Abstract
Purpose: To examine the molecular mechanism of cellular EGFR activation during hyperthermia treatment.
Materials and methods: EGR activities in tumour cells were quantified through the use of a recently developed split-luciferase-based EGFR reporter system which allowed us to monitor EGFR activation in vitro as well as in vivo in a non-invasive manner.
Results: We found that hyperthermia treatment of MDA-MB231 breast cancer cells resulted in a strong induction of EGFR activity in tissue culture as well as in xenograft tumours. Furthermore, we found that this induction is mediated by the heat shock protein Hsp90. Administration of the specific Hsp90 inhibitor geldanamycin as well as RNAi directed against HSP90 effectively inhibited EGFR activation, suggesting an essential role for Hsp90 in hyperthermia-induced EGFR activation. In addition, cells treated with geldanamycin were sensitised to heat treatment, suggesting that adding Hsp90 inhibitors to hyperthermia regimens might have a beneficial effect for cancer treatment.
Conclusions: Our bioluminescent imaging reporter provided a powerful tool to examine hyperthermia-induced EGFR activation in vitro as well as in vivo. Hsp90 was found to be a key factor mediating heat-induced EGFR activation in tumour cells.
Introduction
Hyperthermia is an experimental cancer treatment modality that has shown great promise in a number of clinical trials Citation[1–3]. At the cellular level, hyperthermia has a direct cell-killing effect dependent on the temperature and the duration of heat exposure Citation[4]. In the clinic it is primarily used in an adjuvant setting to sensitise cells to other treatment modalities such as irradiation or chemotherapy Citation[5]. In these settings, temperatures in the range of 38°–43°C are used. The responses taking effect in this temperature range include changes in tumour perfusion and reoxygenation, cellular induction of the heat shock response, cell cycle checkpoint response, immunological stimulation, and inhibition of radiation-induced DNA damage repair Citation[6]. At the molecular level, a great number of genes are known to be up- or down-regulated by heat, most prominently the family of heat-shock proteins Citation[7]. However, a detailed understanding of the biological mechanisms that are directly involved in tumour cell response to hyperthermia is not well understood Citation[8]. Such understanding is clearly necessary to enhance the efficacy of cancer treatment regimens involving hyperthermia.
The epidermal growth factor receptor (EGFR) is over-expressed and/or constitutively activated in a wide variety of solid tumours. In addition, EGFR is known to be activated in response to radiotherapy Citation[9–11] and chemotherapy Citation[12]. It has been proposed that its activation might contribute to resistance to treatment Citation[12], Citation[13].
Heat shock proteins play key roles in cellular response to hyperthermia. Heat shock protein Hsp90 is a molecular chaperone implicated in protein maturation and folding. Many of its numerous clients are involved in cellular stress response as well as carcinogenesis. It has been reported that inhibition of Hsp90 could deplete nascent EGFR receptor levels Citation[14]. Inhibition of Hsp90 by geldanamycin has been shown to sensitise cervical Citation[15] or melanoma cancer cells to radiation treatment. In addition, Ito et al. Citation[16] showed that Hsp90 inhibition sensitises cells to hyperthermia which had been administered very elegantly using thermosensitive ferromagnetic particles and inductive heating. However, the cellular mechanisms and pathways which are involved in the sensitisation effect of Hsp90 inhibition are yet to be identified. Another member of the heat shock protein family, Hsp72, has been shown to induce EGFR phosphorylation upon heat shock Citation[17]. Such relationships between HSPs and EGFR activation are important because of potential roles of EGFR in mediating tumour resistance to hyperthermia treatment.
In the present study, a split-luciferase-based reporter system was employed to assess hyperthermia-induced EGFR signalling in a non-invasive and highly sensitive manner. Using this new technique our results indicated that EGFR is activated strongly by hyperthermia treatment. In addition we identified Hsp90 as a key mediator of EGFR activation in cancer cells. The addition of geldanamycin, a potent inhibitor of HSP90, significantly attenuated heat-induced EGFR activation and sensitised cells to hyperthermia, thus providing a rationale for adding Hsp90 inhibitors to cancer treatment regimens involving hyperthermia.
Material and methods
Cell culture
MDA-MB-231 and 293T cells were obtained from the American Type Culture Collection (Manassas, VA). Head and neck cancer cells UMSCC10 and 1483 were kindly provided by Barbara Frederick of University of Colorado Denver School of Medicine, Aurora, CO. All cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal calf serum (FCS).
Generating cell lines carrying the EGFR reporter system
The split-luciferase reporter system for EGFR visualisation was established in our laboratory; details of the system have been published elsewhere Citation[11]. Recombinant lentiviral vectors encoding the fusion proteins EGFRNLuc and Shc-CLuc were generated using a commercially obtained lentiviral vector pLEX (Open Biosystems, Huntsville, AL). Virus for each reporter construct was produced by transducing 293T cells with pLEX plasmids encoding the fusion proteins together with helper plasmids using a published lentivirus production protocol Citation[18].
To generate cancer cell lines with the reporters, MDA-MB-231, UMSCC10 and 1483 cells were infected with viral vectors encoding the EGFR reporter fusion proteins (EGFR-NLuc and Shc-CLuc). Single cell clones expressing both reporter genes were then obtained by limited dilutions and subsequent testing for luciferase signal. Luciferase positive cell clones were further expanded and tested for induction by EGF ligand at a concentration of 50 ng/mL. Cell clones with low background and high EGF induction were selected for further experiments.
Heat treatment and bioluminescent imaging
For heat induction experiments, cells were plated in 6-well or 12-well tissue culture plates and serum starved overnight. For heating, tissue culture dishes were sealed with parafilm and placed in a water bath of the respective temperature (typically 43°C) for 30 min. Ten minutes prior to imaging, cells were incubated with luciferin (Xenogen) at a concentration of 6 mg/mL. Bioluminescent imaging was carried out using a Xenogen IVIS200 imager. Photographic and bioluminescent images were acquired with an exposure time of 1–2 min at medium (4 × 4) or large (16 × 16) binning, pseudocoloured and superimposed over photographic images which had been taken simultaneously.
Growth curves and clonogenic assay
To establish a growth curve, cells were seeded in four 12-well plates and subjected to heat and/or geldanamycin treatment. For each time point, cells of three separate wells were trypsinised and counted four times using a manual counting chamber.
For the clonogenic assay, cells were subjected to heat (43°C for 30 min) and/or geldanamycin (1 µM, preincubated for 2 h). Cells were counted and 200 cells plated into 10-cm dishes 4 h after treatment. For all groups triplicate experiments were performed. After 10 days, colonies were stained with crystal violet and counted. Seeding efficiency of control cells was 84%.
Western blot analysis was performed using standard procedures as described. The following primary antibodies were used: EGFR (Cell Signaling Technology, Beverly, MA): 1:1000; EGFR-P, Tyr1068 (Zymed Laboratories, San Francisco, CA):1:1000; HSP90 (Cell Signaling Technology): 1:1000; β-actin (Lab Vision, Fremont, CA) 1:1000.
RNAi experiments were performed by transfecting SMARTpool oligos (Fisher Scientific, Pittsburgh, MA) directed against either HSP90AA1 or HSP90AB1 or both using DharmaFECT transfection reagent (Fisher Scientific) into MDA-MB231-EGFR-Shc cells. Protein knock down was verified using western blot analysis 72 h after transfection.
Animal procedures and in vivo imaging
Athymic nude mice were obtained from the National Cancer Institute through Charles River and maintained in the Vivarium of the Anschutz Medical Campus in accordance with University of Colorado Institutional Animal Care and Use Committee guidelines. Xenograft tumours were established by subcutaneous injection of 5 × 105 cells suspended in a 50 µL Matrigel/PBS solution (1:3) in each hind leg. For imaging, mice were intraperitoneally (i.p.) injected with 200 µL of luciferin at a concentration of 15 mg/mL in H2O 10 min prior to image acquisition, and anaesthetised using isofluorane inhalation. For unilateral heat treatment of tumours we used a custom-made mouse restraint which allows dipping one leg into a water bath (43°C) while keeping the rest of the body above the water surface. The restraint consisted of an aluminium foil, isolated, ventilated plastic tube with a leg cut out mounted on a Plexiglas mounting plate. Cool breathing air was supplied using a large cone-shaped aluminium funnel. In order to maintain thermoregulation mice were not anaesthetised during heat treatment.
Results
The split-luciferase system to detect EGFR activation
In order to detect EGFR activation in response to hyperthermia we employed a bi-fragment luciferase reconstitution system previously developed in our laboratory Citation[11]. This system is based on the assumption that luciferase activity is reconstituted when the N- and C-terminal fragments of luciferase which are fused to EGFR and its downstream binding partner Shc, respectively, come in close proximity to each other as a result of EGFR activation and its subsequent binding to Shc. In the presence of luciferin, the substrate of luciferase, bioluminescence is emitted which can be captured using a bioluminescence imaging device (see for schematic demonstration). We have shown previously that this reporter functions well in tumour cell lines as well as in xenografts Citation[11].
Figure 1. Heat-induced EGFR activation in vitro. (A) Diagram of our EGFR-bioluminescent imaging reporter: EGFR and Shc proteins are fused to the N- and C-terminal fragments of luciferase. Upon activation of the EGFR pathway, e.g. by ligand binding, homo-dimers containing EGFR are formed and Shc is recruited to the complex, resulting in Shc phosphorylation and downstream signalling. Binding of EGFR-N-Luc to Shc-CLuc brings the two fragments in close proximity to each other, thus reconstituting luciferase activity. In the presence of the substrate luciferin, light is emitted which can be detected using a highly sensitive camera. (B) Induction of reporter activities in MDA-MB-231-EGFR-Shc-Luc cells. Left panel: cells were exposed to 43°C in a water bath for 30 min and imaged using an IVIS 200 imager at 3 h and 24 h after heating. Shown are luciferase signals of representative wells. Middle panel: graph of luciferase signals expressed as photon counts/s. Shown are average signal values of three individual wells, error bars represent standard error of the mean. Right panel: western blot analysis of total EGFR, mitogen-activated phosphokinase (MAPK), Shc levels the phosphorylated fraction of EGFR and MAPK in heated MDA-MB231 cells at 3 h and 24 h after heat treatment. Beta actin levels are shown as loading control.
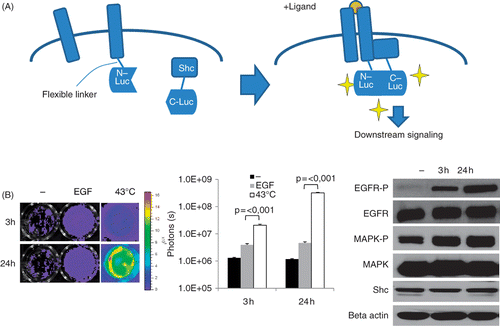
Hyperthermia strongly induces EGFR activation
The EGFR positive breast cancer cell line MDA-MB-231 was transduced with the EGFR reporter and tested for signal induction after incubation with the ligand EGF. As can be seen in (left and mid panels), incubation with EGF at a saturated level (50 ng/mg) for 30 min led to a 3–5-fold increase in luciferase signals, demonstrating that the luciferase reporter was capable of reporting EGFR activation upon ligand binding.
To test whether EGFR signalling is influenced by heat treatment we subjected cells to hyperthermia by incubation of the sealed culture dishes in a 43°C water bath for 30 min. This treatment resulted in a very strong EGFR activation reaching about 50-fold levels after 24 h compared to induction with the receptor ligand EGF alone (, left and middle panel). To exclude direct non-specific heat effects on luciferase activity itself we expressed full length luciferase in MDA-MB-231 cells and subjected them to the same heat treatment which did not result in any induction of the luciferase signal (data not shown).
Due to the rapid heat-induced EGFR induction in response to heat, we suspected that this effect was a result of post-translational regulation rather than increased EGFR expression. To test this hypothesis we performed western blot analysis showing that EGFR total protein levels were not induced by heat, however, increased phosphorylation of EGFR was detected (, right panel). Furthermore, a downstream factor mitogen-activated protein kinase (MAPK) was phosphorylated, indicating true EGFR activation (, right panel).
In order to rule out cell type-specific effects, two head and neck cancer cell lines, 1483 and UMSCC10A were stably transfected with our EGFR reporter and tested for heat-induced EGFR induction. These cell lines became equally induced by heat, however with slightly different kinetics and sensitivity to various temperatures. (left panel) shows the temperature dependence of EGFR activation in all three EGFR reporter cell lines. Temperatures in the range of 41°–44°C, commonly used in clinical hyperthermia procedures, was used. The head and neck cancer cell lines UMSCC10A and 1483 appeared to be more resistant to heat treatment than the MDA-MB-231 cell line which became induced at lower temperatures. Maximum induction for the MDA-MB-231 and the H&N cell lines was reached at 43°C and 44°C, respectively. As shown in (right panel), heat-induced signal induction was a fast process detectable at 2 h after hyperthermia treatment. Its peak level was reached at 24 h and induction was detectable up to 7 days post-hyperthermia treatment.
Figure 2. Further characterization of heat-induced EGFR activation in vitro. (A) Temperature dependence of heat-induced EGFR induction. Left panel shows quantitative analysis of luciferase signals of three different cell lines 24 h after heat treatment in a water bath at 42°, 43° and 44°C, respectively. Left panel shows the time course of EGFR induction by heat. Data are derived from three different cell lines equipped with the EGFR reporter after hyperthermia of 43°C for 30 min over a time period of 9 days. (B) Inhibition of heat-induced EGFR activation by EGFR inhibitors: MDA-MB-231 cells were incubated for 1 h with EGF and/or EGFR inhibitors as indicated, subjected to hyperthermia and imaged using a Xenogen IVIS imager. Shown is a quantitative analysis with corresponding bioluminescent images of representative wells. Error bars represent standard error of the mean of three individual wells. Cet, cetuximab; Gef, gefitinib. Right panel: western blot analysis of EGFR and MAPK and their phosphorylated versions 24 h after heat treatment of MDA-MB-231 cells with or without the presence of the EGFR inhibitor gefitinib (1 uM).
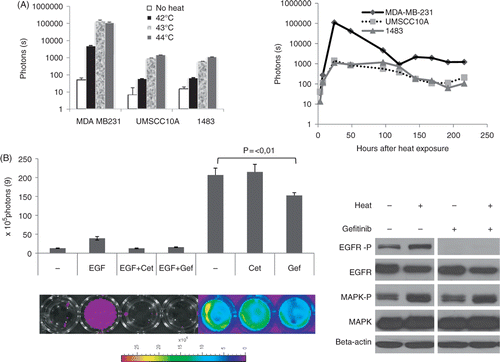
To test whether heat-induced EGFR activation could be inhibited using commonly used EGFR inhibitors, cells were pre-incubated with either the EGFR antibody cetuximab or with the small molecule inhibitor gefitinib, before treatment with either EGF or heat. As can be seen in , both inhibitors, cetuximab and gefitinib, are effective in inhibiting luciferase induction after treatment with the EGFR ligand EGF, again demonstrating the functionality of our reporter system. However, in heated cells, the monoclonal antibody cetuximab (125 nM) did not show any effect on EGFR signalling, whereas the small molecule inhibitor gefitinib (0.5 µm) could decrease activation by about one third, suggesting that EGFR is activated in a ligand-independent manner through an intracellular mechanism. Interestingly, western blot analysis (, right panel) showed that, in heated MDA 231 cells, gefitinib is effective in inhibiting EGFR phosphorylation but not the downstream phosphorylation events as shown by the example of MAPK. These results suggest that heat-induced activation of the EGFR pathway is independent of EGFR ligand binding.
Heat induction is dependent on Hsp90
To test whether Hsp90 function is required for heat-induced EGFR activation, MDA-MB-231 EGFRShc-Luc cells were preincubated with geldanamycin (GA), a potent and highly selective inhibitor of Hsp90. Cells were imaged and luciferase intensity quantified at 7 h after heat treatment. As can be seen in , EGFR activation was suppressed in GA treated cells, suggesting that Hsp90 might – directly or indirectly – be involved in the heat induced EGFR activation.
Figure 3. Key role of HSP90 in mediating heat induced EGFR activation. (A) Effect of geldanamycin on EGFR induction. Geldanamycin was preincubated 1 h prior to heating cells at 43°C for 30 min. Shown are bioluminescent images of representative wells acquired 7 h after heating using a Xenogen IVIS imager (left) and the quantitative analysis of the same experiment (right). GA, geldanamycin. (B) Knockdown of Hsp90. Cells were transfected with SMARTpool oligos (Fisher Scientific) directed against HSP90AA1, AB1 or non-targeting (scramble). After 2 days, cells were heated at 43°C for 30 min and imaged 6 h after heating. Left: western blot analysis of total Hsp90 protein levels after transfection with Hsp90 oligos as indicated. Middle: bioluminescent image of representative wells. Right: quantitative analysis of the same experiment. (C) Geldanamycin sensitizes cells to hyperthermia treatment. Left: representative bright field images of MDA-MB-231 cells 72 h after heat and/or geldanamycin (0.5 µM) treatment as indicated. Size bar = 200 µm. Middle: growth curve of MDA-MB-231 cells after heat and/or geldanamycin (0.5 µM) treatment. For each time-point, cells of three different wells of each treatment group were counted using a cell counting chamber. GA, geldanamycin. Right: clonogenic assay: After treatment with hyperthermia for 30 min in the presence or absence of geldanamycin, 200 cells of each treatment group were seeded into 10-cm dishes. Colonies were counted after 10 days. Error bars indicate standard error of the mean of three individual experiments.
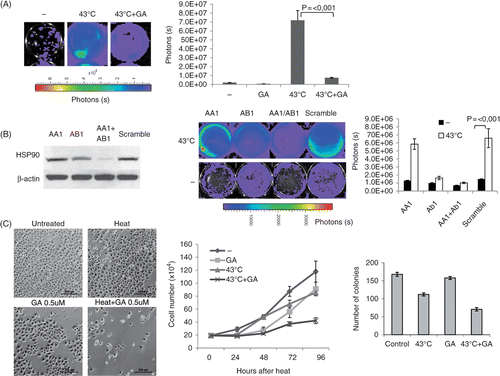
In order to back up these findings, RNAi knockdown of Hsp90 was employed. RNAi oligos (Dharmacon/Fisher Scientific) directed against the inducible and constitutive forms of Hsp90 (HSP90AA1 and HSP90AB1) were transfected into MDA-MB-231 EGFR-Shc cells. As can be seen in (left panel), double knock down of both HSP90AA1 and HSP90AB1 effectively depleted total Hsp90 protein levels. Cells were subjected to hyperthermia 72 h after transfection, and analysed for EGFR induction 6 h following heat treatment. As can be seen in (middle and right panel), heat-induced EGFR activation was substantially inhibited in Hsp90 knock down cells supporting the results obtained with geldanamycin. These data suggest that Hsp90 is required for the massive up-regulation of EGFR signalling in response to hyperthermia.
Hsp90 inhibition sensitises cells to hyperthermia treatment
We went on to test whether a compromised EGFR stress response mediated by Hsp90 inhibition would sensitise cells to heat treatment and enhance its cytotoxic effect. Cells were heated with or without addition of geldanamycin (0.5 uM) and a growth curve was established by counting cells of three individual wells per group in a counting chamber every day. In addition, cell density and status was evaluated by visual inspection using bright field microscopy. As can be seen in , heat treatment alone had a moderate effect on the growth curve compared to the non-treated control. However, the combined treatment of hyperthermia and geldanamycin led to a significant flattening of the growth curve, suggesting that inhibition of Hsp90 sensitises cells to hyperthermia by down-regulating EGFR activity. In order to quantify the heat sensitisation effect of HSP inhibition on clonogenicity, clonogenic assays were performed (see Materials and methods for details). Heat treatment and GA treatment alone caused a reduction of clonogenicity of 32.3% and 6.2% compared to control cells, respectively. In contrast, the combined treatment of hyperthermia and geldanamycin resulted in a 58% reduction of clonogenicity, suggesting a supra-additive effect of the combined versus the individual treatment.
EGFR induction in response to hyperthermia treatment in vivo
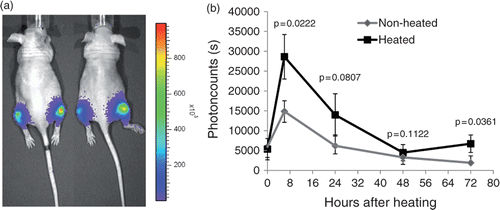
To study heat-induced EGFR activation in vivo, we established xenograft tumours by injecting 1 × 106 cells of the MDA-MB-231 EGFR-Shc cell line into both hind legs of female athymic nude mice. When tumours reached a size of 7–8 mm in diameter, we subjected the right hind leg of the mouse to hyperthermia of 43°C for 30 min. Mice were imaged daily over a time period of 4 days.
As can be seen in , tumours of heated legs showed a profound induction in the EGFR signal compared to the non-treated side. The effect was maintained throughout the observation period and peaked at 24 h after treatment.
Figure 4. Activation or EGFR after hyperthermia in vivo. Xenograft tumours were established by injecting 5 × 105 MDA-MB-231 EGFR/Shc-Luc cells into both hind legs of athymic nude mice. When tumours reached a diameter of about 7 mm, mice (n = 5) were placed in a custom-made restraining device with the right hind leg dipping into a 37°C water bath for 30 min. After treatment, mice were imaged over a time period of 4 days. Left: representative bioluminescent pseudocoloured image superimposed over a regular photograph. Right: quantitative analysis of the same experiment. Error bars indicate standard error of the mean.
Discussion
By use of a recently developed split-luciferase-based EGFR reporter system, we were able to monitor EGFR activation after heat treatment in vitro, and for the first time visualise EGFR activation in in vivo xenografts in response to hyperthermia. We showed that the Hsp90 inhibitor geldanamycin was very effective in down-regulating heat-induced EGFR activation.
Interestingly, the small-molecule EGFR inhibitor gefitinib caused only a slight decrease in heat-induced EGFR activation, while the monoclonal EGFR antibody cetuximab did not show any effect on heat-induced EGFR activation. However, both of them were potent in inhibiting EGFR activation induced by addition of the ligand EGF. As cetuximab only affects the ectodomain of the EGFR receptor and thus its binding capacity to EGF, whereas gefitinib interferes with ATP binding at the intracellular kinase domain, we concluded that Hsp90 mediated EGFR activation is ligand-independent and acts via a different, intracellular mechanism that has yet to be identified.
In addition, conditioned medium of heated cells did not result in induction of the luciferase signal (data not shown), suggesting that Hsp90 mediated EGFR activation is independent from extracellular signalling.
RNAi experiments confirmed the importance of Hsp90 in mediating a ligand-independent EGFR activation. The functional importance of EGFR activation during hyperthermia was further demonstrated by the observation that geldanamycin treatment sensitised cells to hyperthermia resulting in flattening of the growth curve as well as clonogenicity. In fact, the data from the clonogenic assay suggest a supra-additive effect of the combined versus the individual treatment. Although we cannot provide direct evidence that Hsp90 depletion sensitises cells to heat treatment by down-regulating EGFR activity, our data strongly suggest that the reduced levels of EGFR transactivation caused by Hsp90 depletion are related to the sensitising effect of Hsp90 inhibition on cell survival of heated cells. Interestingly, in our in vivo studies the non-heated tumour in the contralateral leg became induced to some extent after treatment. Because in well perfused tumours and extremities, heat is conveyed via the blood stream quite effectively and due to the proximity of the mouse to the water surface, a subtle hyperthermia effect in regions other than the treated region might occur. In addition, distant effects of locoregional treatment modalities such as radiotherapy have been described as bystander effect, which is not well understood. However, it is reasonable to believe that such effects might be present in hyperthermia treatment as well and that they are responsible for the EGFR induction of the contralateral tumour in our experiments.
Conclusion
Taken together, we believe we have clearly demonstrated EGFR activation in cellular response to hyperthermia both in vitro and in vivo. Our discovery of Hsp90 as a critical mediator of EGFR transactivation and the sensitising effect of its depletion also provides perspectives on how the effectiveness of hyperthermia treatment could be enhanced mechanistically.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Vernon CC, Hand JW, Field SB, Machin D, Whaley JB, van der Zee J, van Putten WL, van Rhoon GC, van Dijk JD, Gonzalez Gonzalez D, et al. Radiotherapy with or without hyperthermia in the treatment of superficial localized breast cancer: Results from five randomized controlled trials. International Collaborative Hyperthermia Group. Int J Radiat Oncol Biol Phys 1996; 35: 731–744
- van der Zee J, Gonzalez Gonzalez D, van Rhoon GC, van Dijk JD, van Putten WL, Hart AA. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: A prospective, randomised, multicentre trial. Dutch Deep Hyperthermia Group. Lancet 2000; 355: 1119–1125
- Hildebrandt B, Wust P, Rau B, Schlag P, Riess H. Regional hyperthermia for rectal cancer. Lancet 2000; 356: 771–772
- Dewey WC. Arrhenius relationships from the molecule and cell to the clinic. Int J Hyperthermia 1994; 10: 457–483
- Bull JM. An update on the anticancer effects of a combination of chemotherapy and hyperthermia. Cancer Res 1984; 44: S4853–S4856
- Dewhirst MW, Vujaskovic Z, Jones E, Thrall D. Re-setting the biologic rationale for thermal therapy. Int J Hyperthermia 2005; 21: 779–790
- Li GC, Mivechi NF, Weitzel G. Heat shock proteins, thermotolerance, and their relevance to clinical hyperthermia. Int J Hyperthermia 1995; 11: 459–488
- Hildebrandt B, Wust P. The biologic rationale of hyperthermia. Cancer Treat Res 2007; 134: 171–184
- Todd DG, Mikkelsen RB, Rorrer WK, Valerie K, Schmidt-Ullrich RK. Ionizing radiation stimulates existing signal transduction pathways involving the activation of epidermal growth factor receptor and ERBB-3, and changes of intracellular calcium in A431 human squamous carcinoma cells. J Recept Signal Transduct Res 1999; 19: 885–908
- Reardon DB, Contessa JN, Mikkelsen RB, Valerie K, Amir C, Dent P, Schmidt-Ullrich RK. Dominant negative EGFR-CD533 and inhibition of MAPK modify JNK1 activation and enhance radiation toxicity of human mammary carcinoma cells. Oncogene 1999; 18: 4756–4766
- Li W, Li F, Huang Q, Frederick B, Bao S, Li CY. Noninvasive imaging and quantification of epidermal growth factor receptor kinase activation in vivo. Cancer Res 2008; 68: 4990–4997
- Wang F, Liu R, Lee SW, Sloss CM, Couget J, Cusack JC. Heparin-binding EGF-like growth factor is an early response gene to chemotherapy and contributes to chemotherapy resistance. Oncogene 2007; 26: 2006–2016
- Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int J Radiat Biol 2007; 83: 781–791
- Sakagami M, Morrison P, Welch WJ. Benzoquinoid ansamycins (herbimycin A and geldanamycin) interfere with the maturation of growth factor receptor tyrosine kinases. Cell Stress Chaperones 1999; 4: 19–28
- Bisht KS, Bradbury CM, Mattson D, Kaushal A, Sowers A, Markovina S, Ortiz KL, Sieck LK, Isaacs JS, Brechbiel MW, et al. Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res 2003; 63: 8984–8995
- Ito A, Saito H, Mitobe K, Minamiya Y, Takahashi N, Maruyama K, Motoyama S, Katayose Y, Ogawa J. Inhibition of heat shock protein 90 sensitizes melanoma cells to thermosensitive ferromagnetic particle-mediated hyperthermia with low Curie temperature. Cancer Sci 2009; 100: 558–564
- Evdonin A, Kinev A, Tsupkina N, Guerriero V, Raynes DA, Medvedeva N. Extracellular HspBP1 and Hsp72 synergistically activate epidermal growth factor receptor. Biol Cell 2009; 101: 351–360
- Klages N, Zufferey R, Trono D. A stable system for the high-titer production of multiply attenuated lentiviral vectors. Mol Ther 2000; 2: 170–176