Abstract
Fever commonly occurs in acute lung injury (ALI) and ALI occurs in 25% of victims of heat stroke. We have shown in mouse models of ALI that exposure to febrile-range hyperthermia (FRH), 39.5°C, increases non-cardiogenic pulmonary oedema. In this study we studied the direct effects of FRH on endothelial barrier integrity using human microvascular endothelial cells (HMVEC-Ls). We analysed the effect of exposure to culture temperatures between 38.5° and 41°C with and without tumour necrosis factor-α (TNF-α) up to 250 U/mL for 6–24 h. We found that exposure to 2.5–250 U/mL TNF-α increased HMVEC-L permeability by 4.1–15.8-fold at 37°C. Exposure to 39.5°C alone caused variable, modest, lot-specific increases in HMVEC-L permeability, however raising culture temperature to 39.5°C in the presence of TNF-α increased permeability an additional 1.6–4.5-fold compared with cells incubated with the same TNF-α concentration at 37°C. Permeability occurred without measurable cytotoxicity and was reversible upon removal of TNF-α and reduction in temperature to 37°C. Exposure to 39.5°C or TNF-α each stimulated rapid activation of p38 and ERK but the effects were not additive. Treatment with inhibitors of ERK (U0126) or p38 (SB203580) each reduced TNF-α-induced permeability in 39.5°C monolayers to levels in 37°C cells, but did not alter TNF-α-induced permeability in the 37°C cells. These results demonstrate that FRH directly increases paracellular pathway opening through a process that requires ERK and p38 MAPKs. A better understanding of this mechanism may provide new understanding about how fever may contribute to the pathogenesis of ALI and provide new therapeutic targets to improve clinical outcomes.
Introduction
Acute respiratory distress syndrome (ARDS)/acute lung injury (ALI) is characterised by neutrophil-rich inflammation, epithelial injury, and loss of endothelial barrier, resulting in non-cardiogenic pulmonary oedema, surfactant deficiency, diffuse atelectasis, and intrapulmonary shunting Citation[1]. While definitive therapies for ARDS have not yet been developed, refined supportive care such as low tidal volume ventilation Citation[2] and restrictive fluid management Citation[3] have improved outcomes. Despite these interventions, mortality remains ∼40% Citation[4], Citation[5]. Our previous studies using a mouse model of febrile-range hyperthermia (FRH), ∼2°C increase in core temperature, identified fever as another potentially important and modifiable pathogenic factor in acute lung injury Citation[6], Citation[7]. Concurrent exposure to FRH, core temperature ∼39.5°C, increased neutrophil-mediated lung injury and worsened survival in mice receiving intratracheal instillation of bacterial endotoxin Citation[7], Citation[8] or exposure to >95% oxygen Citation[6]. The enhanced lung injury in both models was characterised by pulmonary oedema and extravasation of protein into the bronchoalveolar space, indicating a loss of endothelial barrier function.
The pulmonary vascular endothelium is a dynamic, semi-selective barrier that regulates transport of fluid and macromolecules between blood and the lung interstitium. A variety of physical and biochemical stimuli alter the endothelial barrier, leading to increased permeability for macromolecules and, if widespread, reduced gas-exchange capacity. Pro-inflammatory agonists, such as tumour necrosis factor-α (TNF-α) promote macromolecular flux through the endothelial paracellular pathway by opening intercellular gaps in endothelia Citation[9], Citation[10]. This process is associated with reorganisation of the actin cytoskeleton Citation[10–13] and altered integrity of the zonula adherens protein complex Citation[14–17].
We previously showed that exposing human pulmonary artery endothelial cells to FRH, 39.5°C incubation temperature, increased capacity for transendothelial neutrophil migration and augmented TNF-α-induced transendothelial macromolecular flux Citation[18]. More recently, we found that FRH exerted similar effects on neutrophil migration through human pulmonary microvascular endothelial cells (HMVEC-Ls) Citation[19], which originate from the site in the pulmonary vasculature where neutrophil emigration and macromolecular flux occur in vivo Citation[20]. In this study we showed that FRH stimulates ERK and p38 mitogen-activated protein (MAP) kinase activation as well as cytoskeletal rearrangement, and that both MAP kinases are required for increased PMN transmigration in FRH-exposed HMVEC-Ls; however, the impact of FRH on macromolecular flux has not yet been defined.
ERK and p38 have been identified as mediators of agonist-induced opening of the endothelial paracellular pathway to macromolecules; however, their participation is both agonist-specific and species-specific. For example, ERK has been implicated in paracellular pathway opening in bovine pulmonary artery endothelial cells (PAECs) treated with thrombin Citation[21], phorbol myristate acetate (PMA) Citation[22], and 4-hydroxy-2-nonenal Citation[23] as well as in human umbilical endothelial cells (HUVECs) treated with vascular endothelial growth factor Citation[24], 8-bromo 3′5′-cyclic guanosine monophosphate Citation[25], and hydrogen-peroxide Citation[26]. However, it has not been shown to facilitate opening of the paracellular pathway in hydrogen peroxide-treated Citation[27] or transforming growth factor-ß (TGF-ß)-treated Citation[28] bovine PAECs or hydrogen peroxide-treated Citation[29] bovine lung microvascular endothelial cells. p38 MAP kinase mediates paracellular pathway opening in human PAECs treated with the microtubule destabiliser, nocodazole Citation[30], hydrogen peroxide-treated rat PAECs Citation[31], bovine lung microvascular endothelial cells Citation[29], and bovine PAECs Citation[27], TGF-ß-treated and 4-hydroxy-2-nonenal-treated Citation[23] bovine PAECs, but not cyclic GMP-treated HUVECs Citation[25]. However, the contribution of ERK and p38 MAP kinases to the effects of FRH on endothelial cell paracellular pathway opening are not yet known.
In this study, we analysed the effects of FRH on macromolecular flux through HMVEC-L monolayers and helped define the mechanism of this interaction by evaluating the contribution of ERK and p38 MAP kinase activation in these effects. We found that increasing temperature by 1.5°C to 2.5°C increased HMVEC-L permeability. The effect of FRH on permeability was reversible, dependent on both ERK and p38 MAPKs and associated with the formation of actin stress fibres and intercellular gaps.
Materials and methods
Endothelial cell culture
Primary HMVEC-Ls were obtained from Lonza (CC-2527, Basle, Switzerland) and grown in EGM-2MV medium (CC-3202, Lonza) with hEGF, hydrocortisone, foetal bovine serum, GA-1000 (gentamicin, amphotericin-B) VEGF, hFGF-B, and R3-IGF-1 or from PromoCell (Heidelberg, Germany) and grown in ECM-MV medium (PromoCell). HMVEC-Ls were isolated from lung tissue that is immediately adjacent to the outer lining of the lung. Prior to primary isolation, all large vessels were removed, and the cells were isolated from arterioles, venules, and capillaries. These primary cells are used between passages 4 and 9 for permeability assays.
In vitro permeability assay
Transwell inserts with 3 µm pore size were precoated with Matrigel (BD Biosciences, Franklin Lakes, NJ). 2 × 106 HMVEC-Ls were plated in each Matrigel-coated insert and incubated at 37°C for 48 h to allow formation of confluent monolayers. Permeability was analysed by monitoring transendothelial flux of a 10 kDa fluorescent dextran marker (Cascade blue; Invitrogen, Grand Island, NY) added to the lower chamber at a final concentration of 100 µg/mL. Baseline barrier function of each monolayer was confirmed by monitoring Cascade blue flux after 30 min incubation at 37°C. Medium was changed to remove the Cascade blue, TNF-α at various concentrations (0.25 to 250 U/mL) was added, and monolayers were incubated for 6 or 24 h at temperatures between 37° and 41°C. The monolayers were then transferred to 37°C, Cascade blue was again added to the lower chamber and transendothelial flux of Cascade blue measured over 30 min. In some experiments, monolayers were pretreated with inhibitors of ERK (10 µM UO126) or p38 MAPK (10 µM SB203580) for 30 min prior to the addition of TNF-α. In other experiments, reversibility of paracellular pathway opening was analysed by first opening the pathway by incubating HMVEC-L monolayers with 2.5 U/mL TNF-α at 37°C or with 0.25 U/mL TNF-α at 39.5°C for 6 h, then washing out the TNF-α, returning all monolayers to 37°C and sequentially measuring transendothelial flux of Cascade blue.
Cytotoxicity assay
Cytotoxicity was monitored in parallel HMVEC-L monolayers established in 96-well culture plates using a colorimetric assay that measured reduction of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) to a formazan dye (CellTiter 96™; Promega; Madison, WI) according to the manufacturer's protocol and quantifying product formation by measuring absorption at 490 nm.
Analysis of cytoskeletal changes by immunocytochemistry and confocal microscopy
Confluent HMVEC-L monolayers were established on multi-chamber glass slides (Lab-tek™, Nalge-Nunc; Rochester, NY), incubated with or without 0.25 U/mL TNF-α for 6 h at 37° or 39.5°C, fixed with 4% paraformaldehyde, and stained using selective fluorescent probes with very high affinity for F-actin and nuclei (Alexa Fluor 488 conjugated phalloidin and DAPI respectively) according to manufacturer protocols (Invitrogen). Monolayers were visualised using an Olympus microscope and FluoView confocal software (Olympus America, Central Valley, PA). Actin polymerisation was analysed by quantifying relative intensities of F-actin staining from low power images and normalised to cell number by the National Institutes of Health ImageJ 1.37v software.
Western blot
HMVEC-Ls were incubated with or without TNF-α at 37°C or 39.5°C for between 10 min and 6 h. Cell extracts were lysed in RIPA buffer (Teknova; Hollister, CA) containing 10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate and protease inhibitors (Roche Applied Science, Indianapolis, IN), 50 mM NaF, 20 mM beta-glycerophosphate, 1 mM sodium vanadate, and phosphatase inhibitor cocktails (Sigma, St Louis, MO). For immunoblotting, 20 µg total protein was resolved by sodium dodecyl sulphate polyacrylamide gel electrophoresis, electrostatically transferred to polyvinylidene difluoride membrane (Stratagene, Santa Clara, CA) and blocked with 5% non-fat dry milk in TTBS (25 mM Tris pH 7.4, 0.5 M NaCl, 0.05% Tween-20). For phospho-ERK (pERK) and phospho-p38 (p-p38) immunoblotting, membranes were probed with anti-pERK (sc-101760, Santa Cruz, CA) and anti-p-p38 antibody (9211, Cell Signaling; Beverly, MA), respectively, in 20% membrane blocking solution (Zymed, Invitrogen) followed by horseradish peroxidase conjugated secondary antibody (Santa Cruz) and bands were developed with a chemiluminescence detection system (Renaissance™; New England Nuclear; Boston, MA). The membrane was then stripped and re-probed for total ERK or p38 (Santa Cruz). The bands were quantified by direct imaging using a gel documentation system (Fuji LAS-1000) and the pERK/total ERK and p-p38/total p38 ratios were calculated.
Statistical methods
Data are presented as mean ± SE. Differences among groups were analysed by applying a Tukey range test to a one-way ANOVA. Differences with p < 0.05 were considered significant.
Results
FRH augments TNF-α-induced opening of HMVEC-L paracellular pathway to macromolecular flux
We analysed the effect of FRH on endothelial injury using a sensitive permeability assay in which transendothelial flux of 10 kDa Cascade blue dextran was measured under static conditions at 37°C (). The effect of incubating at 39.5°C in the absence of TNF-α to alter HMVEC-L permeability was lot- and passage-dependent (), varying from no effect to a 5.6-fold increase when HMVEC-Ls were incubated at 39.5°C as compared with 37°C for 6 h Citation[18], Citation[32]. Two of the three lots of HMVEC-Ls were isolated from lungs of non-smokers (). The three lots of HMVEC-Ls exhibited different responses to FRH exposure in the absence of a pro-inflammatory agonist, ranging from no change to >5-fold increase in permeability, FRH exerted more consistent effects on permeability in HMVEC-Ls co-treated with TNF-α (). Augmentation of endothelial permeability by FRH was evident over a wide range of TNF-α concentrations and persisted during 24 h of continued incubation at 39.5°C with TNF-α (), essentially shifting the dose response curve by an order of magnitude. To determine whether the effect on permeability was related to the magnitude of hyperthermia, monolayers were incubated with 0.25 U/mL TNF-α for 6 h at temperatures between 38.5° and 41°C or at 42°C for 2 h with recovery at 37°C for 4 h. Permeability was almost twice as great in TNF-α-treated cells incubated at 38.5°C compared with TNF-α-treated cells at 37°C (). Further increases in incubation temperature up to 41°C did not cause a further increase in endothelial permeability (). We complemented the dextran flux experiments by analysing the effect of TNF-α and FRH on HMVEC-L morphology. Post-confluent HMVEC-L monolayers were established and the cells treated with or without 0.25 U/mL TNF-α at 37° or 39.5°C for 6 h, fixed and stained with Alexa Fluor 488-coupled phalloidin and visualised by confocal microscopy (). The HMVEC-Ls incubated without TNF-α at 39.5°C exhibited extensive stress fibre formation but no intercellular gaps (lower left panel). The cells exposed to TNF-α at 37°C developed some stress fibres and intercellular gaps (white arrows). The cells incubated with TNF-α at 39.5°C exhibited stress fibre formation similar to the TNF-α-free 39.5°C cells and extensive intercellular gap formation exceeding that in the cells incubated at 37°C with TNF-α. Quantification of F-actin demonstrated that FRH alone increased F-actin polymerisation correlating with an associated increase in stress fibre formation and this effect was further enhanced when cells were treated with TNF-α at 39.5°C ().
Figure 1. Effect of FRH on permeability in TNF-α-treated HMVEC-Ls. HMVEC-Ls were incubated with the indicated concentration of TNF-α for 6 h (A) or 24 h (B) at 37° or 39.5°C, the TNF-α was removed and transendothelial flux of 10 kDa Cascade blue dextran over 30 min at 37°C was measured. (C) HMVEC-Ls were incubated with 0.25 U/mL TNF-α for 6 h at the indicated temperature, the TNF-α was removed and 10 kDa Cascade blue flux measured. Mean ± SE, n = 21. * and † denote changes with TNF-α-free controls and 37°C cells, respectively. (D) HMVEC-Ls grown on chamber slides were incubated for 6 h without or with 0.25 U/mL TNF-α at 37°C or 39.5°C, fixed and stained with phalloidin coupled with Alexafluor488, counterstained with DAPI, and visualised by fluorescent confocal microscopy. Representatives of four monolayers are shown for each treatment. Intercellular gaps are noted by the arrows. (E) F-actin staining intensity from panel D quantified and expressed relative to 37°C without TNF-α. Mean ± SE, n = 4. † and ‡ denote p < 0.05 versus 37°C with and without TNF-α, respectively.
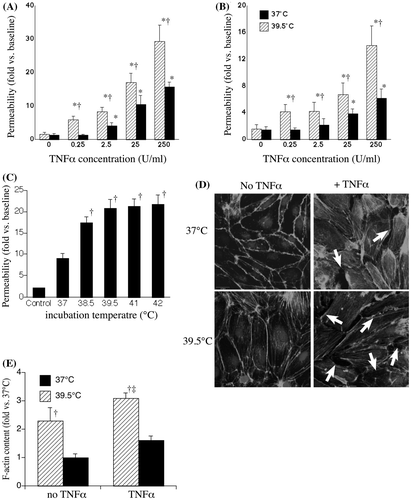
Table I. Effect of FRH exposure alone on permeability in two lots of HMVEC-Ls.
Paracellular pathway in HMVEC-L monolayers incubated at 39.5°C with TNF-α was reversible and occurred without detectable cytotoxicity
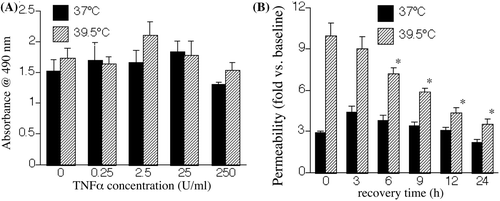
Since FRH and TNF-α have been shown to exert synergistic pro-apoptotic effects on lung epithelium cells Citation[8], we sought to analyse whether the increase in transendothelial macromolecular flux in HMVEC-Ls co-exposed to TNF-α and FRH was caused by cytotoxicity. In confluent monolayers, cultured under the same conditions as the monolayers used in the permeability assay in , incubation with TNF-α up to 250 U/mL for 24 h failed to cause cytotoxicity as measured by the MTS assay (), a general indicator of cell health. In addition, we failed to detect trypan blue uptake in similarly treated monolayers (data not shown). To further analyse for cytotoxicity and to determine whether paracellular pathway opening in cells exposed to FRH and TNF-α was reversible, we treated monolayers for 6 h with either 2.5 U/mL TNF-α at 37°C or 0.25 U/mL TNF-α at 39.5°C to induce similar changes in permeability. We then washed out the TNF-α, transferred all cells to 37°C and followed permeability by sequential measurements of transendothelial dextran flux (). We found permeability in the 39.5°C TNF-α-treated HMVEC-L monolayers to reverse beginning within 6 h after removal of TNF-α and reduction in culture temperature to 37°C. In these experiments, treatment with TNF-α caused greater permeability at 39.5°C than 37°C, but reversibility appeared to be more rapid in the 39.5°C monolayers. Following 6 h incubation with TNF-α, permeability was 3.4-fold higher in cells incubated at 39.5° than cells incubated at 37°C. By 12 h of recovery after TNF-α washout and transfer of all cells to 37°C, permeability in the 39.5°C monolayers was only 1.4-fold greater than the 37°C HMVEC-Ls.
Figure 2. Reversibility and cytotoxicity of FRH on TNF-α-induced paracellular pathway opening. (A) HMVEC-Ls were incubated with the indicated concentration of TNF-α for 6 h at 37°C or 39.5°C and cytotoxicity was determined by measuring using the MTS assay and detected by measuring absorption at 490 nm. (B) HMVEC-Ls were incubated for 6 h with either 2.5 U/mL TNF-α at 37°C or 0.25 U/mL TNF-α at 39.5°C, the TNF-α was removed, all HMVEC-L monolayers returned to 37°C and 10 kDa Cascade blue dextran flux measured immediately and then sequentially during recovery. Mean ± SE, n = 9. * denotes p < 0.05 versus 39.5°C at time 0.
Contribution of ERK and p38 MAP kinase to FRH-augmented endothelial permeability
We have previously shown that incubating HMVEC-L monolayers at 39.5°C increases capacity for neutrophil transmigration and these effects are attenuated by treatment with either the ERK pathway inhibitor, U0126, or the p38 MAP kinase inhibitor, SB203580 Citation[20]. To determine whether ERK and p38 MAP kinase were similarly involved in mediating the effects of FRH on endothelial paracellular pathway opening to macromolecules, we analysed how treatment with U0126 or SB203580 altered macromolecular flux in HMVEC-L monolayers incubated with or without TNF-α at 37° or 39.5°C (). Treatment with U0126 and SB203580 each reduced 10 kDa Cascade blue dextran flux in HMVEC-L monolayers incubated with TNF-α at 39.5°C to levels exhibited in 37°C TNF-α-treated monolayers. Phalloidin staining showed that treatment with U0126 or SB203580 each reduced stress fibre and intercellular gap formation in HMVEC-Ls treated with TNF-α and FRH ().
Figure 3. Contribution of ERK and p38 MAPK to FRH-augmented endothelial permeability. (A) HMVEC-Ls were pre-treated with 10 µM UO126 or p38 MAPK SB203580 for 30 min at 37°C, then incubated with 0.25 U/mL TNF-α for 6 h at the indicated temperature, the TNF-α was removed and 10 kDa Cascade blue dextran flux measured. Mean ± SE, n = 9. * and † denote p < 0.05 versus TNF-α-free controls and 37°C cells, respectively. (B) HMVEC-Ls grown on chamber slides were pretreated with medium alone or 10 µM UO126 or p38 SB203580 for 30 min, incubated for 6 h with 0.25 U/mL TNF-α at 39.5°C, fixed and stained with Alexa Fluor 488-coupled phalloidin and DAPI and visualised by fluorescent confocal microscopy. Representatives of four monolayers are shown for each treatment. (C) HMVEC-Ls were incubated with or without 0.25 ng/mL TNF-α at 37°C or 39.5°C for 10 min to 6 h, lysed, and total- and phospho-p38 and ERK protein were analysed by immunoblot. Representative blots from two similar, independent experiments are shown.
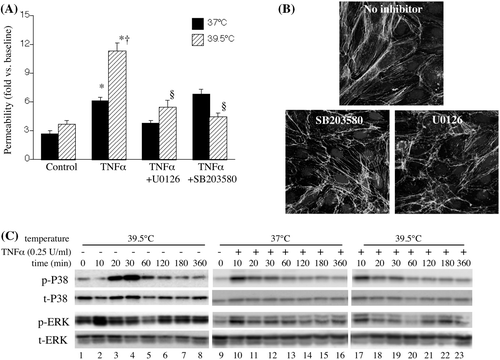
Having demonstrated that ERK and p38 MAP kinase signalling pathways are both required for FRH-augmented paracellular pathway opening, we analysed the effect of FRH on TNF-α-induced ERK and p38 signalling. As we have previously found Citation[20], exposure to 39.5°C stimulated p38 MAP kinase activation that peaked after 20–30 min exposure (). Low concentrations of TNF-α (0.25 U/mL) stimulated more rapid, but less sustained p38 activation, but there was no detectable difference in the pattern of p38 activation between 37°C and 39.5°C TNF-α-treated HMVEC-Ls. Exposing HMVEC-Ls to 39.5°C stimulated biphasic activation of ERK with peaks at 10 min and 2–3 h. Treatment with TNF-α stimulated a single early peak of ERK activation at 37°C and a biphasic ERK activation pattern in 39.5°C HMVEC-Ls.
Discussion
We, and others, have shown that exposure to FRH augments the neutrophil-mediated lung injury caused by exposure to hyperoxia Citation[6] or intratracheal instillation of lipopolysaccharide (LPS) Citation[7], Citation[8]. The pattern of injury in the FRH-exposed mice was characterised by extensive fluid and protein extravasation Citation[6], Citation[7], suggesting a loss of pulmonary vascular barrier function. We have previously shown in human PAECs that TNF-α-induced opening of the paracellular pathway to macromolecules is enhanced at 39.5°C Citation[18]; however, these cells obtained from pulmonary artery do not represent the pulmonary microvasculature, which is the site from which cells and macromolecules enter the lung interstitium Citation[33]. Thus, in the current work, we have extended our previous work by showing that 1) FRH enhances TNF-α-induced paracellular pathway opening in pulmonary microvascular endothelial cells which represent the site in the pulmonary vasculature at which protein leak occurs in ALI; 2) exposure to modest FRH (38.5°C) is sufficient to enhance paracellular pathway opening; 3) the effects of FRH on paracellular pathways occurs without detectable cytotoxicity and is rapidly reversible; and 4) ERK and p38 MAPKs are activated by exposure to FRH and are required for FRH-augmented paracellular pathway opening.
Endothelial paracellular pathway opening is activated by agonists that act through several different receptors, including G-protein-coupled receptors (e.g. thrombin) Citation[34], receptor tyrosine kinases (e.g. VEGF) Citation[24], serine/threonine kinase receptors (e.g. TGF-β) Citation[35], and receptors lacking intrinsic kinase activity (e.g. TNF-α, LPS, TGF-β) Citation[9], Citation[36], each of which activate different post-receptor signalling pathways. Endothelial permeability is also induced by subjecting monolayers to stresses, including stretch Citation[37], hypoxia Citation[38], and heat Citation[39], or by cross-linking ICAM-1 Citation[40]. Opening of the endothelial paracellular pathway is a complex process involving changes in tyrosine phosphorylation status of adhesion junction proteins Citation[9], Citation[34], Citation[41], calcium-dependent contraction of actin-myosin filaments Citation[42], and cytoskeletal rearrangement Citation[43]. In the current study, we focused on ERK and p38 MAP kinase. The signalling pathways activated by endothelial permeability-inducing agonists converge on these two MAP kinases, including the agonists mentioned above Citation[21], Citation[37], Citation[38], Citation[44–46]. Phosphorylation of Hsp27 by the p38 substrate, MK2, promotes actin reorganisation that leads to stress fibre formation Citation[37]. While increased permeability caused by p38 activation has been attributed to Hsp27 phosphorylation and subsequent actin cytoskeletal changes Citation[37], Liu et al. Citation[47] showed that overexpressing phosphomimetic mutant Hsp27 in rat PAECs caused stress fibre formation, but blocked rather than enhanced p38-dependent hypoxia-induced increases in permeability Citation[47]. Lu et al. Citation[35] showed that TGF-β caused hyperpermeability in human PAECs and this effect was not blocked by pre-treating cells with siRNA targeting Hsp27, providing additional evidence that p38 can reduce endothelial barrier function through Hsp27-independent pathways.
Borbiev et al. Citation[21], Citation[44] identified one potential Hsp27-independent mechanism of p38- and ERK-mediated hyperpermeability by demonstrating that p38 and ERK each activate phosphorylation of the myosin inhibitory protein, caldesmon, promoting its dissociation from the myosin complex and allowing contraction of the actin-myosin complex. Lu et al. Citation[35] showed that SB203580 not only blocked TGF-β-induced hyperpermeability but also blocked activation of RhoA GTPase, an activator of cytoskeletal rearrangement, suggesting another alternative p38-dependent pathway to endothelial hyperpermeability.
We previously showed that exposing HMVEC-Ls to FRH activated both MAP kinases and that pretreatment with U0126 and SB203580 each attenuated FRH-induced opening of the monolayers to neutrophil transmigration [20]. In the present study, we showed that incubating HMVEC-Ls at 39.5°C stimulated ERK and p38 activation () and stress fibre formation () in the absence of exogenous TNF-α, but induced little to no macromolecular permeability (, ). Furthermore, although exposure to TNF-α and FRH synergistically induced HMVEC-L permeability, we failed to detect a similar synergy of TNF-α and FRH for ERK and p38 activation. Nonetheless, treatment with either U0126 or SB203580 abrogated the effects of FRH on endothelial permeability but had little effect on permeability in HMVEC-Ls stimulated with TNF-α at 37°C (). Collectively, these results suggest FRH acts through ERK and p38 to enhance TNF-α-induced permeability, but the specific molecular effects of FRH are not yet known.
In models of pulmonary oxygen toxicity Citation[18] and pneumonia Citation[7], co-exposure caused an increase in neutrophil recruitment to the lung and permeability pulmonary oedema. We, and others, have found that FRH exerts multiple effects that converge to increase lung injury, including G-CSF-dependent expansion of circulating neutrophils Citation[48], enhanced expression of neutrophil-attracting chemokines Citation[6], Citation[7], Citation[49], Citation[50], increases in endothelial and neutrophil capacity for transmigration Citation[18], Citation[19], and increased epithelial cell death Citation[7], Citation[8]. The results of the current study shows that temperature increases of 1.5° to 2.5°C, also acts directly on the pulmonary microvascular endothelium to open the paracellular pathway to macromolecular flux, which is requisite for pulmonary interstitial oedema formation. Temperature increases of this magnitude are commonly exceeded during febrile illnesses, especially in the critically ill Citation[51], in individuals exposed to high ambient temperature or performing sustained exercise Citation[52] and in the tissue surrounding thermal ablation zones in patients receiving radioablation therapy for lung cancer Citation[53]. Of clinical importance, the hyperpermeability caused by co-exposure to TNF-α and FRH was reversible following removal of TNF-α and return to 37°C.
Conclusions
We have shown that exposure to elevated temperatures commonly reached during critical illness can cause reversible hyperpermeability in the pulmonary microvascular endothelium. The results of this study suggest that normalising temperature and/or blocking p38 or ERK signalling pathways may be beneficial in preventing, reducing, and/or reversing acute lung injury when the alveolar epithelial barrier is not intact, and further argues for the importance of clinical trials of fever suppression in patients with acute lung injury.
Declaration of interest: This work was supported by US National Institutes of Health grants GM069431 (I.S.S.) and GM066855, HL69057 and HL085256 (J.D.H.), and by US Veterans Association Merit Review grants to J.D.H. and I.S.S. The authors alone are responsible for the content and writing of the paper.
References
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000; 342: 1334–1349
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342: 1301–1308
- Wheeler AP, Bernard GR, Thompson BT, Schoenfeld D, Wiedemann HP, deBoisblanc B, et al. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. N Engl J Med 2006; 354: 2213–2224
- Phua J, Badia JR, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, et al. Has mortality from acute respiratory distress syndrome decreased over time? A systematic review. Am J Respir Crit Care Med 2009; 179: 220–227
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med 2005; 353: 1685–1693
- Hasday J, Garrison A, Singh I, Standiford T, Ellis G, Rao S, et al. Febrile-range hyperthermia augments pulmonary neutrophil recruitment and amplifies pulmonary oxygen toxicity. Am J Pathol 2003; 162: 2005–2017
- Rice P, Martin E, He J-R, Frank M, DeTolla L, Hester L, et al. Febrile-range hyperthermia augments neutrophil accumulation and enhances lung injury in experimental gram-negative bacterial pneumonia. J Immunol 2005; 174: 3676–3685
- Lipke AB, Matute-Bello G, Herrero R, Kurahashi K, Wong VA, Mongovin SM, et al. Febrile-range hyperthermia augments lipopolysaccharide-induced lung injury by a mechanism of enhanced alveolar epithelial apoptosis. J Immunol 2010; 184: 3801–3813
- Angelini DJ, Hyun SW, Grigoryev DN, Garg P, Gong P, Singh IS, et al. TNF{alpha} increases tyrosine phosphorylation of vascular endothelial-cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am J Physiol Lung Cell Mol Physiol 2006; 291: L1232–1245
- Goldblum S, Ding X, Campbell-Washington J. TNF-a induces endothelial cell F-actin depolymerization, new actin synthesis, and barrier dysfunction. Am J Physiol 1993; 264: C894–905
- Goldblum SE, Ding X, Brann TW, Campbell-Washington J. Bacterial lipopolysaccharide induces actin reorganization, intercellular gap formation, and endothelial barrier dysfunction in pulmonary vascular endothelial cells: Concurrent F-actin depolymerization and new actin synthesis. J Cell Physiol 1993; 157: 13–23
- Goldblum SE, Ding X, Funk SE, Sage EH. SPARC (secreted protein acidic and rich in cysteine) regulates endothelial cell shape and barrier function. Proc Natl Acad Sci USA 1994; 91: 3448–3452
- Shasby DM, Shasby SS, Sullivan JM, Peach MJ. Role of endothelial cell cytoskeleton in control of endothelial permeability. Circ Res 1982; 51: 657–661
- Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: Protein interactions and their implications for cadherin function. J Cell Biochem 1996; 61: 514–523
- Ridley AJ, Hall A. Signal transduction pathways regulating Rho-mediated stress fibre formation: Requirement for a tyrosine kinase. Embo J 1994; 13: 2600–2610
- Kemler R. From cadherins to catenins: Cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet 1993; 9: 317–321
- Yamada KM, Geiger B. Molecular interactions in cell adhesion complexes. Curr Opin Cell Biol 1997; 9: 76–85
- Hasday JD, Bannerman D, Sakarya S, Cross AS, Singh IS, Howard D, et al. Exposure to febrile temperature modifies endothelial cell response to tumor necrosis factor-α. J Appl Physiol 2001; 90: 90–98
- Tulapurkar ME, Hasday JD, Singh IS, Prolonged exposure to hyperthermic stress augments neutrophil recruitment to lung during the extended post-exposure period. Int J Hyperthermia 2011;27:717–725
- Tulapurkar ME, Almutairy EA, Shah NG, He JR, Puche AC, Shapiro P, et al. Febrile-range hyperthermia modifies endothelial and neutrophil functions to promote extravasation. Am J Respir Cell Molec Med 2012;46:807–814
- Borbiev T, Verin AD, Birukova A, Liu F, Crow MT, Garcia JG. Role of CaM kinase II and ERK activation in thrombin-induced endothelial cell barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 2003; 285: L43–54
- Verin AD, Liu F, Bogatcheva N, Borbiev T, Hershenson MB, Wang P, et al. Role of ras-dependent ERK activation in phorbol ester-induced endothelial cell barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 2000; 279: L360–370
- Usatyuk PV, Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J Biol Chem 2004; 279: 11789–11797
- Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, II, Duran WN. VEGF increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric oxide. Am J Physiol Heart Circ Physiol 2003; 284: H92–100
- Varma S, Breslin JW, Lal BK, Pappas PJ, Hobson RW, II, Duran WN. p42/44MAPK regulates baseline permeability and cGMP-induced hyperpermeability in endothelial cells. Microvasc Res 2002; 63: 172–178
- Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H(2)O(2)-mediated permeability: Role of MAPK and occludin. Am J Physiol Cell Physiol 2000; 279: C21–30
- Niwa K, Inanami O, Ohta T, Ito S, Karino T, Kuwabara M. p38 MAPK and Ca2+ contribute to hydrogen peroxide-induced increase of permeability in vascular endothelial cells but ERK does not. Free Radic Res 2001; 35: 519–527
- Goldberg PL, MacNaughton DE, Clements RT, Minnear FL, Vincent PA. p38 MAPK activation by TGF-beta1 increases MLC phosphorylation and endothelial monolayer permeability. Am J Physiol Lung Cell Mol Physiol 2002; 282: L146–154
- Usatyuk PV, Vepa S, Watkins T, He D, Parinandi NL, Natarajan V. Redox regulation of reactive oxygen species-induced p38 MAP kinase activation and barrier dysfunction in lung microvascular endothelial cells. Antioxid Redox Signal 2003; 5: 723–730
- Birukova AA, Birukov KG, Gorshkov B, Liu F, Garcia JG, Verin AD. MAP kinases in lung endothelial permeability induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol 2005; 289: L75–84
- Hirano S, Rees RS, Yancy SL, Welsh MJ, Remick DG, Yamada T, et al. Endothelial barrier dysfunction caused by LPS correlates with phosphorylation of Hsp27 in vivo. Cell Biol Toxicol 2004; 20: 1–14
- Shah NG, Tulapurkar ME, Almutairy EA, Hasday JD. Febrile-range hyperthermia augments TNF‐α induced permeability in human microvascular endothelial cells in the lung (hMVEC−L). Am J Respir Crit Care Med 2009; 179: A4014
- Pober JS. Warner-Lambert/Parke-Davis award lecture. Cytokine-mediated activation of vascular endothelium. Physiology and pathology. Am J Pathol 1988; 133: 426–433
- Konstantoulaki M, Kouklis P, Malik AB. Protein kinase C modifications of VE-cadherin, p120, and beta-catenin contribute to endothelial barrier dysregulation induced by thrombin. Am J Physiol Lung Cell Mol Physiol 2003; 285: L434–442
- Lu Q, Harrington EO, Jackson H, Morin N, Shannon C, Rounds S. Transforming growth factor-beta1-induced endothelial barrier dysfunction involves Smad2-dependent p38 activation and subsequent RhoA activation. J Appl Physiol 2006; 101: 375–384
- Bannerman DD, Goldblum SE. Endotoxin induces endothelial barrier dysfunction through protein tyrosine phosphorylation. Am J Physiol 1997; 273: L217–226
- Damarla M, Hasan E, Boueiz A, Le A, Pae HH, Montouchet C, et al. Mitogen activated protein kinase activated protein kinase 2 regulates actin polymerization and vascular leak in ventilator associated lung injury. PLoS ONE 2009; 4: e4600
- Kayyali US, Pennella CM, Trujillo C, Villa O, Gaestel M, Hassoun PM. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J Biol Chem 2002; 277: 42596–42602
- Friedl J, Turner E, Alexander HR, Jr. Augmentation of endothelial cell monolayer permeability by hyperthermia but not tumor necrosis factor: Evidence for disruption of vascular integrity via VE-cadherin down-regulation. Int J Oncol 2003; 23: 611–616
- Sumagin R, Kuebel JM, Sarelius IH. Leukocyte rolling and adhesion both contribute to regulation of microvascular permeability to albumin via ligation of ICAM-1. Am J Physiol Cell Physiol 2011; 301: C804–813
- Goldblum SE, Young BA, Wang P, Murphy-Ullrich JE. Thrombospondin-1 induces tyrosine phosphorylation of adherens junction proteins and regulates an endothelial paracellular pathway. Mol Biol Cell 1999; 10: 1537–1551
- Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: Role of myosin light chain phosphorylation. J Cell Physiol 1995; 163: 510–522
- Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 2001; 91: 1487–1500
- Borbiev T, Birukova A, Liu F, Nurmukhambetova S, Gerthoffer WT, Garcia JG, et al. p38 MAP kinase-dependent regulation of endothelial cell permeability. Am J Physiol Lung Cell Mol Physiol 2004; 287: L911–918
- Nwariaku FE, Rothenbach P, Liu Z, Zhu X, Turnage RH, Terada LS. Rho inhibition decreases TNF-induced endothelial MAPK activation and monolayer permeability. J Appl Physiol 2003; 95: 1889–1895
- Wang Q, Doerschuk CM. The p38 mitogen-activated protein kinase mediates cytoskeletal remodeling in pulmonary microvascular endothelial cells upon intracellular adhesion molecule-1 ligation. J Immunol 2001; 166: 6877–6884
- Liu T, Guevara OE, Warburton RR, Hill NS, Gaestel M, Kayyali US. Modulation of Hsp27 alters hypoxia-induced endothelial permeability and related signaling pathways. J Cell Physiol 2009; 220: 600–610
- Ellis G, Carlson D, Hester L, Bagby G, Singh IS, Hasday J. G-CSF, but not corticosterone mediates circulating neutrophilia induced by febrile-range hyperthermia. J Appl Physiol 2005; 98: 1799–1804
- Maity TK, Henry MM, Tulapurkar ME, Shah NG, Hasday JD, Singh IS. Distinct, gene-specific effect of heat shock on heat shock factor-1 recruitment and gene expression of CXC chemokine genes. Cytokine 2011; 54: 61–67
- Singh IS, Gupta A, Nagarsekar A, Cooper Z, Manka C, Hester L, et al. Heat shock co-activates interleukin-8 transcription. Am J Respir Cell Molec Biol 2008; 39: 235–242
- Laupland KB, Shahpori R, Kirkpatrick AW, Ross T, Gregson DB, Stelfox HT. Occurrence and outcome of fever in critically ill adults. Crit Care Med 2008; 36: 1531–1535
- Cheuvront SN, Kenefick RW, Montain SJ, Sawka MN. Mechanisms of aerobic performance impairment with heat stress and dehydration. J Appl Physiol 2010; 109: 1989–1995
- Dupuy DE. Image-guided thermal ablation of lung malignancies. Radiology 2011; 260: 633–655