Abstract
Purpose: The objective of the present study was to examine the consequences of a mild hyperthermia in human tumour cell lines deficient and proficient in the DNA mismatch repair system (MMR) to advance our understanding on the relationship between MMR and heat shock proteins (HSPs).
Materials and methods: The human colon carcinoma cell lines HCT116 (parent cells), HCT116 + ch2 (MMR-deficient), and HCT116 + ch3 (MMR-proficient) were used. Cells were incubated at 41°C and 42°C for 1 h and then at 37°C for 4 and 24 h. The expression of Hsp27 and Hsp72 was evaluated by immunocytochemistry. Hsp27, Hsp72, hMLH1 and hMSH2 levels were assessed by western blotting in nuclear and cytoplasmic fractions. The alkaline comet assay was used to evaluate the DNA damage.
Results: The mild hyperthermia significantly increased the protein expression levels of Hsp27 and Hsp72 in all cell lines, which was higher in the cytoplasm and nucleus of HCT116 + ch3 cells. We also observed that heat induced translocation of hMLH1 and hMSH2 proteins from the nucleus to the cytoplasm in HCT116 + ch3 cells. The comet assay revealed that HCT116 parent cells were more resistant to heat-induced DNA damage. However, the MMR-proficient and deficient cell lines repaired the DNA damage at the same rate.
Conclusions: The present study demonstrates that hyperthermia induced the nuclear accumulation of Hsp27 and Hsp72 and affected the subcellular localisation of hMLH1 and hMSH2 in HCT116 + ch3 cells. Our findings suggest that the MMR system is not a direct determining factor for the different heat shock response in HCT116 cells.
Introduction
In order to enhance the effectiveness of chemotherapy or radiotherapy, local, regional or whole-body hyperthermia may be used in clinical oncology (colorectal cancer, breast cancer, melanoma, cervical cancer, etc.) Citation[1]. In addition, the combination of hyperthermia with gene therapy, in which hyperthermia induces transgene expression, represents a promising strategy Citation[2]. Exposure of cells to a transient, non-lethal elevation of temperature causes the activation of cellular stress responses. A mild heat shock (39°–42°C) induces the expression of heat shock proteins (HSPs). HSPs are molecular chaperones, highly conserve across prokaryotes and eukaryotes, which points to their importance in cellular protection. The best known HSPs are members of the HSPB (small HSP: Hsp27, α-A-crystallin, α-B-crystallin), DNAJ (Hsp40), HSPA (Hsp70), HSPC (Hsp90), HSPH (Hsp110) and chaperonin families (Hsp60 and CCT) Citation[3]. HSPs may be expressed under normal physiological conditions at basal levels; these are known as constitutive or cognate HSPs performing multiple house-keeping functions. Inducible HSPs are expressed after stressful situations (heat shock, anoxia, fever, inflammation, antineoplasic drugs, etc.). HSPs are abundantly expressed in cancer cells and they participate in oncogenesis. Moreover they may also limit the response to cancer therapy Citation[4].
Heat shock leads to denaturation and misaggregation of proteins, inhibition of DNA-synthesis and transcription, inhibition of mRNA-processing and translation, and also to blockage of the progression through the cell cycle. Exposure to heat cause unfolding of a number of nuclear proteins, which results in changes in protein associations Citation[5]. Vanderwaal et al. have found a number of proteins that increase their binding to DNA after heat shock, inhibiting the access of the repair proteins to the damaged site(s) Citation[6]. Thus the effects of hyperthermia on DNA have been thought to be due to secondary effects on proteins involved in DNA replication, chromosome segregation and DNA repair. After heat shock, the proteins that bind nuclear matrix associated DNA, appear to mask DNA strand breaks, delaying repair and altering DNA supercoiling that triggers DNA repair Citation[7]. In addition, heat stress has been reported to induce DNA double strand breaks by the formation of γH2AX foci Citation[8]. If the cell is able to induce a competent HSP response, HSPA1A (Hsp72) and HSPB1 (Hsp27) can accumulate in the nucleus contributing to DNA polymerase-beta reactivation and stimulation which results in thermotolerance. Takahashi et al. have shown that thermotolerance was partially suppressed in DNA polymerase-beta (-/-) mouse fibroblasts and also in the presence of a HSP inhibitor Citation[9]. It has been reported that some HSPs may contribute to different mechanisms of DNA repair as part of their molecular chaperone capabilities, interacting with DNA repair proteins Citation[10]. Kotoglou et al. found that HSPA1A translocates to the nuclei/nucleoli during heat stress, binds to PARP-1 and/or XRCC1 at BRCT domains (BRCA1 C-terminus), and protects HeLa cells from increased single strand breaks Citation[11]. Hunt et al. showed increased levels of spontaneous and radiation-induced chromosomal aberrations in mouse fibroblasts with HSPA1A knockout, suggesting that this protein may be involved in maintaining genome stability Citation[12].
The mismatch repair (MMR) system is one of the DNA repair mechanisms that contribute to the maintenance of genome integrity and to prevention of carcinogenesis. MMR constitutes a post-replicative machinery that corrects base–base mismatches generated principally during replication by DNA polymerases. If DNA mismatches (arising from different circumstances) are not corrected genomic instability may be originated, especially at repeated-sequence motifs known as microsatellites Citation[13]. A significant proportion of cancers of the colon, endometrium and other organs exhibit microsatellite instability (MSI) Citation[14]. This phenotype of MSI is caused by defects in MMR in the hereditary non-polyposis colorectal cancer (HNPCC) and in a variety of sporadic cancers. Expression of the hMLH1 gene is essential for competent MMR and maintenance of MSI Citation[15]. Several human MMR proteins have been identified based on their homology to Escherichia coli MMR proteins (hMSH2, hMSH3, hMSH6, hMLH1, hPMS1, hPMS2, hMLH3). hMSH2 heterodimerises with hMSH6 or hMSH3 to form hMutSα or hMutSβ, while hMLH1 heterodimerises with hPMS2, hPMS1 or hMLH3 to form hMutLα, hMutLβ, or hMutLγ, respectively. hMutSα repairs base–base mismatches and insertion/deletion loops (IDLs) of one or two extrahelical nucleotides, whereas the repair of larger IDLs is initiated by hMutSβ. The hMutSα-hMutLα complex remains bound at the mismatch and initiates the repair reaction by looping out the DNA Citation[16]. Our group has reported the co-localisation of HSPB1 and HSPA1A with the MMR proteins hMLH1 and hMSH2, in peripheral blood lymphocytes from healthy persons exposed to hyperthermia, with cisplatin or doxorubicin Citation[17]. However, our understanding on the relationship between MMR and HSPs remains to be achieved.
The objective of the present study was to examine the consequences of the heat shock treatment in a human colon adenocarcinoma model of hMLH1-deficient and hMLH1-proficient cells. HCT116 cells are deficient in MMR due to the lack of hMLH1 transcript expression Citation[18] and HCT116 + ch3 cells were generated by introducing a single copy of normal human chromosome 3 into HCT116 cells Citation[19]. We characterised the basal and heat-induced expression (41° or 42°C) of HSPB1, HSPA1A, hMLH1 and hMSH2 in nuclear and cytoplasmic extracts. Our results show that hyperthermia is able to induce the expression of HSPB1 and HSPA1A in hMLH1-proficient and deficient HCT116 cells, with nuclear accumulation of HSPA1A, in some cases. In addition, the alkaline comet assay, which constitutes a rapid assay for the screening of mutagen sensitivity, was employed to detect single and double strand breaks caused by the heat shock. Our study provides the first evidence of the differential expression of HSPB1 and HSPA1A in HCT116 and HCT116 + ch3 cells after a mild hyperthermia. The results of the present study and their implications are further discussed.
Materials and methods
Cell lines
A genetically matched pair of a MLH1-deficient human colorectal adenocarcinoma subline (HCT116 + ch2) and its MLH1-proficient counterpart (HCT116 + ch3) was used. They were derived from the human colorectal adenocarcinoma cell line HCT116 containing a hemizygous for a C→A mutation in hMLH1 gene located on chromosome 3, generating a nonsense codon (number 252), producing a truncated but no full-length hMLH1 protein Citation[19]. The HCT116 + ch3 subline was created by microcell chromosome transfer of a single normal human chromosome 3 into HCT116 human colon carcinoma cells (competent in MMR function) Citation[20]. HCT116 + ch2 subline, complemented with chromosome 2, was used as control (MMR incompetent). All cell lines were kindly donated by C.R. Boland and M. Koi (Baylor University Medical Center, Dallas, TX, USA). The cell lines were maintained in Iscove's modified Dulbecco's medium (Gibco-Life Technologies, New York, USA) supplemented with 10% foetal bovine serum (Gibco). The chromosome-complemented lines were maintained in medium containing 400 µg/mL geneticin (Gibco).
Experimental hyperthermia
The cells were incubated at 41°C or 42°C for 1 h in a water bath. One group was collected immediately after a heat shock (time 0, T0), and the other groups were allowed to recover 4 h and 24 h at 37°C (time 4 and 24, T4 and T24, respectively) and then collected for further studies. Three experiments were performed in duplicate.
Immunocytochemistry
Cells (1 × 105) were incubated on glass cover-slips. Experimental treatments were done as mentioned above. Cells were fixed in 4% buffered formalin at room temperature for 20 min. Membrane permeabilisation was done with 0.5% Triton X-100 in cold PBS (pH 7.4) for 5 min. Antigen unmasking for HSPA1A was carried out in 0.01 M citrate buffer (pH 6.0) at 100°C for 25 min. The antibodies used were: mouse monoclonal antibody (MAb) against HSPB1 (Stressgen, Ann Arbor, MI, USA) and rabbit polyclonal antibody against HSPA1A (Stressgen). The cells were incubated with the primary antibodies overnight at 4°C in humidity chambers at the following dilutions: HSPB1, 1:200; HSPA1A, 1:250. As secondary antibody we used a labelled polymer conjugated with goat anti-rabbit and goat anti-mouse immunoglobulins (DAKO EnVision System Peroxidase, Carpinteria, CA, USA). Diaminobenzidine (0.5 mg/mL)/hydrogen peroxide (0.01%) was used as chromogen substrate. Slides were lightly counterstained with haematoxylin and observed with a Nikon Eclipse E200 microscope (Kanagawa, Japan). The immunostaining was evaluated according to the percentage of positive cells (cytoplasmic and nuclear staining) counting 200 cells per sample under double blindness throughout the study. Non-immune IgG isotype and no primary antibody controls were also included as negative controls.
Immunoblotting
After treatments, the cells were washed in PBS and resuspended in lysis buffer for nuclear and cytoplasmic protein extraction as reported by Andrews et al. Citation[21]. The proteins were separated by 10% SDS-PAGE. A sample protein of 20 µL (3.5 × 105 cells) was loaded. All protein level data were compared in linear conditions of signal identity. One lane was loaded with molecular weight markers (Rainbow Marker, Amersham, Buckinghamshire, UK). Detection of the HSPB1, HSPA1A, hMLH1 and hMSH2 bands on nitrocellulose paper was performed using the specific antibodies: mouse MAb against HSPB1 (Stressgen) at 1:2000 dilution in blocking buffer (5% BSA in PBS–0.5% Tween 20), mouse MAb against HSPA1A (Stressgen) at 1:1000 dilution, mouse MAb against hMLH1 (Pharmingen, San Diego, CA, USA) at 6 µg/mL dilution, and mouse MAb against hMSH2 (Calbiochem, San Diego, California, USA) at 0.5 µg/mL dilution. After overnight incubation with the primary antibody, the membranes were incubated with a biotinylated rabbit antibody to mouse immunoglobulins (DAKO, 1:2500), then with peroxidase-labelled streptavidin-biotin complex (DAKO, 1:5000). Washed membranes were then incubated with chemiluminescence reagents (Dupont, NEN, Boston, MA, USA) following manufacturer's instructions. Protein loading was controlled with MAb anti- α-tubulin (Sigma-Aldrich, St. Louis, MO, USA, 1:12000) and MAb anti- HDAC1 (Sigma, 1:10000) antibodies. The light was captured using LAS-4000 imaging system (Fujifilm Life Science, Stamford, CT, USA). Quantification of blots was performed using NIH Image version 1.62 program (National Institutes of Health, Washington, USA). The relative amount of each protein was calculated as a function of the corresponding internal loading control. To prevent false results in the quantification of nuclear proteins by the potential cross-contamination, the values of the cytoplasmic marker (α-tubulin) performed in the nuclear fraction were discounted.
Alkaline comet assay
This method was performed according to the procedure described by Olive et al. Citation[22]. To prevent additional DNA damage, the assay was done in the dark and at 4°C. After electrophoresis, the agarose gels were silver stained as reported by Nadin et al. Citation[23]. Comets were evaluated in duplicate samples and under double blindness, using the 20 x objective of a Nikon optic microscope, counting 40 randomly selected cells per slide (that is, 80 cells/each sample). A visual score based on extend of migration was used: 0, very low migration; 1, 5–10% of migrated DNA; 2, 11–30% of migrated DNA; 3, 31–60% of migrated DNA; 4, 61–95% of migrated DNA; and 5, >95% of migrated DNA Citation[24]. To facilitate the management of the data, an average of DNA migration was calculated as: [(% of cells with score 1)×1 + (% of cells with score 2)×2 + (% of cells with score 3)×3 + (% of cells with score 4)×4 + (% of cells with score 5)×5] /100 Citation[25]. As positive control, we used cells treated with 60 µM of hydrogen peroxide for 1 h.
Statistical analysis
Statistical analyses were performed by one-way analysis of variance with Bonferroni post hoc test. All data analyses were done using the PRISM program (GraphPad, San Diego, CA, USA). P-values < 0.05 were considered statistically significant.
Results
Hsp27 (HSPB1) and Hsp72 (HSPA1A) expression in HCT116 hMLH1-deficient and proficient cells after heat shock treatment
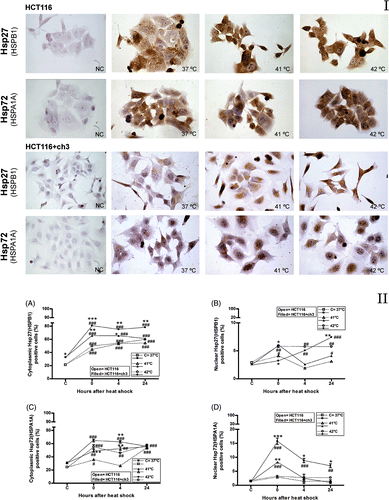
The expressions of HSPB1 and HSPA1A after the heat shock treatments (41° or 42°C) were evaluated by immunocytochemistry. HCT116 and HCT116 + ch3 cells constitutively expressed HSPB1 and HSPA1A and these proteins significantly increased in response to heat treatments, particularly at 42°C (, panel I). HCT116 + ch3 (hMLH1-proficient cells) expressed higher constitutive cytoplasmic levels of HSPB1 than HCT116 (hMLH1-deficient cells) (, panel II). The hyperthermia significantly increased the cytoplasmic and the nuclear expression of HSPB1 in HCT116+ch3 cells, especially after 42°C treatment, reaching higher levels than in HCT116 cells ( and 1B, panel II, respectively). Although the cytoplasmic expression of HSPA1A significantly increased after heat treatments in both cell lines, HCT116 + ch3 expressed higher levels than HCT116 cells (, panel II). In addition, we observed a higher nuclear expression of HSPA1A in HCT116 + ch3 cells after hyperthermia (41° or 42°C) with a peak immediately after the treatment (, panel II). The results obtained for HCT116 + ch2 cells were similar to the parental HCT116 cell line (data not shown).
Figure 1. Immunocytochemistry for Hsp27 (HSPB1) and Hsp72 (HSPA1A) proteins in hMLH1-deficient (HCT116) and hMLH1-proficient (HCT116 + ch3) cell lines. Panel I. Images of immunostained cells. NC, negative control (non-immune IgG control). Control group (37°C) and cells exposed to heat treatment at 41°C or 42°C for 1 h. Images were taken with a 40 × objective. Panel II. Immunostaining quantification. (A, C) Percentage of cells with positive immunostaining for cytoplasmic HSPB1and HSPA1A, respectively. (B, D) Percentage of cells with nuclear positive immunostaining for HSPB1 and HSPA1A, respectively. C = control at 37°C. Cells were exposed to heat treatment at 41° or 42°C for 1 h, and then incubated at 37°C at the indicated times (0 = immediately after heat shock). Points represent mean ± SEM calculated from three independent experiments. Comparison with the control group (#) and between cell lines (*),#*P < 0.05, ##**P < 0.01, ###***P < 0.001.
Differential nuclear and cytoplasmic expression of MMR proteins and heat shock proteins by immunoblotting
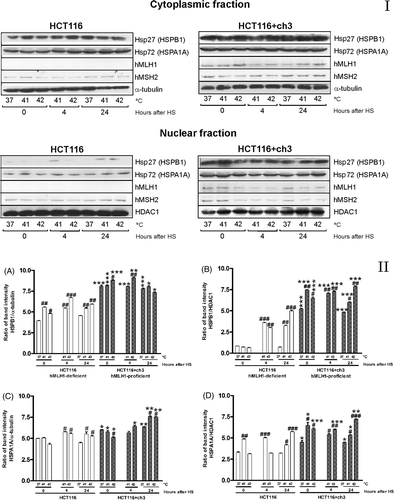
Nuclear and cytoplasmic fractions were separated to examine Hsp27 (HSPB1) and Hsp72 (HSPA1A) behaviour after 41° or 42°C hyperthermia. Under non-stressed conditions (37°C), HCT116 + ch3 cells exhibited higher nuclear and cytoplasmic expression levels of HSPB1 and HSPA1A than HCT116 cells (). A mild hyperthermia (41° or 42°C) significantly increased the cytoplasmic expression of HSPB1 in HCT116 cells at the indicated times, and in HCT116 + ch3 the expression levels of the protein especially increased after 42°C treatment (, panel II). However, the cytoplasmic expression of HSPA1A in HCT116 showed a moderate increased after hyperthermia (, panel II). In HCT116 + ch3 cells, HSPA1A expression increased in the cytoplasm 24 h after heat shock at 41° or 42°C (, panel II). The hyperthermia caused a significant nuclear accumulation of HSPB1 and HSPA1A proteins in both cell lines, particularly 24 h after heating at 42°C (, panel II).
Figure 2. Immunoblots of Hsp27 (HSPB1), Hsp72 (HSPA1A), hMLH1 and hMSH2 proteins in the cytoplasmic and nuclear fractions of HCT116 and HCT116 + ch3 cells exposed to heat treatment. Panel I. Cells were heated at 41° or 42°C for 1 h, and then the cytoplasmic and nuclear protein fractions were extracted and analysed by western blot: immediately after hyperthermia (0), 4 and 24 h after heating. Control unheated cells were also included (37°C). HS: heat shock. As loading standard for cytoplasmic proteins we used α-tubulin and for nuclear proteins we used HDAC1. Data are representative of three independent experiments. Panel II. (A, C) Ratio of band intensity for cytoplasmic HSPB1and HSPA1A, respectively. (B, D) Ratio of band intensity of nuclear HSPB1 and HSPA1A, respectively. The ratio of band intensity to three independent experiments, as shown in Panel I, was quantified by densitometry as described in the text. The means ± SEM are plotted. Statistical differences were calculated with respect to the unheated control group (37°C,#) and between hMLH1-proficient and hMLH1-deficient cells (*). #*P < 0.05, ##**P < 0.01, ###***P < 0.001.
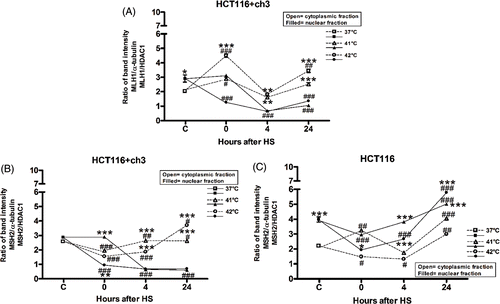
We also studied the cytoplasmic and nuclear expression of MMR proteins, hMLH1 and hMSH2, before and after hyperthermia. The hMLH1 protein was not detected in nuclear and cytoplasmic fractions of HCT116 cells (hMLH1-deficient, see description of cell lines, Materials and methods). HCT116 + ch3 cells, without a heat stress stimulus (37°C), expressed hMLH1 and hMSH2 proteins in the nucleus and less in the cytoplasm (, panel I). The expression of hMLH1 protein induced by heat shock in HCT116 + ch3 cells decreased in the nucleus as a function of time until 24 h post-hyperthermia increasing simultaneously in the cytoplasmic fraction (). We found a similar behaviour in HCT116 + ch3 cells for hMSH2 protein, 4 and 24 h after both heat treatments at 41° or 42°C, suggesting that the protein was translocated from the nucleus to the cytoplasm (). However, the expression of hMSH2 protein resulted higher in the nucleus than in the cytoplasm in HCT116 cells at the same post-heating incubation times (). Similar results were found in HCT116 + ch2 and in HCT116 cells (data not shown).
Figure 3. De-localisation of hMLH1 and hMSH2 under heat shock in HCT116 and HCT116 + ch3 cells. (A) hMLH1 protein expression in cytoplasmic and nuclear compartment in HCT116 + ch3 tumour cells. (B, C) hMSH2 protein expression in cytoplasmic and nuclear compartment in HCT116 + ch3 and HCT116 tumour cells, respectively. Cells were exposed to a heat treatment at 41° or 42°C for 1 h, and then the cytoplasmic and nuclear protein fractions were analysed by western blot immediately after hyperthermia (0), 4 and 24 h post-treatment. HS: heat shock. The ratio of band intensity corresponds to three independent experiments as shown in . The means ± SEM are plotted. Statistical differences were calculated with respect to the unheated control group (C = 37°C,#) and between nuclear and cytoplasmic fractions (*). #*P < 0.05, ##**P < 0.01, ###***P < 0.001.
Effects of hyperthermia in the DNA migration in MMR proficient and deficient cells
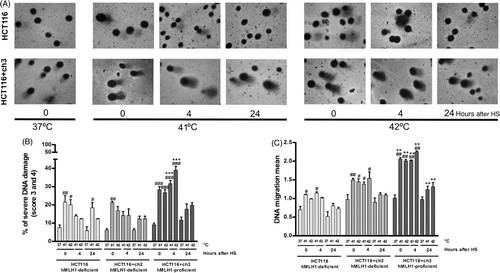
The alkaline comet assay was used to test how hyperthermia may influence the DNA migration of human colon carcinoma cells with and without proficiency of the MMR system. The DNA damage was detected with the alkaline comet assay as a function of the DNA migration mean (see Materials and methods) and of the DNA damage intensity: none (percentage of cells with score 0), low (percentage of cells with score 1 and 2), severe (percentage of cells with score 3 and 4), and total (percentage of cells with score 5). Basal DNA damage (without hyperthermia) was similar between MMR-proficient and MMR-deficient cells (). Hyperthermia caused a rapid increase in both DNA migration mean and the percentage of cells with severe DNA damage, irrespectively of the heating level (41° or 42°C), in HCT116 and HCT116 + ch2 cells. These parameters were more extensively observed in HCT116 + ch3 cells, which remained at high levels during the following 4 h (Figures 4A, B and C). In addition, 4 h after hyperthermia (41° and 42°C), HCT116 + ch3 cells showed a higher percentage of cells with total DNA damage (data not shown). However, we did not find statistically significant differences between HCT116 and HCT116 + ch3 cells in terms of the percentage of cells with low DNA damage (data not shown). Moreover, at 24 h post-hyperthermia, the DNA migration mean and the percentage of cells with severe DNA damage fell down to basal levels suggesting a proficient DNA repair process independently of the MMR status. To be more objective, we calculated the DNA repair capacity (DRC) as the percentage of cells with DNA migration in the control range Citation[26], and no statistically significant differences between HCT116 and HCT116 + ch3 cells were found (data not shown).
Figure 4. Effects of loss of MMR on the DNA damage induced by hyperthermia. MMR-deficient cells (HCT116 and HCT116 + ch2) and MMR-proficient cells (HCT116 + ch3) were exposed 1 h to 41° or 42°C or then collected to study the DNA damage by the comet assay immediately after hyperthermia (0), 4 and 24 h post-treatment. Control unheated cells were included (37°C). (A) Images of comets taken with 40 × objective. Note the increment of the comet tail length as a result of the DNA damage induced by the heat treatment in MMR-proficient cells (HCT116 + ch3). (B) DNA migration mean. (C) Percentage of cells with severe DNA damage (score 3: 31–60% and score 4: 61–95%). HS: heat shock. Statistical differences were calculated with respect to the unheated control group (#) and between hMLH1-proficient and hMLH1-deficient cells (*). #*P < 0.05, ##**P < 0.01, ###***P < 0.001. Data are representative of at least three independent experiments.
Discussion
Several tumour cell lines constitutively express high levels of Hsp27 (HSPB1) and Hsp72 (HSPA1A), which protect them from the microenvironment deleterious factors, critical for cell proliferation and survival, and are associated with drug resistance Citation[27], Citation[4]. However, the effects of a mild hyperthermia on the expression of Hsp27 (HSPB1), Hsp72 (HSPA1A) and MMR proteins hMLH1 and hMSH2 in MMR-proficient (HCT116 + ch3) and deficient (HCT116 and HCT116 + ch2) colon cancer cells have not been established.
In a first step, we characterised the constitutive and heat shock induced (41° and 42°C) expression of HSPB1 and HSPA1A by immunocytochemistry. As most cells lines, HCT116, HCT116 + ch2 and HCT116 + ch3 cells constitutively expressed HSPB1 and HSPA1A. Hyperthermia induced the synthesis of both HSPs, which were over-expressed in the cytoplasm and nucleus of HCT116 + ch3 cells, especially after heating at 42°C. Then, nuclear and cytoplasmic extracts were separated and quantified by western blot. Under non-stressed condition (37°C), HCT116 + ch3 cells exhibited higher nuclear and cytoplasmic expression levels of HSPB1 and HSPA1A than HCT116 cells. We corroborated that the mild hyperthermia (41° or 42°C) increased the cytoplasmic and nuclear expression of HSPB1and HSPA1A, more extensively in HCT116 + ch3 cells (principally at 42°C treatment). There are some reports showing that HSPA (Hsp70) can translocate to the nucleus and accumulate there under heat shock or in other harmful conditions Citation[28–30], but the mechanism involved remains unknown. Previous studies suggested a possible relationship between HSPA levels and DNA damage Citation[31], Citation[32] and it has been suggested that HSPB1 and HSPA have a role in the base excision repair of DNA damage Citation[33]. In a previous work, using cultured peripheral blood lymphocytes (PBL) from healthy individuals, we have reported that a heat shock before cisplatin or doxorubicin treatment induced nuclear accumulation of HSPB1 and HSPA1A, which was also associated with elevated DNA repair capacity and with higher expression of hMLH1 and hMSH2 proteins Citation[17].
We next explored in HCT116 cells the heat-stress induced expression of hMLH1 and hMSH2 proteins. Interestingly our data showed that in HCT116 + ch3 cells (MMR-proficient) hMLH1and hMSH2 proteins were released from the nucleus into the cytoplasm in response to heat shock. Previous studies showed that temperatures of 41°C or higher will drive out of the nucleus the repair protein MRE11 (component of the MRN complex involved in homologous recombination and non-homologous end joining) and lead to a subsequent sensitisation to ionising radiation Citation[34], Citation[35]. This effect could be related to heat dissociating MRE11 from its functional complex MRN with other DNA repair proteins RAD50 and NBS1 Citation[36]. In addition, heat induces an association between MRE11 and HSPA (Hsp70) Citation[36]. In the present study we observed that delocalisation of hMLH1 and hMSH2 proteins was associated with increased nuclear and cytoplasmic expression levels of Hsp27 and Hsp72. Further studies will be necessary to determine whether an interaction between HSPs and MMR proteins may cause the delocalisation of the DNA repair proteins from the nucleus. In HCT116 cells, the hyperthermia increased the expression of hMSH2 protein principally in the nuclear fraction, which may accomplish a function out of the MMR system. For example, it has been reported that during cisplatin treatment, hMSH2 binds ATR (key regulator of the DNA damage response) and recruits it to the DNA damage site for activation Citation[37]. Recently, it has been demonstrated that MLH1 can move from the nucleus to the cytoplasm after irradiation or cisplatin treatment, by a nuclear export sequence in the C-terminal part of the molecule that facilitates the export via the CRM1/exportin pathway Citation[38]. In addition, using small interfering RNA to deplete hMSH2, it has been reported that hMLH1 interaction with PCNA on the chromatin is dependent upon functional hMSH2, as this interaction is abolished in cells with depleted hMSH2 Citation[39].
It is generally accepted that proteins are the major cellular target of the hyperthermia. Exposure to heat leads to alterations in chromatin structure, which reduce the accessibility to the DNA repair machinery, leading to the perturbations of DNA double-strand break repair Citation[40], Citation[41]. Thus, another possible target involved in hyperthermic cell killing is the DNA. Heat can indirectly lead to DNA base modifications such as oxidative base damage Citation[42], abasic DNA sites Citation[43], deamination of cytosine Citation[44] and reactive nitrogen species induced by heat treatment Citation[45]. However, the genotoxicity of heat remains controversial. Here, we employed the alkaline comet assay to evaluate changes in the DNA damage and repair by effect of hyperthermia in hMLH1-proficient and deficient cells. This technique detects DNA single and double-strand breaks and alkali-labile lesions and has many applications in radiation biology, in estimation of oxidative damages and DNA crosslinks, in apoptosis, and in genotoxicity induced by chemical compounds Citation[46]. Untreated hMLH1-proficient and deficient cells showed similar DNA migration mean (basal DNA damage). After a moderate heat shock (41° or 42°C) the DNA migration mean and the percentage of severe and total DNA damage increased, resulting higher in HCT116 + ch3 cells. Therefore, HCT116 cells (MMR-deficient) were more resistant to the heat treatment and they tolerated better the effects of mild hyperthermia. Loss of MMR is important with respect to the emergence of drug resistance since MMR-deficient cells are resistant to cisplatin because of an apparent reduced ability of the cell to sense the presence of adducts in DNA Citation[47]. Heat-induced DNA double strand breaks were found using techniques to detect γH2AX (histone H2AX phosphorylated at serine 139) foci Citation[8]. Hunt showed that heat shock (43°C for 30 or 60 min) induced γH2AX foci but not DNA double strand breaks detectable by pulse field gel electrophoresis Citation[48]. Other authors reported that even if double strand breaks are induced by heat, they do not contribute to heat-induced killing Citation[49], Citation[50].
Finally, 24 h after hyperthermia, HCT116 and HCT116 + ch3 cells showed similar DNA repair capacities. Although hyperthermia may mask the DNA damage from repair pathways due (in part) to the heat-induced association of the nucleolar protein, nucleophosmin (NPM) with matrix attachment region (MAR) DNA Citation[51], the DNA damage induced by the heat treatment decreased near to the basal levels in MMR-proficient and deficient cells. The higher nuclear expression of Hsp27 and Hsp72 after hyperthermia, especially in HCT116 + ch3 cells, may be indicative of their participation in the repair of the heat-induced DNA damage. However, additional experiments that exceed the scope of this paper are needed to determine whether these HSPs would alter the kinetics of the MMR system. Hunt supports a model for heat-induced chromatin alterations that correlates with activation of ATM in the absence of DNA damage Citation[48]. Other authors have reported that there is no relation between double strand break repair deficiency and heat sensitivity using radiosensitive mutants deficient, in either non-homologous end joining or in homologous recombination Citation[49]. However, it has been demonstrated that short-duration severe heat shocks inhibit the repair of UV-induced DNA damage and that the comet assay could detect the repair inhibition after 1–3 min heat shocks Citation[52].
Therefore, our data suggest that the status of the DNA MMR system is not a direct determining factor for the different heat shock responses in HCT116 cells. Other DNA repair mechanisms must be implicated. However, the mild hyperthermia affected the subcellular distribution of hMLH1 and hMSH2 in HCT116 + ch3 cells. Further studies are required to determine possible functional interactions between HSPs and MMR proteins, the significance and mechanisms of hMLH1 and hMSH2 redistribution and their implications in drug sensitivity.
Acknowledgements
We thank C.R. Boland and M. Koi for kindly providing the cell lines.
Declaration of interest: This work was supported by the National Research Council of Argentina and by a grant from the National Agency for Scientific and Technological Promotion of Argentina (PICT 1047, Préstamo BID). The authors alone are responsible for the content and writing of the paper. Darío Cuello-Carrión and Mayra L. Sottile, contributed equally to this work.
Related Research Data
References
- Hurwitz MD. Today's thermal therapy: Not your father's hyperthermia: Challenges and opportunities in application of hyperthermia for the 21st century cancer patient. Am J Clin Oncol 2011; 33: 96–100
- Walther W, Stein U. Heat-responsive gene expression for gene therapy. Adv Drug Deliv Rev 2009; 61: 641–649
- Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009; 14: 105–111
- Ciocca DR, Calderwood SK. Heat shock proteins in cancer: Diagnostic, prognostic, predictive and treatment implications. Cell Stress Chaperones 2005; 10: 86–103
- Roti Roti JL. Heat-induced alterations of nuclear protein associations and their effects on DNA repair and replication. Int J Hyperthermia 2007; 23: 3–15
- Vanderwaal RP, Roti Roti JL. Heat induced ‘masking’ of redox sensitive component(s) of the DNA-nuclear matrix anchoring complex. Int J Hyperthermia 2004; 20: 234–239
- Roti Roti JL. Cellular responses to hyperthermia (40–46°C): Cell killing and molecular events. Int J Hypertthermia 2008; 24: 3–15
- Takahashi A, Matsumoto H, Nagayama K, Kitano M, Hirose S, Tanaka H, et al. Evidence for the involvement of double-strand breaks in heat-induced cell killing. Cancer Res 2004; 64: 8839–8845
- Takahashi A, Yamakawa N, Mori E, Ohnishi K, Yokota S, Sugo N, et al. Development of thermotolerance requires interaction between polymerase-beta and heat shock proteins. Cancer Sci 2008; 99: 973–978
- Nadin SB, Ciocca DR. Participation of heat shock proteins in DNA repair mechanisms in cancer. DNA Repair: Damage, Repair Mechanisms and Aging, F Columbus. Nova Science Publishers, New York 2010; 165–186
- Kotoglou P, Kalitzakis A, Vezyraki P, Tzavaras T, Michalis LK, Dantzar F, et al. Hsp70 translocates to the nuclei and nucleoli, binds to XRCC1 and PARP-1, and protects HeLa cells from single-strand DNA breaks. Cell Stress Chaperones 2009; 14: 391–406
- Hunt CR, Dix DJ, Sharma GG, Pandita RK, Gupta A, Funk M, et al. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol 2004; 24: 899–911
- Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol 2003; 21: 1174–1179
- Jiricny J. Mismatch repair: The praying hands of fidelity. Curr Biol 2000; 10: 788–790
- Hoeijmakers JHJ. Genomic maintenance mechanisms for preventing cancer. Nature 2001; 411: 366–374
- Li G-M. Mechanisms and functions of DNA mismatch repair. Cell Res 2008; 18: 85–98
- Nadin SB, Vargas-Roig LM, Drago G, Ibarra J, Ciocca DR. Hsp27, Hsp70 and mismatch repair proteins hMLH1 and hMSH2 expression in peripheral blood lymphocytes from healthy subjects and cancer patients. Cancer Lett 2007; 252: 131–146
- Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994; 368: 258–261
- Boyer JC, Umar A, Risinger JI, Lipford JR, Kane M, Yin S, et al. Microsatellite instability, mismatch repair deficiency, and genetic defects in human cancer cell lines. Cancer Res 1995; 55: 6063–6070
- Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, et al. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N’-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res 1994; 54: 4308–4312
- Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res 1991; 19: 2499
- Olive PL, Wlodek D, Durand RE, Banáth JP. Factors influencing DNA migration from individual cells subjected to gel electrophoresis. Exp Cell Res 1992; 198: 259–267
- Nadin SB, Vargas-Roig LM, Ciocca DR. A silver staining method for single-cell gel assay. J Histochem Cytochem 2001; 49: 1183–1186
- Anderson D, Yu T-W, Phillips BJ, Shemezer P. The effect of various antioxidants and other modifying agents on oxygen-radical-generated DNA damage in human lymphocytes in the COMET assay. Mutat Res 1994; 307: 261–271
- Branham MT, Nadin SB, Vargas-Roig LM, Ciocca DR. DNA damage induced by paclitaxel and DNA repair capability of peripheral blood lymphocytes as evaluated by the alkaline comet assay. Mutat Res 2004; 560: 11–17
- Schemezer P, Rajaee-Behbahani N, Risch A, Thiel S, Rittgen W, Drings P, et al. Rapid screening assay for mutagen sensitivity and DNA repair capacity in human peripheral blood lymphocytes. Mutagenesis 2001; 16: 25–30
- Yaglom JA, Gabai VL, Sherman MY. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res 2007; 67: 2373–2381
- Chughtai ZS, Rassadi R, Matusiewicz N, Stochaj U. Starvation promotes nuclear accumulation of the Hsp70 Ssa4p in yeast cells. J Biol Chem 2001; 276: 20261–20266
- Lepock JR, Frey HE, Heynen ML, Senisterra GA, Warters RL. The nuclear matrix is a thermolabile cellular structure. Cell Stress Chaperones 2001; 6: 136–147
- Szekely L, Jiang WQ, Pokrovskaja K, Wiman KG, Klein G, Ringertz N. Reversible nucleolar translocation of Epstein-Barr virus-encoded EBNA-5 and Hsp70 proteins after exposure to heat shock or cell density congestion. J Gen Virol 1995; 76: 2423–2432
- Calini V, Urani C, Camatini M. Overexpression of Hsp70 is induced by ionizing radiation in C3H 10T1/2 cells and protects from DNA damage. Toxicol In Vitro 2003; 17: 561–566
- Niu P, Liu L, Gong Z, Tan H, Wang F, Yuan J, et al. Overexpressed heat shock protein 70 protects cells against DNA damage caused by ultraviolet C in a dose-dependent manner. Cell Stress Chaperones 2006; 11: 162–169
- Mendez F, Kozin E, Bases R. Heat shock protein 70 stimulation of the deoxyribonucleic acid base excision repair enzyme polymerase beta. Cell Stress Chaperones 2003; 8: 153–161
- Zhu WG, Seno JD, Beck BD, Dynlacht JR. Translocation of MRE11 from the nucleus to the cytoplasm as a mechanism of radiosensitization by heat. Radiat Res 2001; 156: 95–102
- Xu M, Myerson RJ, Xia Y, Whitehead T, Moros EG, Straube WL, et al. The effects of 41°C hyperthermia on the DNA repair protein, MRE11, correlate with radiosensitization in four human tumour cell lines. Int J Hyperthermia 2007; 23: 343–351
- Dynlacht JR, Xu M, Pandita RK, Wetzel EA, Roti Roti JL. Effects of heat shock on the Mre11/Rad50/Nbs1 complex in irradiated or unirradiated cells. Int J Hyperthermia 2004; 20: 144–156
- Pabla N, Ma Z, McIlhatton MA, Fishel R, Dong Z. hMSH2 recruits ATR to DNA damage sites for activation during DNA damage-induced apoptosis. J Biol Chem 2011; 286: 10411–10418
- Brieger A, Adam R, Passmann S, Plotz G, Zeuzem S, Trojan J. A CRM1-dependent nuclear export pathway is involved in the regulation of MutLα subcellular localization. Genes Chromosomes Cancer 2011; 50: 59–70
- Mastrocola AS, Heinen CD. Nuclear reorganization of DNA mismatch repair proteins in response to DNA damage. DNA Repair (Amst) 2010; 9: 120–133
- Laszlo A. The effects of hyperthermia on mammalian cell structure and function. Cell Prolif 1992; 25: 59–87
- Kampinga HH, Dynlacht JR, Dikomey E. Mechanism of radiosensitization by hyperthermia (> or = 43°C) as derived from studies with DNA repair defective mutant cell lines. Int J Hyperthermia 2004; 20: 131–139
- Bruskov VI, Malakhova LV, Masalimov ZK, Chernikov AV. Heat-induced formation of reactive oxygen species and 8-oxoguanine, a biomarker of damage to DNA. Nucleic Acids Res 2002; 30: 1354–1363
- Warters RL, Brizgys LM. Apurinic site induction in the DNA of cells heated at hyperthermic temperatures. J Cell Physiol 1987; 133: 144–150
- Lindahl T, Nyberg B. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 1974; 13: 3405–3410
- Hall DM, Buettner GR, Matthes RD, Gisolfi CV. Hyperthermia stimulates nitric oxide formation: Electron paramagnetic resonance detection of NO-heme in blood. J Appl Physiol 1994; 77: 548–553
- McKelvey-Martin VJ, Green MHL, Schemezer P, Pool-Zobel BL, De Méo MP, Collins A. The single cell gel electrophoresis assay (comet assay): A European review. Mutat Res 1993; 288: 47–63
- Lin X, Ramamurthi K, Mishima M, Kondo A, Christen RD, Howell SB. p53 modulates the effect of loss of DNA mismatch repair on the sensitivity of human colon cancer cells to the cytotoxic and mutagenic effects of cisplatin. Cancer Res 2001; 61: 1508–1516
- Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res 2007; 67: 3010–3017
- Kampinga HH, Laszlo A. DNA double strand breaks do not play a role in heat-induced cell killing. Cancer Res 2005; 65: 10632–10633
- Laszlo A, Fleischer I. The heat-induced gamma-H2AX response does not play a role in hyperthermic cell killing. Int J Hyperthermia 2009; 25: 199–209
- Vanderwaal RP, Maggi LB, Jr, Weber JD, Hunt CR, Roti Roti JL. Nucleophosmin redistribution following heat shock: A role in heat-induced radiosensitization. Cancer Res 2009; 69: 6454–6462
- Roti Roti JL, Pandita RK, Mueller JD, Novak P, Moros EG, Laszlo A. Severe, short-duration (0–3 min) heat shocks (50–52°C) inhibit the repair of DNA damage. Int J Hyperthermia 2010; 26: 67–78