kong et al. Citation[15]), used MPPC as the lysolipid, whereas this study used a formulation that included MSPC. This difference in lysolipid resulted in a slightly higher peak DOX release temperature of the formulation used herein, which could have an effect on kinetics and magnitude of drug delivery. It is also possible that the FaDu tumour evolved resistance to HT-LTSL-DOX through successive passages. The present study does not include a free doxorubicin control group, and the earlier studies Citation[7], Citation[15] did not include FaDu in vitro sensitivity to DOX, which makes it difficult to compare the results of the two studies. We have included a free doxorubicin control into another study that will be reported separately to better relate to earlier work.
Future directions
It is clear that an assessment of in vitro tumour doubling time may be useful, and analogous in vivo measurements could become practical in the clinic with the recent development of 18F-thymidine as a PET tracer for monitoring proliferation Citation[60]. This type of information may prove useful for patient selection and/or establishment of best treatment schedules for each patient.
The prior observation from our group showing that this treatment exhibits anti-vascular effects suggests that monitoring of perfusion after therapy would be important for assessment of efficacy Citation[16]. In addition, changes in vascular permeability due to HT may be responsible for some of the difference in efficacy among different tumour types Citation[17]. Therefore, measuring pre-treatment changes in permeability in response to HT may predict whether HT-LTSL-DOX will be effective in a given tumour type. Additional treatment to increase vascular permeability may also be of value.
Monitoring of drug delivery may still bear fruit as well. Recently, we reported that LTSL, loaded with doxorubicin and manganese (serving as MR contrast agent), could be used to spatially quantify drug release with MRI Citation[27] and that such quantification of drug dose distribution correlates with treatment outcome, within a single rat tumour line Citation[61]. Development of LTSL with both therapeutic and imaging capabilities may prove useful for clinical application.
Conclusion
HT-LTSL-DOX resulted in the best anti-tumour effect in each of the five tumour types, despite variations in the efficacy of this drug formulation between the five tumour types tested. These variations in efficacy are mostly correlated to in vitro doubling time. The implication of this finding is that for HT-LTSL-DOX, assessment of the rate of tumour growth or proliferation may be valuable in influencing design of clinical treatment regimens.
Acknowledgements
The authors thank Hong Yuan, Miriam Wahl, and David Adams for providing expert advice, and Ashley Manzoor and Eui-jung Moon for stimulating discussions.
Declaration of interest: Mark Dewhirst is a consultant and Chair of the Scientific Advisory Board for Celsion Corporation, which licensed this drug from Duke for clinical development. David Needham is the inventor of the LTSL, which was licensed from Duke University for clinical development. This work was supported by NIH grants GM40162 (to D.N.) and CA42745 (to M.W.D.). Drug was kindly supplied by the Celsion Corporation.
Related Research Data
References
- Allen TM, Cullis PR. Drug delivery systems: Entering the mainstream. Science 2004; 303: 1818–1822
- Drummond DC, Zignani M, Leroux JC. Current status of pH-sensitive liposomes in drug delivery. Progress in Lipid Research 2000; 39: 409–460
- Jensen SS, Andresen TL, Davidsen J, Hoyrup P, Shnyder SD, Bibby MC, Gill JH, Jorgensen K. Secretory phospholipase A2 as a tumor-specific trigger for targeted delivery of a novel class of liposomal prodrug anticancer etherlipids. Mol Cancer Ther 2004; 3: 1451–1458
- Lindner LH, Eichhorn ME, Eibl H, Teichert N, Schmitt-Sody M, Issels RD, Dellian M. Novel temperature-sensitive liposomes with prolonged circulation time. Clin Cancer Res 2004; 10: 2168–2178
- Hossann M, Wiggenhorn M, Schwerdt A, Wachholz K, Teichert N, Eibl H, Issels RD, Lindner LH. In vitro stability and content release properties of phosphatidylglyceroglycerol containing thermosensitive liposomes. Biochim Biophys Acta 2007; 1768: 2491–2499
- Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. Design of liposomes for enhanced local release of drugs by hyperthermia. Science 1978; 202: 1290–1293
- Needham D, Anyarambhatla G, Kong G, Dewhirst MW. A new temperature-sensitive liposome for use with mild hyperthermia: Characterization and testing in a human tumor xenograft model. Cancer Res 2000; 60: 1197–1201
- Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: A novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin Pharmacother 2009; 10: 333–343
- Matteucci ML, Anyarambhatla G, Rosner G, Azuma C, Fisher PE, Dewhirst MW, Needham D, Thrall DE. Hyperthermia increases accumulation of technetium-99m-labeled liposomes in feline sarcomas. Clin Cancer Res 2000; 6: 3748–3755
- Kong G, Dewhirst MW. Hyperthermia and liposomes. Int J Hyperthermia 1999; 15: 345–370
- Kong G, Braun RD, Dewhirst MW. Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature. Cancer Res 2001; 61: 3027–3032
- Anyarambhatla GR, Needham D. Enhancement of the phase transition permeability of DPPC liposomes by incorporation of MPPC: A new temperature-sensitive liposome for use with mild hyperthermia. J Liposome Res 1999; 9: 491–506
- Mills JK, Needham D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim Biophys Acta Oct 15, 2005; 1716(2)77–96
- Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: Review of animal and human studies. Clin Pharmacokinet 2003; 42: 419–436
- Kong G, Anyarambhatla G, Petros WP, Braun RD, Colvin OM, Needham D, Dewhirst MW. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: Importance of triggered drug release. Cancer Res 2000; 60: 6950–6957
- Chen Q, Tong S, Dewhirst MW, Yuan F. Targeting tumor microvessels using doxorubicin encapsulated in a novel thermosensitive liposome. Mol Cancer Ther 2004; 3: 1311–1317
- Chen Q, Krol A, Wright A, Needham D, Dewhirst MW, Yuan F. Tumor microvascular permeability is a key determinant for antivascular effects of doxorubicin encapsulated in a temperature sensitive liposome. Int J Hyperthermia 2008; 24: 475–482
- Hauck ML, LaRue SM, Petros WP, Poulson JM, Yu D, Spasojevic I, Pruitt AF, Klein A, Case B, Thrall DE, et al. Phase I trial of doxorubicin-containing low temperature sensitive liposomes in spontaneous canine tumors. Clin Cancer Res 2006; 12: 4004–4010
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). 2000. A phase I dose escalation tolerability study of ThermoDox™ (thermally sensitive liposomal doxorubicin) in combination with radiofrequency ablation (RFA) of primary and metastatic tumors of the liver; 2007. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00441376 (accessed 14 February 2010)
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). 2000. A phase I dose escalation study of heat activated liposome delivery of doxorubicin and radiofrequency ablation of primary and metastatic tumors of the liver; 2004. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00093444 (accessed 14 February 2010)
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). 2000. A phase III, randomized, double-blinded, dummy-controlled study of the efficacy and safety of ThermoDox® (thermally sensitive liposomal doxorubicin) in combination with radiofrequency ablation (RFA) compared to RFA alone in the treatment of non-resectable hepatocellular carcinoma; 2008. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00617981 (accessed 14 February 2010)
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). 2000. A phase I, dose escalation and pharmacokinetics study of temperature sensitive liposome encapsulated doxorubicin(ThermoDox™) and hyperthermia in patients with local-regionally recurrent breast cancer; 2006. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00346229?term=ThermoDox&rank=4 (accessed 14 February 2010)
- Mayer LD, Bally MB, Cullis PR. Uptake of adriamycin into large unilamellar vesicles in response to a pH gradient. Biochim Biophys Acta 1986; 857: 123–126
- Madden TD, Harrigan PR, Tai LC, Bally MB, Mayer LD, Redelmeier TE, Loughrey HC, Tilcock CP, Reinish LW, Cullis PR. The accumulation of drugs within large unilamellar vesicles exhibiting a proton gradient: A survey. Chem Phys Lipids 1990; 53: 37–46
- Mayer LD, Tai LC, Ko DS, Masin D, Ginsberg RS, Cullis PR, Bally MB. Influence of vesicle size, lipid composition, and drug-to-lipid ratio on the biological activity of liposomal doxorubicin in mice. Cancer Res 1989; 49: 5922–5930
- Wright A. Drug Loading And Release from Thermally Sensitive Liposome. Duke University, Durham, NC 2006
- Viglianti BL, Abraham SA, Michelich CR, Yarmolenko PS, MacFall JR, Bally MB, Dewhirst MW. In vivo monitoring of tissue pharmacokinetics of liposome/drug using MRI: Illustration of targeted delivery. Magnetic Resonance in Medicine 2004; 51: 1153–1162
- DeLean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: Application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol 1978; 235: E97–102
- Prescott DM, Charles HC, Sostman HD, Page RL, Thrall DE, Moore D, Oleson JR, Dewhirst MW. Manipulation of intra- and extracellular pH in spontaneous canine tumours by use of hyperglycaemia. Int J Hyperthermia 1993; 9: 745–754
- Allard P, Zoubeidi A, Nguyen LT, Tessier S, Tanguay S, Chevrette M, Aprikian A, Chevalier S. Links between Fer tyrosine kinase expression levels and prostate cell proliferation. Mol Cell Endocrinol 2000; 159: 63–77
- Aslakson CJ, Rak JW, Miller BE, Miller FR. Differential influence of organ site on three subpopulations of a single mouse mammary tumor at two distinct steps in metastasis. Int J Cancer 1991; 47: 466–472
- NCI/NIH Developmental Therapeutics Program DS. NCI/NIH Developmental Therapeutics Program, Discovery Series. NCI/NIH; 2010. Available from: http://dtp.nci.nih.gov/docs/misc/common_files/cell_list.html (accessed 14 February 2010)
- Perez LA, Dombkowski D, Efird J, Preffer F, Suit HD. Cell proliferation kinetics in human tumor xenografts measured with iododeoxyuridine labeling and flow cytometry: A study of heterogeneity and a comparison between different methods of calculation and other proliferation measurements. Cancer Res 1995; 55: 392–398
- Rodriguez R, Ritter MA, Fowler JF, Kinsella TJ. Kinetics of cell labeling and thymidine replacement after continuous infusion of halogenated pyrimidines in vivo. Int J Radiat Oncol Biol Phys 1994; 29: 105–113
- Jones E, Alvarez Secord A, Prosnitz LR, Samulski TV, Oleson JR, Berchuck A, Clarke-Pearson D, Soper J, Dewhirst MW, Vujaskovic Z. Intra-peritoneal cisplatin and whole abdomen hyperthermia for relapsed ovarian carcinoma. Int J Hyperthermia 2006; 22: 161–172
- Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, Felix R, Schlag PM. Hyperthermia in combined treatment of cancer. Lancet Oncol 2002; 3: 487–497
- Tilly W, Gellermann J, Graf R, Hildebrandt B, Weissbach L, Budach V, Felix R, Wust P. Regional hyperthermia in conjunction with definitive radiotherapy against recurrent or locally advanced prostate cancer T3 pN0 M0. Strahlenther Onkol 2005; 181: 35–41
- Working PK, Newman MS, Huang SK, Mayhew E, Vaage J, Lasic DD. Pharmacokinetics, Biodistribution and Therapeutic Efficacy of Doxorubicin Encapsulated in Stealth® Liposomes (Doxil®). J Liposome Res. 1994; 4(1)667–687
- Huang SK, Stauffer PR, Hong K, Guo JW, Phillips TL, Huang A, Papahadjopoulos D. Liposomes and hyperthermia in mice: Increased tumor uptake and therapeutic efficacy of doxorubicin in sterically stabilized liposomes. Cancer Res Apr 15, 1994; 54(8)2186–2191
- Dunn J. Doxorubicin-induced cardiomyopathy. J Pediatr Oncol Nurs 1994; 11: 152–160
- Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res 1994; 54: 4564–4568
- Kong G, Braun RD, Dewhirst MW. Hyperthermia enables tumor-specific nanoparticle delivery: Effect of particle size. Cancer Res 2000; 60: 4440–4445
- Karino T, Koga S, Maeta M. Experimental studies of the effects of local hyperthermia on blood flow, oxygen pressure and pH in tumors. Jpn J Surg 1988; 18: 276–283
- Needham D, Dewhirst MW. The development and testing of a new temperature-sensitive drug delivery system for the treatment of solid tumors. Adv Drug Deliv Rev 2001; 53: 285–305
- Brown GA, McPherson JP, Gu L, Hedley DW, Toso R, Deuchars KL, Freedman MH, Goldenberg GJ. Relationship of DNA topoisomerase II alpha and beta expression to cytotoxicity of antineoplastic agents in human acute lymphoblastic leukemia cell lines. Cancer Res 1995; 55: 78–82
- Giaccone G, Gazdar AF, Beck H, Zunino F, Capranico G. Multidrug sensitivity phenotype of human lung cancer cells associated with topoisomerase II expression. Cancer Res 1992; 52: 1666–1674
- Kasahara K, Fujiwara Y, Sugimoto Y, Nishio K, Tamura T, Matsuda T, Saijo N. Determinants of response to the DNA topoisomerase II inhibitors doxorubicin and etoposide in human lung cancer cell lines. J Natl Cancer Inst 1992; 84: 113–118
- Fry AM, Chresta CM, Davies SM, Walker MC, Harris AL, Hartley JA, Masters JR, Hickson ID. Relationship between topoisomerase II level and chemosensitivity in human tumor cell lines. Cancer Res 1991; 51: 6592–6595
- Szulawska A, Czyz M. [Molecular mechanisms of anthracyclines action]. Postepy Hig Med Dosw 2006; 60: 78–100
- Nielsen D, Maare C, Skovsgaard T. Cellular resistance to anthracyclines. Gen Pharmacol 1996; 27: 251–255
- Duan Z, Feller AJ, Toh HC, Makastorsis T, Seiden MV. TRAG-3, a novel gene, isolated from a taxol-resistant ovarian carcinoma cell line. Gene 1999; 229: 75–81
- Smith V, Raynaud F, Workman P, Kelland LR. Characterization of a human colorectal carcinoma cell line with acquired resistance to flavopiridol. Mol Pharmacol 2001; 60: 885–893
- van Brussel JP, van Steenbrugge GJ, Romijn JC, Schroder FH, Mickisch GH. Chemosensitivity of prostate cancer cell lines and expression of multidrug resistance-related proteins. Eur J Cancer 1999; 35: 664–671
- El-Kareh AW, Secomb TW. Two-mechanism peak concentration model for cellular pharmacodynamics of Doxorubicin. Neoplasia 2005; 7: 705–713
- Owen CS. Dependence of proton generation on aerobic or anaerobic metabolism and implications for tumour pH. Int J Hyperthermia 1996; 12: 495–499
- Wike-Hooley JL, van den Berg AP, van der Zee J, Reinhold HS. Human tumour pH and its variation. Eur J Cancer Clin Oncol 1985; 21: 785–791
- Prescott DM, Charles HC, Poulson JM, Page RL, Thrall DE, Vujaskovic Z, Dewhirst MW. The relationship between intracellular and extracellular pH in spontaneous canine tumors. Clin Cancer Res 2000; 6: 2501–2505
- Atema A, Buurman KJ, Noteboom E, Smets LA. Potentiation of DNA-adduct formation and cytotoxicity of platinum-containing drugs by low pH. Int J Cancer 1993; 54: 166–172
- Born R, Eichholtzwirth H. Effect of different physiological conditions on the action of adriamycin on Chinese hamster cells in vitro. Brit J Cancer 1981; 44: 241–246
- Muzi M, Spence AM, O’Sullivan F, Mankoff DA, Wells JM, Grierson JR, Link JM, Krohn KA. Kinetic analysis of 3′-deoxy-3′-18F-fluorothymidine in patients with gliomas. J Nucl Med 2006; 47: 1612–1621
- Ponce AM, Viglianti BL, Yu D, Yarmolenko PS, Michelich CR, Woo J, Bally MB, Dewhirst MW. Magnetic resonance imaging of temperature-sensitive liposome release: Drug dose painting and antitumor effects. J Natl Cancer Inst 2007; 99: 53–63