Abstract
Purpose: The anti-cancer effect of β-lapachone (β-lap) is positively related to the cellular activity of NAD(P)H:quinone oxidoreductase (NQO1). Heat shock has been reported to elevate cellular NQO1. The effect of heating on the NQO1 expression in human osteosarcoma cells (HOS) and the response of the cells to the combined treatment with β-lap and hyperthermia was investigated.
Materials and methods: The effects of β-lap alone, hyperthermia alone and in combination to cause clonogenic death and apoptosis in HOS cells were elucidated. The effect of heating on the NQO1expression was evaluated with western blot analysis. The effect of β-lap on the cell cycle distribution was elucidated with flow cytometry and to cause DNA damage was determined by assessing the γH2AX foci formation.
Results: Treatment of HOS cells with β-lap at 42°C was markedly more effective than that at 37°C in causing clonogenic cell death. Heating caused a long-lasting up-regulation of NQO1 in the cells, and sensitised the cells to β-lap. The γH2AX foci formation was increased immediately after β-lap treatment and preheating increased the β-lap-induced γH2AX foci formation.
Conclusions: The sensitivity of HOS cells to β-lap was increased not only during heating but also after heating as demonstrated by the increase in the clonogenic cell death and γH2AX foci formation. The increase in β-lap sensitivity after heating appeared to be due to the heat-induced elevation of NQO1 activity.
Introduction
β-Lapachone (β-lap; 3,4-dihydro-2,2-dimethyl-2H-naptho[1,2-b]puran-5,6-dione), a quinine-containing compound originally obtained from lapacho trees in South America, is a novel anticancer drug expressing strong cytotoxicity towards a variety of animal and human cancer cells Citation[1]. Boothman et al. Citation[2–5] reported that β-lap is a bioreductive pro-drug which undergoes two-electron reduction mediated by NAD(P)H:quinine oxidoreductase (NQO1) using NADH and NADPH as electron sources. It was further suggested that the two-electron reduced form of β-lap is unstable and thus it is rapidly oxidised to the original β-lap form, creating futile cycling between oxidised and reduced forms of β-lap. Consequently, the cellular contents of NADH, NADPH and ATP are severely depleted, and cytoplasmic Ca2+ is elevated. This chaotic intracellular environment activates Ca2+-dependent calpain or similar proteases, resulting in degradations of vital proteins, including p53 and poly (ADP-ribose) polymerase-1 (PARP-1), thereby leading to caspase-independent apoptosis Citation[2–5]. Indications are that the one-electron reduced β-lap (i.e. the semi-quinone form of β-lap) formed during the oxidation process of the two-electron reduced β-lap induces redox cycling reactions, which subsequently generate reactive oxygen species (ROS) and cause DNA damage leading to caspase-dependent cell death Citation[2–5]. β-Lap is currently in phase II clinical trials for the treatment of pancreatic adenocarcinoma in combination with gemcitabine. Recently, β-lap-containing poly (ethylene glycol)-block-poly (D,L-lactide) (PEG-PLA) polymer micelles has been developed to overcome the poor aqueous solubility of β-lap in treating NQO1-expressing human tumours Citation[6] or polymer implant Citation[7].
The NQO1, also known as DT-diaphorase, (EC 1.6.99.2), catalyses two-electron reduction of endogenous and exogenous quinones, quinone imines and nitrogen oxides using H+ from NADH and NADPH Citation[8–14]. The two-electron reduced toxic compounds are subsequently conjugated with glutathione or glucuronic acid, and excreted from the cells Citation[9], Citation[10], Citation[15]. Therefore, NQO1 has been regarded as a detoxifying enzyme with cancer prevention property. On the other hand, the NQO1-mediated two-electron reduction of certain bioreductive drugs such as β-lap, mitomycin C (MMC) Citation[8–12], Citation[16–18] and EO9 Citation[8], Citation[13], Citation[14], Citation[19], Citation[20] triggers a series of molecular changes in the cells leading to cell death. Therefore, the sensitivity of cancer cells to these bioreductive drugs including β-lap is positively related to the NQO1 activity in the cells. Importantly, the level of NQO1 in many human tumours is significantly greater than that in adjacent normal tissues Citation[21], Citation[22], indicating that β-lap may be toxic preferentially to tumours relative to normal tissues.
It has been known that DNA double strand breaks (DSBs) caused by various stresses lead to focal phosphorylation of H2AX at serine 139 resulting in generation of γH2AX Citation[23]. It was reported that β-lap causes DNA damage, thereby inducing γH2AX foci formation Citation[24], and we have recently demonstrated that β-lap-induced γH2AX foci formation is markedly increased when the cells are pre-treated with ionising radiation Citation[25] or cisplatin Citation[26]. Takahash et al. Citation[27] observed that heat shock induced γH2AX foci, and concluded that the heat-induced γH2AX foci formation was due to DNA DSB. On the other hand, Hunt et al. Citation[28] reported that the heat-induced γH2AX foci formation was due not to DNA DSB but to the heat-induced chromatin alteration. Furthermore, the heat-induced radiosensitisation was found to be due to heat-induced perturbations of the formation of γH2AX/MDC1/53BP1 complex, an early DNA damage signalling pathway in after irradiation Citation[28], Citation[29]. Importantly, the heat-induced γH2AX formation did not appear to play a role in heat-induced cell death Citation[30].
Hyperthermia is an effective anti-cancer modality for human tumours particularly when it is used in combination with radiotherapy or chemotherapy Citation[31–33]. While heating at temperature higher than 42–43°C causes tumour cell death and vascular destruction, heating at relatively mild temperatures, e.g. 40–42°C, increases the cellular sensitivity of cancer cells to radiotherapy or various chemotherapy drugs, and increases tumour blood perfusion, which not only increases drug delivery to tumour cells but also improves tumour oxygenation Citation[34]. We have reported that β-lap and heating at 41–42°C caused synergistic cell death Citation[35], Citation[36]. Furthermore, we observed that tumour cells become sensitive to β-lap after heating due to the heat-induced elevation of NQO1 activity in the cancer cells Citation[35], Citation[36].
Human osteosarcoma (OS) is the most common primary malignant bone tumour with an estimated incidence of 4–5 per million people. OS is usually a disease of young people, and it frequently occurs in the second decade with about 60% of patients under the age of 25 Citation[37]. Although the introduction of adjuvant chemotherapy in conjunction with surgical treatment has improved the long-term survival rate of OS patients in recent years, OS still remains a disease difficult to cure due in part to inherent or acquired resistance to certain chemotherapeutic agents Citation[37], Citation[38]. Therefore, a more effective and novel approach is strongly required for the management of patients with OS. Liu et al. Citation[39] reported that β-lap induced necrotic death in a human osteosarcoma cell line (U2-OS). The purpose of the present study was to further reveal the effect of β-lap on OS cells and explore the feasibility of treating OS patients with β-lap alone or in combination with hyperthermia. We determined the influence of heating on various cellular and molecular changes caused by β-lap treatment such as clonogenic death, perturbation of cell cycle progression and γH2AX foci formation in OS.
Methods
Cells and β-lapachone
Human osteosarcoma cells (HOS cells) were obtained from Riken Cell Bank (Tsukuba, Japan) and cultured in T-type plastic culture flasks with MEM Eagle's medium (Gibco, Grand Island, NY) containing 10% heat-inactivated foetal bovine serum (FBS) (JRH Bioscience, Lenexa, KS), 5% non-essential amino acids (Sigma-Aldrich), penicillin (100 U/mL) and streptomycin (100 µg/mL) (Gibco, St. Louis, MO) in 5% CO2 incubator under humidified air at 37°C. β-Lap (Biomol International, Plymouth Meeting, PA) was dissolved in dimethyl sulphoxide (DMSO) at 5 mM and stored at −20°C. Immediately before use, the stock β-lap was thawed and diluted to desired concentrations with culture medium.
Effect of β-lap on the clonogenic cell survival
Stock HOS cells were treated with a mixture of 0.25% trypsin and 0.53 mM EDTA at 37°C for 3 min, rinsed twice with culture medium, and plated into 25 cm2 T-type plastic tissue culture flasks with 5 mL of medium. After incubation overnight at 37°C under 5% CO2 atmosphere, the medium was replaced with fresh medium only or medium containing specific amounts of β-lap and the cells were incubated for varying lengths of time at 37°C. The cells were then rinsed twice with medium, and cultured with complete medium for 7–9 days at 37°C. The resultant colonies were fixed with a mixture of methanol and acetic acid (10 : 1 v/v), stained with 1% crystal violet, and the colonies containing more than 50 cells were counted. The role of NQO1 in the cell death caused by β-lap was studied by determining the influence of dicoumarol, an inhibitor of NQO1 Citation[3], Citation[25], Citation[26], Citation[35], Citation[36], on the β-lap-induced clonogenic cell death.
Combined effect of β-lap and heating
HOS cells were plated in 25 cm2 T-type plastic culture flasks, incubated overnight, and the medium was replaced with fresh medium only or medium containing various concentrations of β-lap. The flasks were tightly closed, the cap area was sealed with wax paper, and the flasks were immersed into a water bath pre-heated at 41–44°C. After heating for 1 h, the cells were subjected to various assays such as clonogenic and apoptotic cell death, cell cycle distribution and NQO1 expression.
Western blot analysis for NQO1 expression
HOS cells were heated at 42°C for 1 h as described above, harvested using trypsin solution, rinsed twice with PBS, and treated with lysis buffer as previously reported Citation[25], Citation[26], Citation[35], Citation[36]. The lysates were mounted on 15% polyacrylamide gels (30 µg proteins/each), and electrophoresced. The proteins were transferred to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ), blocked by incubating the membrane with 5% non-fat dry milk for 1 h, and washed with Tris-buffered saline containing Tween 20 (TBST). The membranes were incubated with anti-NQO1 antibody (1 : 100 dilution, sc-16464; Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C and then with secondary horseradish peroxidase (HRP)-conjugated anti-goat antibody (1 : 1000 dilution, Santa Cruz Biotechnology). The signals were visualised on X-ray film using ECL detection reagents (Amersham Bioscience).
Cell cycle analysis
The cell cycle distributions after various treatments were assayed with flow cytometric method Citation[37]. The control or treated cells were harvested with trypsin-EDTA treatment, rinsed, and fixed with 70% methanol at 4°C overnight. After centrifugation, the cells were incubated for 30 min at room temperature with 0.25 mg/mL RNase (Sigma) in PBS, and treated with 50 µg/mL propidium iodide (PI) (Sigma) in darkness at 37°C for 1 h. The stained cells were subjected to cell cycle distribution analysis with a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA).
γH2AX formation by β-lap at different cell cycle phases
We studied whether the γH2AX formation by β-lap is cell cycle dependent. Exponentially growing HOS cells in T-flasks were incubated with 7.5 µM β-lap for 1 h at 37°C, washed with PBS twice, fixed with cold 70% methanol, and kept at −20°C overnight. The cells were centrifuged and rinsed with PBS containing 0.05% Tween 20 (TPBS). The cells were then treated with TPBS containing 4% bovine serum albumin (Sigma) for 15 min, rinsed with TPBS, and incubated with anti-γH2AX monoclonal antibody (JBW301, Upstate Biotechnology, Lake Placid, NY) at 300-fold dilution for 60 min, and rinsed with TPBS. The cells were then incubated for 60 min with the fluorescein isothiocyanate (AlexaFluor 488-conjugated anti-mouse IgG second antibody, Molecular Probes, Eugene, OR) at a 400-fold dilution for 60 min, and rinsed with TPBS. After incubation with 0.25 mg/mL RNase in PBS for 30 min at room temperature, the cells were treated with 50 µg/mL PI in darkness at 37°C for 1 h, subjected to flow cytometric analysis as described above, and the extent of γH2AX expressing in different cell cycle phases was assessed.
Combined effects of β-lap and heating on γH2AX formation
For microscopic studies of the γH2AX foci formation, HOS cells were plated on glass slides (Gold Seal, Portsmouth, NH) in plastic Petri dishes, and the dishes were covered and maintained at 37°C for 1 h. The slides on which the cells were firmly adhered were then placed in 50 mL plastic tubes containing culture medium with or without β-lap, incubated for 1 h at 37°C, rinsed twice with medium, incubated with culture medium for varying lengths of time and the γH2AX formation was assessed. To investigate the γH2AX formation by the combined treatment of heating and β-lap, the plastic tubes in which the slides were placed were filled with medium containing β-lap, closed and sealed with wax-paper. The cells were heated by placing the tubes horizontally in a water bath at 42°C for 1 h; the slides were rinsed twice with PBS and the γH2AX foci formation was assessed as follows. The cells on the slides were fixed with cold methanol for 20 min and acetone for 7 seconds and dried. After treating with 3% skimmed milk in TPBS for 10 min and washing with TPBS, the cells were incubated with anti-γH2AX monoclonal antibody (JBW301) at a 300-fold dilution for 60 min at room temperature and rinsed with TPBS. The cells were further incubated with fluorescein isothiocyanate (AlexaFluor 488-conjugated anti-mouse IgG second antibody) at 400-fold dilution for 60 min at room temperature and rinsed with TPBS. The cells were stained and mounted with anti-fade DAPI solution (Molecular Probes), and the photographs of the cells were taken with a fluorescence microscope (Axiovert 2, Carl Zeiss MicroImaging, Thornwood, NY). All the images were captured using the same parameters to directly compare the samples. The total numbers of cells stained with DAPI (blue fluorescence), and the number of cells expressing γH2AX foci (green fluorescence) were counted. The numbers of γH2AX foci in each cells were also counted by imaging analysis of the microscopic images obtained using a ×40 objective lens. At least 20 cells were registered for each sample Citation[25], Citation[26].
Results
Effect of β-lap treatment on the clonogenic cell survival
shows the surviving fractions of the HOS cells treated with different concentrations of β-lap for 1–8 h. The surviving fraction decreased to about 0.12 after treating with 5 µM β-lap for 2 h. The 2-h treatments with 7.5 µM β-lap or 10 µM β-lap were only slightly more effective than that with 5 µM. However, when treatment was prolonged for longer than 2 h, the differences between the surviving fractions of the cells treated with different concentrations of β-lap became markedly large. For example, the treatment of cells with 5 µM β-lap for 4 h reduced the surviving fraction by slightly more than 1 log while the treatment with 10 µM β-lap for 4 h reduced the surviving faction by more than 5 logs.
Figure 1. Effect of β-lap alone or with dicoumarol on the clonogenic survival of HOS cells. (A) HOS cells were treated with 5, 7.5 and 10 µM β-lap for 1, 2, 4 and 8 h, cultured for 7–9 days, and the surviving fractions were calculated. Averages of 4 experiments with 1 SE are shown. (B) HOS cells were incubated with 50 µM dicoumarol alone, 7.5 µM β-lap alone or in combination for 1 h at 37°C and the clonogenic survivals were determined. Averages of 4 experiments with 1 SE are shown.
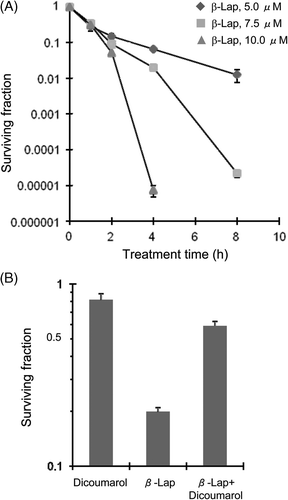
shows that the 1-h incubation of HOS cells with 50 µM dicoumarol, an inhibitor of NQO1 Citation[26], Citation[31], Citation[35], Citation[36], slightly reduced cell survival, and that the 1-h incubation of cells with 7.5 µM β-lap reduced the surviving fraction to 0.25. On the other hand, the 1-h incubation with 7.5 µM β-lap together with 50 µM dicoumarol decreased the surviving fraction only to 0.81 demonstrating that the inhibition of NQO1 activity with dicoumarol suppressed the effect of β-lap to cause clonogenic cell death in HOS cells. This result demonstrated that the cell death caused by β-lap is dependent on NQO1 activity.
Effects of β-lap on cell cycle distribution and γH2AX formation
The changes in cell cycle distribution as a result of the 1-h treatment with 7.5 µM β-lap in HOS cells are shown in . About 40% of HOS cells were in G0/G1 phase before the treatment and the G0/G1 cell population markedly decreased to about 10% at 24 h after the treatment. About 32% of HOS ells were in S-phase before the treatment and the S-phase cell population increased to 55% at 12 h, but it markedly decreased to only 17% at 24 h after the treatment. The G2/M cell population before treatment was 28% and it increased to 67% at 24 h after the treatment as the S-phase cell population decreased following the peak increase at 12 h. As the G2/M cell population decreased 48–72 h after the treatment, the G0/G1 cell population and the apoptotic cells, i.e. subG1 cells, increased. In short, when HOS were treated with 7.5 µM β-lap for 1 h, the cells were initially arrested in S-phase and then in G2/M phases. As the cells were released from the G2/M arrest from 24 h after the treatment, the G0/G1 cell population and apoptotic cell population increased.
Figure 2. Effect of β-lap on cell cycle and γH2AX expression. HOS cells were treated with 7.5 µM β-lap for 1 h, incubated in regular medium at 37°C, harvested 0, 3, 6, 12, 24, 48, 72 h later, and the cell cycle distribution was analysed with flow cytometry method (A and B). γH2AX formation at different cell cycle phases after treatment with 7.5 µM β-lap for 1 h (C). Cells were treated with 7.5 µM β-lap for 1 h at 37°C, incubated with regular medium for 30 min, immunostained for γH2AX and then the cell cycle distribution was assessed by flow cytometry.
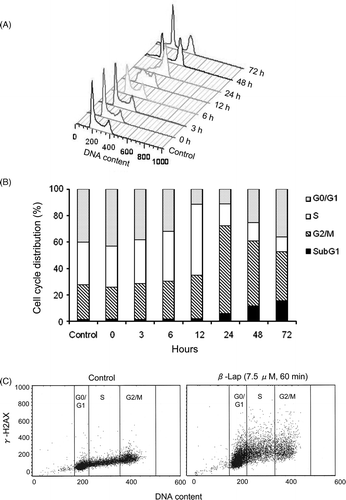
(left) shows that the background γH2AX expression in G2/M cells was as much as 2-fold greater than that in G0/G1 cells. (right) shows that the γH2AX formation significantly increased 30 min after treating the cells with 7.5 µM β-lap for 1 h and that the γH2AX formation increase almost equally in all the cell cycle phases demonstrating the induction of DNA double strand breaks by β-lap in HOS is independent of cell cycle phase.
Effect of heating alone or in combination with β-lap on cell survival
shows the clonogenic survival of HOS cells after heating at different temperatures for 1 h. While no significant clonogenic cell death occurred when the cells were heated at 41°C or 41.5°C for 1 h, the heating at 42°C for 1 h reduced the surviving fraction to about 0.8. The surviving fractions progressively decreased as the heating temperature was raised above 42.5°C. shows the effect of heating on the NQO1 expression in HOS cells as determined with western blot method. A small amount of NQO1 protein could be detected before the heating and the NQO1 protein contents slightly increased 3 h after heating at 42°C for 1 h, and further increased 12–24 h after the heating.
Figure 3. Heat-induced potentiation of the cytotoxicity of β-lap and heat-induced change in the cell cycle distribution. (A) HOS cells were treated with 7.5 µM β-lap for 1 h at different temperatures, washed, cultured for 7–9 days, and the surviving fractions were calculated. Average of 6 experiments is shown. (B) Western blot analysis for NQO1 expression in HOS cells at various times after heating at 42°C for 1 h. (C) Cell survivals were determined after treating the cells with heating at 42°C for 1 h alone, incubation with 7.5 µM β-lap at 37°C for 1 h alone or treating the cells with 7.5 µM β-lap at 42°C for 1 h. Cell survival was also determined after heating the cells at 42°C for 1 h and then treating the cells with 7.5 µM β-lap for 1 h at various times after the heating. The dotted line indicates the expected cell survival when the heating and β-lap treatment reduced the survival of the cells additively. Averages of 6 experiments with 1 SE are shown. (D) Effect of heating on the cell cycle progression is shown. Cells were heated at 42°C for 1 h, incubated in regular medium at 37°C for 0–24 h and the cell cycle distribution was assessed with flow cytometry.
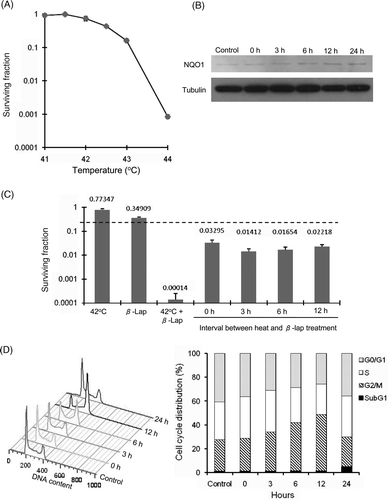
The clonogenic cell deaths caused by heating in combination with β-lap treatment are shown in . The heating at 42°C for 1 h decreased the surviving fraction to 0.77, and the incubation with 7.5 µM β-lap for 1 h at 37°C decreased the surviving fraction to 0.35. When the cells were treated with 7.5 µM β-lap at 42°C for 1 h the surviving fraction decreased to as low as 0.00014. If the combination of heating and β-lap killed the HOS cells in an additive manner, the surviving fraction would have decreased only to 0.27 (i.e. 0.77 × 0.35: dotted line in ) instead of 0.00014. It was therefore apparent that the cells died of synergistic interaction between the β-lap treatment and 42°C heating. When cells were heated at 42°C for 1h and then treated with β-lap at 0–12 h after the heating, the extent of cell death was greater than additive, indicating that pre-heating sensitised the cells to β-lap.
Cell cycle distribution after heat treatment
shows the changes in the distribution of HOS cells in different cell cycle phases after heating at 42°C for 1 h. The cell population in G2/M phase before heating was about 26.6% and it gradually increased to about 43.4% at 12 h after heating. At the same time, the cell populations in G0/G1 phase and S phase slightly decreased, indicating the progression of HOS cells through the G2/M phase was blocked after heating. The heated cells were eventually released from the G2/M block so that the cell cycle distribution at 24 h after heating was similar to that of unheated control cells except for a slight increase in the apoptotic cell population.
Combined effect of heating and β-lap on γH2AX formation
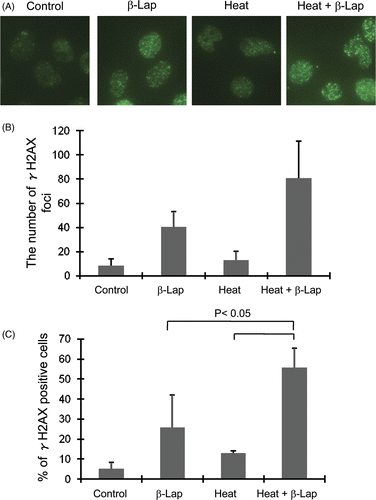
The γH2AX formations in the HOS cells by β-lap alone, heating alone or in combination was investigated with immunohistochemistry method. shows the immunohistochemical staining for γH2AX foci and shows the average number of γH2AX foci in each cell. The γH2AX foci formation 30 min after 1h treatment with 7.5 µM β-lap was significantly greater than that in the control cells while the γH2AX foci formation 24 h after 1 h heating at 42°C was only slightly greater than that in the control cells. The γH2AX foci formation in the cells which were heated at 42°C for 1 h and then treated 24 h later with β-lap was greater than that in the cells treated with β-lap alone or heating alone. The percentage of cells positive with γH2AX in the group preheated and treated with β-lap 24 h later was significantly greater than that in the groups treated with β-lap alone or heating alone ()
Figure 4. Effect of β-lap and heat treatment on the phosphorylation of H2AX. β-Lap: Cells were incubated with 7.5 µM β-lap for 1h, incubated at 37°C for 30 min and the expression of γH2AX was determined. Heat: cells were heated at 42°C for 1 h, incubated at 37°C for 24 h and the expression of γH2AX was determined. Heat + β-Lap: cells were heated at 42°C for 1 h, incubated at 37°C for 24 h, treated with 7.5 µM β-lap for 1 h, incubated at 37°C for 30 min and the expression of γH2AX was determined. (A) Immunofluorescent staining for γH2AX; (B) average number of γH2AX foci in each cell; (C) percentages of γH2AX positive cells.
Discussion
Whereas β-lap has been demonstrated to be cytotoxic against a variety of human and animal cancer cells Citation[1–5], Citation[25], Citation[26], Citation[35], Citation[36], little is known about the effect of β-lap against osteosarcoma, one of the most frequent bone tumours. In the present study, β-lap treatment caused γH2AX formation in a cell cycle independent manner ( and ), triggered S-phase arrest followed by G2/M-phase arrest (), and induced NQO1-dependent clonogenic death in HOS cells (). Simultaneous treatment of HOS cells with heating and β-lap induced synergistic cell death and pre-heating increased the sensitivity of the cells to β-lap applied 0–24 h after the heating ( and ). It appeared that the pre-heating up-regulated the expression of NQO1, which in turn increased the sensitivity of the cells to β-lap ().
The clonogenic death of HOS cells by β-lap in the present study could be suppressed by dicoumarol, an inhibitor of NQO1 Citation[5], Citation[25], Citation[26], Citation[35], Citation[36] () demonstrating the pivotal role of NQO1 in the β-lap-induced death of HOS cells. It has been reported that β-lap causes NQO1-dependent cell death by depleting the cellular contents of NADH, NADPH and ATP, elevating the cytoplasmic Ca2+ and enhancing the calcium-dependent proteolytic pathway Citation[2–5]. It has also been shown that ROS are generated by β-lap, thereby triggering signal pathways for caspase-dependent apoptosis Citation[5]. Which of these two pathways, i.e. calcium-dependent proteolytic pathway or ROS generation, plays the central role in the β-lap-induced cell death may be dependent on various factors such as cell type and concentration of β-lap, although both pathways depend on the NQO1-mediated two-electron reduction of β-lap. In a study with human breast cancer cell line, low concentrations of β-lap caused cell cycle-independent and caspase-mediated apoptosis, whereas high concentrations of β-lap killed the cells through the cell cycle independent activation of a non-caspase proteolytic pathway in human breast cancer cells Citation[5]. In the present study, the 4-h treatment of HOS cells with 10 µM β-lap was profoundly more effective than the 4-h treatments with 5 µM or 7.5 µM β-lap in causing clonogenic cell death (). Such a marked increase in the cytotoxcity of β-lap by a small increment in the concentration of β-lap, i.e. from 5–7.5 µM to 10 µM, indicated that the mechanism responsible for the death of HOS cells caused by β-lap may also be dependent on β-lap concentration as previously reported Citation[5]. When HOS cells were treated with 7.5 µM β-lap for 1 h, the Go/G1 phase cell population gradually decreased, and at the same time the cells were arrested first in S phase and then in G2/M phase (). As the cells were released from the G2/M arrest, beginning 24 h after β-lap treatment, the Go/G1 cell population began to recover and also apoptosis began to increase indicating that the apoptosis caused by β-lap in the HOS cells occurred from the G2/M arrest. This observation was at variance with the report by others that β-lap activated the S phase checkpoint, and induced apoptosis from the S phase arrest in human breast cancer cells Citation[1].
The clonogenic survival of HOS cells decreased by heating in a temperature-dependent manner () and heating caused long-lasting up-regulation of NQO1, as observed in other cell lines Citation[35], Citation[36]. As shown in , the clonogenic death of HOS cells by treating the cells with β-lap at 42°C was markedly greater than that expected to occur when the two treatments interacted additively. The apparent synergism between β-lap and heating in killing HOS cells observed in the present study was in good agreement with the results we observed in other cell lines Citation[35], Citation[36]. The cause of such synergistic cell death upon treating cells with β-lap at elevated temperatures is unclear, but it is a well-known fact that hyperthermia potentiates the effect of various anti-cancer drugs Citation[33]. Therefore, the remarkable increase in the effect of β-lap by heating is not a unique phenomenon for β-lap. However, it is rather interesting that HOS cells were sensitive to β-lap 0–24 h after heating at 42°C for 1 h (). We previously attributed such an increase in the β-lap sensitivity of cancer cells following exposure of the cells to heating Citation[35], Citation[36], ionising radiation Citation[25] and cisplatin Citation[26] to the up-regulation of NQO1 in the cells by the therapeutic stresses. Therefore, it would be reasonable to conclude that the post-heating increase in β-lap sensitivity in HOS was due to the long-lasting elevation of NQO1 expression as shown in . However, in light of the previous report by other investigators that the cell death caused by β-lap was cell cycle phase-dependent Citation[1], the possibility could not be excluded that the increase in the effect of β-lap against the pre-heated HOS cells was due to accumulation of the cells in the cell cycle phase(s) which is preferentially sensitivity to β-lap. As shown in , small changes occurred in the cell cycle distribution within 6 h after heating, while the sensitivity to β-lap maximally increased 3 h after heating in HOS cells (). Furthermore, the β-lap-induced γH2AX formation, i.e. DNA damage, was independent of cell cycle phase (). Therefore, we may conclude that the increase in β-lap sensitivity after heating was not due to heat-induced change in cell cycle distribution but to the heat-induced up-regulation of NQO1.
The background γH2AX expression in G2/M cells was about 2-fold greater than that in Go/G1 cells (). This difference may be related to the cellular DNA content which is 2-fold greater in G2/M cells than that in Go/G1 cells. It was previously reported that β-lap causes DNA damage, thereby inducing γH2AX foci formation Citation[24]. As shown in and , the γH2AX foci formation in HOS cells increased soon after β-lap treatment, indicating that the induction of DNA double strand breaks is an early and upstream event of the signalling pathways of the cell death caused by β-lap. When HOS cells were pre-heated and then treated 24 h later with β-lap for 1 h, the % of cells expressing γH2AX foci was significantly greater (p < 0.05) than that in the cells treated with heating or β-lap alone (). The number of γH2AX foci in each cell also increased when the cells were heated and treated 24 h later with β-lap, although the increase was not statistically significant. Therefore it may be reasonable to conclude that the heat-induced potentiation of the β-lap-induced clonogenic death in HOS cells was a result, at least in part, of the heat-induced potentiation of the β-lap-induced DNA damage. In this regard, it should be noted that whereas the γH2AX foci formation by heat is not due to DNA DBS as recently reported Citation[28–30], the β-lap-induced γH2AX foci formation is due to DNA damage Citation[24]. Therefore, it is highly likely that the increase in β-lap-induced γH2AX foci formation 24 h after heating in the present study was due to an increase in β-lap-induced DNA damage. The thermal radiosensitisation has been attributed to heat-induced perturbation in the earliest event of the cellular response to ionising radiation-induced DNA damage Citation[29]. It would be interesting to reveal whether a similar mechanism is responsible for the heat-induced increase in the sensitisation of HOS cells to β-lap.
In summary, hyperthermia and β-lap synergistically killed HOS cells in the present study. The cell death caused by β-lap was dependent on NQO1 expression, and hyperthermia caused long-lasting elevation of NQO1 expression in HOS cells. Consequently, HOS cells became sensitive to β-lap not only during the heating but also as long as 24 h after the heating. The induction of γH2AX foci by β-lap due to DNA damage was cell cycle independent and pre-heating increased the β-lap-induced γH2AX foci formation. It is concluded that the combination of hyperthermia with β-lap may be an effective regimen for the treatment of osteosarcoma patients.
Declaration of interest: This work was supported by grants from the National Institute of Health of the United States (RO1 CA116721), Korea Health 21 R&D Project (A062254), and the Nuclear R&D Program of KOSEF (2009-0093747).
Related Research Data
References
- Pardee AB, Li YZ, Li CJ. Cancer therapy with beta-lapachone. Curr Cancer Drug Targets 2002; 2: 227–242
- Planchon SM, Pink JJ, Tagliarino C, Bornmann WG, Varnes ME, Boothman DA. Beta-lapachone-induced apoptosis in human prostate cancer cells: Involvement of NQO1/xip3. Exp Cell Res 2001; 267: 95–106
- Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem 2000; 2755416–5424
- Tagliarino C, Pink JJ, Dubyak GR, Nieminen AL, Boothman DA. Calcium is a key signaling molecule in beta-lapachone-mediated cell death. J Biol Chem 2001; 276: 19150–19159
- Pink JJ, Wuerzberger-Davis S, Tagliarino C, Planchon SM, Yang X, Froelich CJ, Boothman DA. Activation of a cysteine protease in MCF-7 and T47D breast cancer cells during beta-lapachone-mediated apoptosis. Exp Cell Res 2000; 255: 144–155
- Blanco E, Bey EA, Dong Y, Weinberg BD, Sutton DM, Boothman DA, Gao J. Beta-lapachone-containing PEG-PLA polymer micelles as novel nanotherapeutics against NQO1-overexpressing tumor cells. J Control Release 2007; 122: 365–374
- Dong Y, Chin SF, Blanco E, Bey EA, Kabbani W, Xie XJ, Bornmann WG, Boothman DA, Gao J. Intratumoral delivery of beta-lapachone via polymer implants for prostate cancer therapy. Clin Cancer Res 2009; 15: 131–139
- Begleiter A, Fourie J. Induction of NQO1 in cancer cells. Methods Enzymol 2004; 382: 320–351
- Rauth AM, Goldberg Z, Misra V. DT-diaphorase: Possible roles in cancer chemotherapy and carcinogenesis. Oncol Res 1997; 9:339–349
- Ross D, Siegel D. NAD(P)H:Quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol 2004; 382: 115–144
- Siegel D, Beall H, Senekowitsch C, Kasai M, Arai H, Gibson NW, Ross D. Bioreductive activation of mitomycin C by DT-diaphorase. Biochemistry 1992; 31: 7879–7885
- Robertson N, Stratford IJ, Houlbrook S, Carmichael J, Adams GE. The sensitivity of human tumour cells to quinone bioreductive drugs: What role for DT-diaphorase?. Biochem Pharmacol 1992; 44:409–412
- Loadman PM, Bibby MC, Phillips RM. Pharmacological approach towards the development of indolequinone bioreductive drugs based on the clinically inactive agent EO9. Br J Pharmacol 2002; 137: 701–709
- Colucci MA, Moody CJ, Couch GD. Natural and synthetic quinones and their reduction by the quinone reductase enzyme NQO1: From synthetic organic chemistry to compounds with anticancer potential. Org Biomol Chem 2008; 6: 637–656
- Duvoix A, Delhalle S, Blasius R, Schnekenburger M, Morceau F, Fougère M, Henry E, Galteau MM, Dicato M, Diederich M. Effect of chemopreventive agents on glutathione S-transferase P1-1 gene expression mechanisms via activating protein 1 and nuclear factor kappa B inhibition. Biochem Pharmacol 2004; 68: 1101–1111
- Begleiter A, Leith MK, Thliveris JA, Digby T. Dietary induction of NQO1 increases the antitumour activity of mitomycin C in human colon tumours in vivo. Br J Cancer 2004; 91: 1624–1631
- Beall HD, Murphy AM, Siegel D, Hargreaves RH, Butler J, Ross D. Nicotinamide adenine dinucleotide (phosphate): Quinone oxidoreductase (DT-diaphorase) as a target for bioreductive antitumor quinones: Quinone cytotoxicity and selectivity in human lung and breast cancer cell lines. Mol Pharmacol 1995; 48: 499–504
- Keyes SR, Fracasso PM, Heimbrook DC, Rockwell S, Sligar SG, Sartorelli AC. Role of NADPH:Cytochrome c reductase and DT-diaphorase in the biotransformation of mitomycin C1. Cancer Res 1984; 44: 5638–5643
- Fitzsimmons SA, Workman P, Grever M, Paull K, Camalier R, Lewis AD. Reductase enzyme expression across the National Cancer Institute tumor cell line panel: Correlation with sensitivity to mitomycin C and EO9. J Natl Cancer Inst 1996; 88: 259–269
- Dehn DL, Winski SL, Ross D. Development of a new isogenic cell-xenograft system for evaluation of NAD(P)H:quinone oxidoreductase-directed antitumor quinones: Evaluation of the activity of RH1. Clin Cancer Res 2004; 10: 3147–3155
- Siegel D, Franklin WA, Ross D. Immunohistochemical detection of NAD(P)H:Quinone oxidoreductase in human lung and lung tumors. Clin Cancer Res 1998; 4: 2065–2070
- Belinsky M, Jaiswal AK. NAD(P)H:Quinone oxidoreductase1 (DT-diaphorase) expression in normal and tumor tissues. Cancer Metastasis Rev 1993; 12: 103–117
- Olive PL, Banáth JP. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int J Radiat Oncol Biol Phys 2004; 58: 331–335
- Bentle MS, Reinicke KE, Bey EA, Spitz DR, Boothman DA. Calcium-dependent modulation of poly(ADP-ribose)poymerase-1 alters cellular metabolisms and DNA repair. J Biol Chem 2006; 281: 33684–33696
- Choi EK, Terai K, Ji IM, Kook YH, Park KH, Oh ET, Griffin RJ, Lim BU, Kim JS, Lee DS, et al. Upregulation of NAD(P)H:Quinone oxidoreductase by radiation potentiates the effect of bioreductive beta-lapachone on cancer cells. Neoplasia 2007; 9: 634–642
- Terai K, Dong GZ, Oh ET, Park MT, Gu Y, Song CW, Park HJ. Cisplatin enhances the anticancer effect of beta-lapachone by upregulating NQO1. Anticancer Drugs 2009; 20: 901–909
- Takahashi A, Matsumoto H, Nagayama K, Kitano M, Hirose S, Tanaka H, Mori E, Yamakawa N, Yasumoto J, Yuki K, et al. Evidence for the involvement of double-strand breaks in heat-induced cell killing. Cancer Res 2004; 64: 8839–8845
- Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, Gupta A, Rief N, Horikoshi N, Baskaran R, et al. Hyperthermia activates a subset of ataxia-telangiectasis mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res 2007; 67: 2010–3017
- Laszlo A. Fleischer I. Heat-induced perturbations of DNA damage signaling pathways are modulated by molecular chaperones. Cancer Res 2009; 69: 2042–2049
- Laszlo A, Fleischer I. The heat-induced gamma-H2AX response does not play a role in hyperthermic cell killing 2009; 25: 199–209
- Jone EL, Oleson JR, Prosnitz LR, Samulski TV, Vujaskovic Z, Yu D, Sanders LL, Dewhirst MW. Randomized trial of hyperthermica and radiation for superficial tumors. J Clin Oncol 2005; 23: 3079–3085
- van der Zee J, Gonzalez Gonalez D, van Rhoon GC, van Dijk JD, van Putten WL, Hart AA. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in coally advanced pelvic tumours: A prospective radomised, multicentre trial. Dutch Deep Hyperthermia Group. Lancet 2000; 355: 1119–11125
- Issels RD. Hyperthermia adds to chemotherapy. Eur J Cancer 2008; 44: 2546–2554
- Song CW, Park HJ, Griffin RJ. Improvement of tumor oxygenation by mild hyperthermia. Radiat Res 2001; 155: 515–528
- Park HJ, Choi EK, Choi J, Ahn KJ, Kim EJ, Ji IM, Kook YH, Ahn SD, Williams B, Griffin R, Boothman DA, Lee CK, Song CW. Heat-induced up-regulation of NAD(P)H:Quinone oxidoreductase potentiates anticancer effects of beta-lapachone. Clin Cancer Res 2005; 11: 8866–8871
- Song CW, Chae JJ, Choi EK, Hwang TS, Kim C, Lim BU, Park HJ. Anti-cancer effect of bio-reductive drug beta-lapachone is enhanced by activating NQO1 with heat shock. Int J Hyperthermia 2008; 24: 161–169
- Fletcher CDM, Unni KK, Mertens F. Conventional osteosarcoma. World Health Organization classification of tumors. Pathology and genetics of tumours of soft tissue and bone, AK Raymond, AG Ayala, S Knuutila. IARC Press, Lyon 2002; 264–270
- Bacci G, Longhi A, Versari M, Mercuri M, Briccoli A, Picci P. Prognostic factors for osteosarcoma of the extremity treated with neoadjuvant chemotherapy: 15-year experience in 789 patients treated at a single institution. Cancer 2006; 106: 1154–1161
- Liu TJ, Lin SY, Chau YP. Inhibition of poly (ADP-ribose) polymerase activation attenuates beta-lapachone-induced necrotic cell death in human osteosarcoma cells. Toxicol Appl Pharmacol 2002; 182: 116–125
- Park HJ, Lyon JC, Ohtsubo T, Song CW. Cell cycle progression and apoptosis after irradiation in an acidic environment. Cell Death Diff 2000; 7: 202–207