Abstract
Local hyperthermia is an effective treatment modality to augment radio- and chemotherapy-based anti-cancer treatments. Although the effect of hyperthermia is pleotropic, recent experiments revealed that homologous recombination, a pathway of DNA repair, is directly inhibited by hyperthermia. The hyperthermia-induced DNA repair deficiency is enhanced by inhibitors of the cellular heat-shock response. Taken together, these results provide the rationale for the development of novel anti-cancer therapies that combine hyperthermia-induced homologous recombination deficiency with the systemic administration of drugs that specifically affect the viability of homologous recombination deficient cells and/or inhibit the heat-shock response, to locally sensitise cancer cells to DNA damaging agents.
Introduction
Efficient repair mechanisms protect DNA of cancer cells, thereby reducing the effectiveness of DNA damage-based therapies. Agents that inhibit DNA repair processes could thus considerably potentiate the cytotoxicity of DNA damage in cancer therapy.
Hyperthermia is one agent that augments DNA damage-based anti-cancer therapies, suggesting that DNA repair mechanisms may be among its targets. Early studies focused on temperatures above 43°C, which cause a large number of macromolecular changes and affect functions of all cellular compartments Citation[1–3]. The more clinically relevant temperatures below 43°C mainly induce protein unfolding and exposure of hydrophobic groups leading to aggregation. As a result, innumerable cellular pathways are disturbed by elevated temperature, but pathways responsible for sensing and repairing DNA damage, as well as for general heat-shock response, are of special interest in cancer research. This review summarises the recent findings that link the molecular effects of hyperthermia to DNA repair and heat-stress response, providing a framework for development of novel anti-cancer strategies.
DSB repair mechanisms
Cells in our bodies suffer damage to their DNA every day, but efficient repair systems maintain its integrity. DNA double-stranded breaks (DSBs) are particularly hazardous since they can lead to genomic rearrangements. DSBs occasionally arise during normal cellular metabolism and their mis-repair can promote carcinogenesis. On the other hand, an overload of DSBs is highly toxic to proliferating cells and this potential is exploited in many effective anti-cancer therapies. Ionising radiation (IR) induces DNA base damage, single-stranded breaks (SSBs) and DSBs – either directly by an ionising particle, such as an electron, or indirectly, when SSBs are generated in close proximity on both DNA strands. DSBs can be indirectly induced by various chemotherapeutic agents, including inhibitors of DNA helicases, DNA intercalating, alkylating or cross-linking agents and nucleotide analogues Citation[4–8].
Efficient DSB repair mechanisms have evolved that help to protect cells against potentially carcinogenic DNA rearrangements. However, DSB repair also counteracts the efficacy of DNA damage-based anti-cancer therapies. Two mechanistically distinct DSB repair pathways are non-homologous end joining (NHEJ) and homologous recombination (HR) (). NHEJ directly rejoins the broken DNA ends and repairs most DSBs in mammalian G1 cells. It involves, among others, the Ku70/Ku80 complex Citation[9], DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and DNA Ligase IV/XRCC4 Citation[10–11]. NHEJ does not utilise the homologous DNA sequence as a template for repair and therefore its activity frequently results in DNA sequence alterations at the repaired break site. One of the earliest NHEJ events is binding of DNA ends by the Ku complex, which later recruits DNA-PKcs. The latter undergoes autophosphorylation-mediated activation Citation[12] and recruits other NHEJ proteins Citation[13]. Broken DNA ends usually harbour various modifications, such as base damage and cross-links, which do not allow direct rejoining and require processing by the Artemis nuclease Citation[14–15] and other DNA metabolising enzymes such as DNA polynucleotide kinase and DNA polymerases Citation[16]. In the final step of NHEJ, DNA ends are ligated by the DNA Ligase IV/XRCC4 complex Citation[16] with help of Cernunnos/XLF, which has an accessory function and is not essential for repair of most DSBs Citation[17–18].
Figure 1. Double-strand break repair by NHEJ and HR. (A) Schematic of NHEJ-mediated DSB repair. DSB ends are recognised by the Ku70/80 heterodimer, resulting in the activation of DNA-PKcs and XRCC4/DNA ligase IV. Processing of DNA ends that cannot be directly ligated (represented by the red star) can be performed by nucleases (Artemis) and DNA polymerases, as well as polynucleotide kinase (not shown). (B) Schematic of HR-mediated DSB repair. DNA ends are organised by the Rad50/MRE11/NBS1 complex, converted by nucleolytic processing to RPA-coated single-strand DNA tails. Mediator proteins such as BRCA2 are involved in loading the core HR protein, Rad51, onto single-strand DNA generating a nucleoprotein filament capable of performing homology recognition on the template DNA. Rad54, another mediator processes Rad51-bound to double-strand DNA such that the intermediate can be handed off to a DNA polymerase to initiate DNA replication (represented by black arrows), which is as an obligatory subsequent step on the way to repair of the DSB.
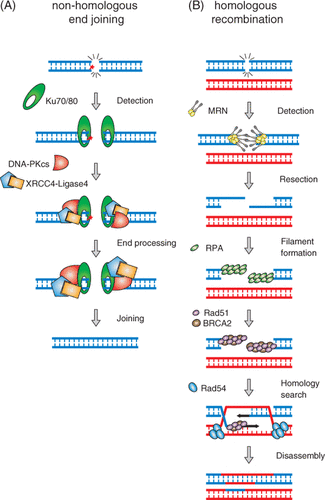
The second major DSB repair pathway is HR (). HR is a genome maintenance process centrally important for accurate genome duplication, DNA damage repair and chromosome segregation. DSB repair by HR is considered to be essentially error free but the requirement of homologous DNA limits its activity to post-replicative chromatin in S and G2 phase cells Citation[19]. HR is essential for cell viability because it allows DNA replication to proceed past SSBs or other DNA damage by promoting strand switch synthesis and repairs otherwise lethal DSBs Citation[20–23]. HR involves a carefully choreographed series of DNA transactions requiring the concerted action of multiple proteins. The catalytic core of HR is a recombinase protein called Rad51 in eukaryotes. Rad51 assembles into a nucleoprotein filament on single-stranded DNA, which is generated at the break site by a combination of proteins with nuclease and helicase activities Citation[23]. This protein-DNA complex is responsible for identifying homologous sequence in the template double-stranded DNA (homology search) and for facilitating the exchange of base-paired partners (joint molecule or D-loop formation). Genetic and biochemical evidence indicate that nucleoprotein filament dynamics and function in strand exchange are controlled in mammalian cells by the activity of proteins including RPA, BRCA2, Rad51 and XRCC3 Citation[23–24]. The biological relevance of these proteins is underscored by the fact that they are essential for viability of mammalian cells Citation[23] and that mutations in BRCA2 are associated with cancer predisposition, in particular with ovarian and breast cancer Citation[25–26].
Hyperthermia and DSB repair
The precise mechanism of hyperthermic radio-sensitisation remains elusive, but hyperthermia appears to be toxic particularly in the S-phase of the cell cycle Citation[27–30]. Early work on DNA damage after hyperthermia in the 40–46°C range showed that although heat does induce chromosomal aberrations in S-phase cells, DNA damage could not be detected by direct measurement or, when detected, could not be attributed to a direct effect on DNA Citation[31]. Thus, the effects of hyperthermia on DNA are not likely caused by direct heat-induced DNA damage, but rather by the effects of heat on proteins involved in DNA replication, chromosome segregations or DNA repair. G1-phase cells are less sensitive to heat and in this phase of the cell cycle no chromosomal aberrations are induced by heat alone Citation[28]. Interestingly, induction of γ-H2AX, a marker for DNA DSBs, by heat also occurred predominantly in S-phase cells undergoing replication Citation[32], but it is disputed whether hyperthermia-induced γ-H2AX in fact represents DNA damage Citation[32]. If hyperthermia does indeed increase the amount of DNA damage in S-phase cells and if this effect is indeed indirect, it could be a result of impaired DNA repair or replication.
Since these processes are tightly coupled during S-phase, especially S-phase-specific HR and replication, the affected process is difficult to pinpoint. Work from van der Waal et al. Citation[33] showed that, upon hyperthermia treatment, binding of replication-associated proteins to the nuclear matrix at the nuclear periphery is enhanced. This enhanced binding directly correlated with hyperthermia-induced cell killing and could be inhibited by blocking replication. Furthermore, heated cells showed defective formation of radiation-induced MRE11 Citation[34]. Relocalisation to the nuclear periphery is not uncommon for DSB proteins and can indicate persistence of DSBs or repair delay Citation[35–37]. These results suggest that radio-sensitisation by hyperthermia requires replication, however, they do not rule out that it is caused by persistent DNA damage due to defective DSB repair. Overall, evidence is limited and mostly still relies on the classical work showing that cellular radio-sensitivity correlates with increased frequency of chromosomal aberrations Citation[28], Citation[31].
Other studies dealing with cellular radio-sensitivity focused on clonogenic survival efficiency using mutant cell lines defective in a single specific DNA repair pathway. This approach is based on the assumption that if heat would inhibit a specific DNA repair pathway, then cells defective in that pathway could no longer be radio-sensitised by heat. Conversely, if hyperthermia treatment could radiosensitise such a mutant cell line, then the underlying defective DNA repair pathway would not likely be targeted by heat. A wide variety of NHEJ mutants were tested, including mutants in key proteins such as Ku70/80, DNA-PK and DNA ligase IV. In none of the mutants the extent of thermal radiosensitisation was decreased Citation[38–40]. Likewise, original studies on genetic mutants of HR, such as XRCC2 and XRCC3 deficient rodent cells and HR deficient chicken cells, also showed normal radiosensitisation when exposed to hyperthermia Citation[41–43]. These results initially excluded HR as a target for heat. However, the issue was recently revised using an isogenic set of mouse embryonic stem cells. HR-deficient cells lacking the HR factor Rad54 could not be further sensitised to IR using heat in the range of clinically relevant temperatures. In addition, similar was true for human cells depleted of XRCC3 Citation[44], suggesting that HR may indeed be targeted by heat. The same observation was made in a human adenocarcinoma cell line, where MRE11, a protein essential in HR, was depleted Citation[45]. These contradictory results suggest that outcomes of clonogenic survival assays might be highly dependent on cell type, genotype and temperature applied and should be interpreted with caution. One confounding factor with clonogenic survival assays is that the end point measured depends on many cellular processes and not solely on the genetically inactivated pathway in the cells used. Unambiguous evidence on the inhibitory effect of elevated temperature on HR came from experiments where HR activity was measured directly and shown to be significantly reduced by heat Citation[44].
Additional experiments that are useful in dissecting how heat affects HR are ones that follow the accumulation of HR proteins at the sites of DNA damage. These experiments allowed further pinpointing of the heat-sensitive step in HR. Most HR proteins accumulate in large numbers at the site of DNA damage into subnuclear structures called foci Citation[19], Citation[37], Citation[46]. These foci are thought to be biologically relevant and indicative of correct repair responses because cell lines defective in focus formation, such as BRCA2 or Rad51 paralogue mutants, are defective in HR, show spontaneous chromosomal instability, and increased sensitivity to DNA damaging agents Citation[47–48]. This implies that DNA damage-induced foci are functional markers of HR and can be used as specific tools to monitor the response of HR to external stimuli Citation[37]. In one type of experiment, DNA damage was inflicted by α-particles which create linear tracks of DSBs Citation[49–50]. Using immunostaining, the accumulation of specific HR proteins at DSBs was subsequently tested in heated cells. While accumulation of proteins that act early in DSB repair, such as MRE11 and RPA, was not affected by hyperthermia, the accumulation of the downstream-acting proteins BRCA2 and Rad51 was abrogated Citation[44]. These results indicated that both recognition and resection of the DSB was executed normally but formation of the D-loop structures, a process requiring both BRCA2 and Rad51, was impaired. The lack of accumulation of Rad51 at sites of DNA damage is also a hallmark of cells harbouring a genetic defect in BRCA2, which suggested that BRCA2 might be rendered dysfunctional upon hyperthermia treatment. Indeed, it was demonstrated that hyperthermia induces degradation of the BRCA2 protein Citation[44]. It must be stressed that these results do not implicate degradation of BRCA2 or impairment of HR as the sole or even most important effect of heat on DNA repair or on other cellular processes relevant for cell killing Citation[38–40], Citation[42], Citation[51]. However, they do provide insight into molecular mechanisms that may be relevant in hyperthermic radiosensitisation.
Induced synthetic lethality
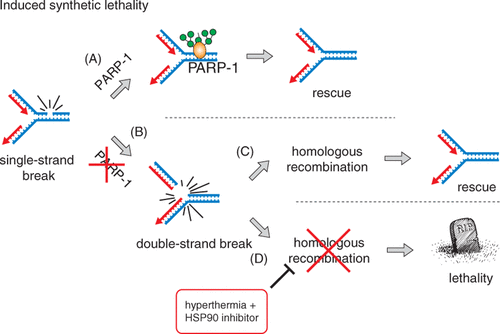
Previous studies have shown that the BRCA2 gene is mutated in certain cases of hereditary breast cancer Citation[25], Citation[26], resulting in a severe HR defect in affected cancer cells. Currently, a number of clinical trials are testing drugs, called poly(ADP-ribose) polymerase 1 (PARP-1) inhibitors that specifically kill tumour cells with defective HR, such as those with mutated BRCA2 Citation[52–54] (). PARP-1 is a DNA repair enzyme that signals the presence of DNA SSBs by attaching polymers of ADP-ribose to various substrates, thereby activating DNA repair pathways. In the absence of PARP-1 activity, unrepaired SSBs encountered by progressing replication forks are converted to DSBs that require HR for repair. Two pioneering studies exploited this HR dependence in a synthetic lethality approach. These studies showed that (cancer) cells harbouring defects in HR are hypersensitive to PARP-1 inhibitors Citation[52], Citation[54]. This formed the foundation for a novel therapeutic approach utilising PARP-1 inhibitors to target rare forms of breast cancer with defects in BRCA1 and BRCA2, important HR factors. Importantly, this strategy, while cytotoxic to HR-deficient (cancer) cells, is much less harmful to normal cells and tissues, which can employ HR to repair DSBs induced indirectly by PARP-1 inhibitors. This approach was recently validated in high-profile clinical trials Citation[53]. The recent insight into the effect of hyperthermia on HR raised the possibility that the combination of hyperthermia and PARP-1 inhibitors could be detrimental to innately HR-proficient (tumour) cells. Indeed, hyperthermia not only sensitised cultured cells to PARP-1 inhibitors, but also xenografted tumours in animals treated with PARP-1 inhibitors 24 h in advance Citation[44]. This new approach, termed ‘induced synthetic lethality’, may provide the rationale for the use of PARP-1 inhibitors not only to treat tumours with rare genetic mutations, but in principle against all tumours that can be locally heated. These results might have direct clinical relevance, because recent trials demonstrated that PARP-1 inhibitors are generally well tolerated Citation[55–56]. An advantage of combining hyperthermia with PARP-1 inhibitors is that such a transient inhibition prevents the risk of selecting for mutations that confer resistance to PARP-1 inhibitors.
Figure 2. Induced synthetic lethality. (A) PARP-1 is important for proper repair of SSBs that occur spontaneously as a consequence of normal cellular metabolism. (B) In the absence of PARP-1 activity, unrepaired SSBs that are encountered by progressing DNA replication forks in S-phase cells are converted to DSBs. This type of DNA lesion is very cytotoxic, as even a single unrepaired DSB can result in cell death. (C) In S-phase, replication-associated DSBs are restored by HR, which involves proteins such as BRCA1/2 and Rad51. Therefore, in repair-competent cells the DSBs arising at the replication fork (due to inhibition of PARP-1) are promptly and accurately repaired by HR, with no further cytotoxic consequences. (D) However, in the absence of HR, either due to genetic mutation or induced by the hyperthermia (and heat-shock protein inhibition), these DSBs are not timely repaired and cause cell death.
The role of heat-shock response in thermo-sensitivity
The cellular response to (heat) stress consists of a cascade of events affecting multiple cellular pathways and components in many compartments, ranging from covalent modifications of chromatin to changes in liquidity of cellular membranes Citation[57]. Hyperthermia also directly induces protein unfolding, especially in the higher temperature range. A prominent role in heat-stress response is played by the so-called heat-shock proteins (HSPs). HSPs are ATP-driven molecular chaperones controlling the fate of numerous proteins by initiating or influencing their proper (re)folding as well as by targeting to proteasomal degradation Citation[58–59]. In response to heat, activation of heat-shock factors (HSFs), via heat-shock elements (HSEs), induces expression of HSPs. Related to the (re)folding activity of HSPs is their function in the heat-stress response; HSPs are believed to protect proteins against heat-induced misfolding and to help in recovery of already unfolded proteins [60]. At the same time, HSPs can directly inhibit caspase-dependent as well as caspase-independent apoptotic pathways Citation[58], likely providing the time necessary for protein re-folding. This two-fold action of HSPs has significant influence on the sensitivity of mammalian cells to heat stress and an even more dramatic role in response to subsequent heat exposure Citation[57]. Importantly, heat-stimulated expression of HSPs protects cells against the effects of a subsequent heat exposure in a mechanism termed thermotolerance. Thermotolerance, an effect lasting for up to a few days after the initial heat exposure, thus dictates the time-schedules of clinical hyperthermia treatments that can only be applied on a once- or twice-weekly basis. From the clinical point of view, neutralising HSP activity in heated cells might thus present a two-fold benefit; it might enhance the cytotoxic potential of the treatment and eliminate thermotolerance.
HSPs are a heterogeneous group of proteins, classified according to their molecular weight to a number of families, including Hsp100, Hsp90, Hsp70 or Hsp60. HSPs do not act alone; they are mostly part of larger, heterogeneous and dynamic protein complexes that allow broad substrate specificity Citation[61–62]. The Hsp70 family plays major role in heat-shock response and thermotolerance, but another heat-induced chaperone family, Hsp90, is rapidly gaining attention in oncology. Hsp90 has been implicated in various processes related to tumourigenesis, such as tumour angiogenesis Citation[63] or protection of overexpressed/mutated oncogene proteins that drive tumour progression. The survival of transformed cells depends, to a large extent, on the activity of these proteins in a mechanism called oncogene addiction. Hsp90 activity maintains the supply of oncogenes encoded proteins for the ‘addicted’ cancer cells by protecting them from degradation and/or misfolding. As a consequence, cancer cells are more dependent on Hsp90 than non-transformed cells, which renders this chaperone a desirable therapy target Citation[64–65].
Over 200 Hsp90 client proteins have been identified, including factors relevant for chromatin remodelling, DNA repair, cellular homeostasis, transcriptional regulation, signalling and tumour immunology Citation[64], Citation[66–69]. The interaction of Hsp90 with its clients is essential not only during stress, but also under normal physiological conditions and Hsp90 is an essential part of numerous protein complexes. Here, we concentrate on results linking the Hsp90 chaperone machinery to DNA repair processes.
The first clues linking Hsp90 to the DNA damage response come from experiments showing that geldanamycin and its derivatives, compounds binding the ATP-pocket of Hsp90 and thereby inhibiting its chaperone activity, enhanced radio-sensitivity of multiple human cancer cell lines, abrogated the G2 and S-phase check-points and interfered with DNA damage signalling Citation[70–77]. Furthermore, inhibition of Hsp90 induced depletion and decreased activation of ATR and CHK1, master-regulators of cellular signalling after SSB induction, but not of ATM, which orchestrates the DSB response Citation[78]. Likewise, Hsp90 was shown to be important for stability of FANCA, a key factor in the Fanconi anaemia-associated DNA repair, and Hsp90 inhibition led to proteasomal degradation of FANCA and down-regulation of the pathway Citation[79]. Inhibition of Hsp90 with the geldanamycin derivative 17 -(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG) reduced the activation of DNA-PKcs and ATM, and impaired the ability of the MRN complex to interact with ATM and form nuclear foci in response to radiation Citation[77]. 17-DMAG repressed DSB repair as measured by dissolution of radiation-induced yH2AX foci and by the neutral comet assay Citation[77] and inhibited nucleotide excision repair in human lung cancer lines Citation[80]. Among clients of Hsp90 are also the HR repair factors Rad51 and BRCA2; Hsp90 inhibition reduced the activity and levels of both proteins in mammalian cell lines Citation[81]. In line with these results, 17-DMAG sensitised glioma cells to PARP-1 inhibitors Citation[82]. The interference of Hsp90 with multiple cellular pathways, including those of special interest in cancer, triggered considerable effort of scientific and pharmaceutical industry environments to design and validate drugs targeting Hsp90 Citation[64].
The heat-shock response as an emerging therapeutic target
Perhaps surprisingly, although the function of Hsp90 in the heat-shock response has been intensively investigated and efficient small-molecule Hsp90 inhibitors are available, the effects of Hsp90 inhibition on thermosensitivity are poorly documented. Results of one recent study by Ito and colleagues Citation[83] showed significant thermosensitisation of human melanoma cells by 17-DMAG in vitro. In the same study, a combination of 17-DMAG and hyperthermia significantly inhibited tumour outgrowth in a mouse melanoma model, as compared to hyperthermia or 17-DMAG treatments alone Citation[83]. Likewise, recent results with rat rhabdomyosarcoma cells showed thermosensitisation by 17-DMAG in cell culture and a xenograft model, by temperatures below 43°C Citation[44]. Inhibition of Hsp90 thus clearly exacerbates effects of hyperthermia and this appears to be the case not only for thermosensitivity, but also for hyperthermia-mediated inhibition of the HR pathway Citation[44]. Recent experiments demonstrated that 17-DMAG strongly stimulates sensitivity of cells to ionising radiation and increases hyperthermia-induced degradation of BRCA2 and inhibition of HR. Most importantly, the triple-agent combination of hyperthermia, PARP inhibitor and 17-DMAG dramatically reduced tumour outgrowth in a xenograft model Citation[44]. Such a combination approach can thus be a powerful tool for which translation to the clinic has great potential since PARP-1 and Hsp90 inhibitors are being tested in clinical trials Citation[53], Citation[55]. Both combinations, hyperthermia with PARP-1 inhibitor and hyperthermia with Hsp90 inhibitors, have yet to be tested clinically.
In addition to the recent results on the effects of heat and Hsp90 inhibition on HR Citation[44], the plethora of substrates of Hsp90 chaperone implies that a combination of heat and Hsp90 inhibition may sensitise (cancer) cells to multiple other anti-cancer agents that induce cytotoxicity by targeting Hsp90 clients. In light of broad involvement of Hsp90 in DNA repair and signalling, it will be interesting to confirm, in vivo, the effects of Hsp90 inhibitors on the hyperthermia/radiotherapy combination that is routinely applied in some medical centres Citation[84]. Cisplatin/hyperthermia combination treatment may, likewise, benefit from the addition of Hsp90 inhibitors.
The data accumulating in previous decades and the more recent results described above may extend the concept of hyperthermia-mediated synthetic lethality. By inhibiting different chaperone proteins, with the addition of heat stress, it might be possible to target their respective client proteins. This could be achieved in two ways. First, some client proteins may be sensitive to elevated temperature and may therefore need the protection of HSPs against heat-induced misfolding and/or degradation. Second, some client proteins may not be temperature-sensitive by their nature, but they might need HSP assistance to perform their normal functions. In conditions of (heat) stress, HSPs are recruited to mis-/unfolded proteins, abandoning their regular partners and thereby impairing their functionality. These two scenarios are likely to develop in concert, amplifying the deleterious treatment effects. However, although Hsp90 inhibitors are in late stages of development and clinical testing Citation[65], very few specific small-molecule inhibitors targeting other chaperones are available Citation[85–86].
Perspective
The demonstration that hyperthermia inhibits HR repair of DNA damage Citation[44] helps to explain why hyperthermia augments therapies using DSB-inducing agents, such as ionising radiation and cisplatin, in multiple types of cancer, including cervical cancer Citation[87–95]. Moreover, the proof-of-principle experiments reviewed above suggest that hyperthermia-induced and Hsp90 inhibitor-enhanced HR deficiency could sensitise cells to other agents inducing DSBs that are repaired by HR. Many applied and potential therapeutic approaches utilise such agents to kill cancer cells. For instance, cells defective in BRCA1 and BRCA2 are severely sensitive to DSBs induced by inhibitors of topoisomerase II Citation[96], which suggests that HR is at least partially responsible for repair of these lesions. Similarly, defects in XRCC2, an important HR factor, sensitise cells to anticancer alkylating agents such as temozolomide, fotemustine and maphosphamide Citation[97].
HR is also responsible for repair of DSBs caused indirectly by DNA cross-linking agents cisplatin, mitomycin C, and melphalan Citation[98]. A recent study showed that DSBs induced by sulphoraphane, a novel compound with anti-cancer properties, are directly rejoined by the HR machinery Citation[99]. Confirmation and optimisation in model systems could bring these approaches based on the induced synthetic lethality concept a step closer to clinical application.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. Our research has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement HEALTH-F2-2010-259893, and from the Netherlands Organisation for Scientific Research (NWO) and the Netherlands Genomics Initiative/NWO.
Related Research Data
References
- Stege GJ, Kampinga HH, Konings AW. Heat-induced intranuclear protein aggregation and thermal radiosensitization. Int J Radiat Biol 1995; 67: 203–209
- Beck BD, Dynlacht JR. Heat-induced aggregation of XRCC5 (Ku80) in nontolerant and thermotolerant cells. Radiat Res 2001; 156: 767–774
- Laszlo A, Davidson T, Harvey A, Sim JE, Malyapa RS, Spitz DR, et al. Alterations in heat-induced radiosensitization accompanied by nuclear structure alterations in Chinese hamster cells. Int J Hyperthermia 2006; 22: 43–60
- Hanada K, Budzowska M, Modesti M, Maas A, Wyman C, Essers J, et al. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. Embo J 2006; 25: 4921–4932
- Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, et al. Alkylation damage in DNA and RNA-repair mechanisms and medical significance. DNA Repair (Amst) 2004; 3: 1389–1407
- Gupta R, Brosh RM, Jr. DNA repair helicases as targets for anti-cancer therapy. Cur Med Chem 2007; 14: 503–517
- Galmarini CM, Popowycz F, Joseph B. Cytotoxic nucleoside analogues: Different strategies to improve their clinical efficacy. Cur Med Chem 2008; 15: 1072–1082
- Wheate NJ, Brodie CR, Collins JG, Kemp S, Aldrich-Wright JR. DNA intercalators in cancer therapy: Organic and inorganic drugs and their spectroscopic tools of analysis. Mini Rev Med Chem 2007; 7: 627–648
- Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res 2008; 18: 114–124
- Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 2006; 124: 301–313
- Smith GC, Divecha N, Lakin ND, Jackson SP. DNA-dependent protein kinase and related proteins. Biochem Soc Symp 1999; 64: 91–104
- Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double strand break repair pathway choice. Mol Cell Biol 2005; 25: 10842–10852
- Meek K, Douglas P, Cui X, Ding Q, Lees-Miller SP. trans Autophosphorylation at DNA-dependent protein kinase's two major autophosphorylation site clusters facilitates end processing but not end joining. Mol Cell Biol 2007; 27: 3881–3890
- Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. Embo J 2006; 25: 3880–3889
- Darroudi F, Wiegant W, Meijers M, Friedl AA, van der Burg M, Fomina J, et al. Role of Artemis in DSB repair and guarding chromosomal stability following exposure to ionizing radiation at different stages of cell cycle. Mutat Res 2007; 615: 111–124
- van Gent DC, van der Burg M. Non-homologous end-joining, a sticky affair. Oncogene 2007; 26: 7731–7740
- Tsai CJ, Kim SA, Chu G. Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc Natl Acad Sci USA 2007; 104: 7851–7856
- Wu PY, Frit P, Malivert L, Revy P, Biard D, Salles B, et al. Interplay between Cernunnos-XLF and nonhomologous end-joining proteins at DNA ends in the cell. J Biol Chem 2007; 282: 31937–31943
- Tashiro S, Walter J, Shinohara A, Kamada N, Cremer T. Rad51 accumulation at sites of DNA damage and in post replicative chromatin. J Cell Biol 2000; 150: 283–291
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature 2000; 404: 37–41
- Eppink B, Wyman C, Kanaar R. Multiple interlinked mechanisms to circumvent DNA replication roadblocks. Exp Cell Res 2006; 312: 2660–2665
- Kowalczykowski SC. Initiation of genetic recombination and recombination dependent replication. Trends Biochem Sci 2000; 25: 156–165
- Wyman C, Kanaar R. DNA double-strand break repair: All's well that ends well. Annu Rev Genet 2006; 40: 363–383
- Wyman C, Kanaar R. Homologous recombination: Down to the wire. Curr Biol 2004; 14: R629–R631
- Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006; 25: 5864–5874
- Shivji MK, Venkitaraman AR. DNA recombination, chromosomal stability and carcinogenesis: Insights into the role of BRCA2. DNA Repair (Amst) 2004; 3: 835–843
- Mackey MA, Dewey WC. Time–temperature analyses of cell killing of synchronous G1 and S phase Chinese hamster cells in vitro. Radiat Res 1988; 113: 318–333
- Dewey WC, Westra A, Miller HH, Nagasawa H. Heat-induced lethality and chromosomal damage in synchronized Chinese hamster cells treated with 5-bromodeoxyuridine. Int J Radiat Biol Relat Stud Phys Chem Med 1971; 20: 505–520
- Mackey MA, Anolik SL, Roti Roti JL. Changes in heat and radiation sensitivity during long duration, moderate hyperthermia in HeLa S3 cells. Int J Radiat Oncol Biol Phys 1992; 24: 543–550
- Mackey MA, Roti Roti JL. A model of heat-induced clonogenic cell death. J Theor Biol 1992; 156: 133–146
- Dewey WC, Sapareto SA, Betten DA. Hyperthermic radiosensitization of synchronous Chinese hamster cells: Relationship between lethality and chromosomal aberrations. Radiat Res 1978; 76: 48–59
- Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res 2007; 67: 3010–3017
- VanderWaal RP, Griffith CL, Wright WD, Borrelli MJ, Roti JL. Delaying S-phase progression rescues cells from heat-induced S-phase hypertoxicity. J Cell Physiol 2001; 187: 236–243
- Dynlacht JR, Xu M, Pandita RK, Wetzel EA, Roti Roti JL. Effects of heat shock on the MRE11/Rad50/NBS1 complex in irradiated or unirradiated cells. Int J Hyperthermia 2004; 20: 144–156
- Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 2009; 23: 912–927
- Kalocsay M, Hiller NJ, Jentsch S. Chromosome-wide Rad51 spreading and SUMOH2A. Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol Cell 2009; 33: 335–343
- Agarwal S, van Cappellen WA, Guenole A, Eppink B, Linsen SE, Meijering E, et al. ATP-dependent and independent functions of Rad54 in genome maintenance. J Cell Biol 2011; 192: 735–750
- Dynlacht JR, Bittner ME, Bethel JA, Beck BD. The non-homologous end-joining pathway is not involved in the radiosensitization of mammalian cells by heat shock. J Cell Physiol 2003; 196: 557–564
- Kampinga HH, Kanon B, Konings AW, Stackhouse MA, Bedford JS. Thermal radiosensitization in heat- and radiation-sensitive mutants of CHO cells. Int J Radiat Biol 1993; 64: 225–230
- Raaphorst GP, Thakar M, Ng CE. Thermal radiosensitization in two pairs of CHO wild-type and radiation-sensitive mutant cell lines. Int J Hyperthermia 1993; 9: 383–391
- Wachters FM, van Putten JW, Maring JG, Zdzienicka MZ, Groen HJ, Kampinga HH. Selective targeting of homologous DNA recombination repair by gemcitabine. Int J Radiat Oncol Biol Phys 2003; 57: 553–562
- Yin HL, Suzuki Y, Matsumoto Y, Tomita M, Furusawa Y, Enomoto A, et al. Radiosensitization by hyperthermia in the chicken B-lymphocyte cell line DT40 and its derivatives lacking nonhomologous end joining and/or homologous recombination pathways of DNA double-strand break repair. Radiat Res 2004; 162: 433–441
- Raaphorst GP, Maude-Leblanc J, Li L. Evaluation of recombination repair pathways in thermal radiosensitization. Radiat Res 2004; 161: 215–218
- Krawczyk PM, Eppink B, Essers J, Stap J, Rodermond H, Odijk H, et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc Natl Acad Sci USA 2011; 108: 9851–9856
- Xu M, Myerson RJ, Hunt C, Kumar S, Moros EG, Straube WL, et al. Transfection of human tumour cells with Mre11 siRNA and the increase in radiation sensitivity and the reduction in heat-induced radiosensitization. Int J Hyperthermia 2004; 20: 157–162
- Essers J, Houtsmuller AB, van Veelen L, Paulusma C, Nigg AL, Pastink A, et al. Nuclear dynamics of Rad52 group homologous recombination proteins in response to DNA damage. Embo J 2002; 21: 2030–2037
- Thacker J. The Rad51 gene family, genetic instability and cancer. Cancer Lett 2005; 219: 125–135
- van Veelen LR, Essers J, van de Rakt MW, Odijk H, Pastink A, Zdzienicka MZ, et al. Ionizing radiation-induced foci formation of mammalian Rad51 and Rad54 depends on the Rad51 paralogs, but not on Rad52. Mutat Res 2005; 574: 34–49
- Aten JA, Stap J, Krawczyk PM, van Oven CH, Hoebe RA, Essers J, et al. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 2004; 303: 92–95
- Stap J, Krawczyk PM, Van Oven CH, Barendsen GW, Essers J, Kanaar R, et al. Induction of linear tracks of DNA double-strand breaks by alpha-particle irradiation of cells. Nat Methods 2008; 5: 261–266
- Iliakis G, Seaner R. A DNA double-strand break repair-deficient mutant of CHO cells shows reduced radio sensitization after exposure to hyperthermic temperatures in the plateau phase of growth. Int J Hyperthermia 1990; 6: 801–812
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–921
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361: 123–134
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434: 913–917
- Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: A phase I dose-escalation study. J Clin Oncol 2007; 25: 5410–5417
- Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res 2008; 14: 7917–7923
- Kampinga HH. Cell biological effects of hyperthermia alone or combined with radiation or drugs: A short introduction to newcomers in the field. Int J Hyperthermia 2006; 22: 191–196
- Calderwood SK, Ciocca DR. Heat shock proteins: Stress proteins with Janus-like properties in cancer. Int J Hyperthermia 2008; 24: 31–39
- Mayer MP. Phosphotyrosine confers client specificity to Hsp90. Mol Cell 2010; 37: 295–296
- Vaughan CK, Neckers L, Piper PW. Understanding of the Hsp90 molecular chaperone reaches new heights. Nat Struct Mol Biol 2010; 17: 1400–1404
- Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol 2003; 15: 419–424
- Calderwood SK. Heat shock proteins in breast cancer progression – A suitable case for treatment?. Int J Hyperthermia 2010; 26: 681–685
- Staufer K, Stoeltzing O. Implication of heat shock protein 90 (Hsp90) in tumor angiogenesis: A molecular target for anti-angiogenic therapy?. Curr Cancer Drug Targets 2010; 10: 890–897
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic Hsp90 complex in cancer. Nat Rev Cancer 2010; 10: 537–549
- Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: Are we there yet?. Clin Cancer Res 2012; 18: 64–76
- Bishop SC, Burlison JA, Blagg BS. Hsp90: A novel target for the disruption of multiple signaling cascades. Curr Cancer Drug Targets 2007; 7: 369–388
- Yamashita T, Oda T, Sekimoto T. Hsp90 and the Fanconi anemia pathway: A molecular link between protein quality control and the DNA damage response. Cell Cycle 2007; 6: 2232–2235
- Whitesell L, Lindquist SL. Hsp90 and the chaperoning of cancer. Nat Rev Cancer 2005; 5: 761–772
- Sangster TA, Queitsch C, Lindquist S. Hsp90 and chromatin: Where is the link?. Cell Cycle 2003; 2: 166–168
- Russell JS, Burgan W, Oswald KA, Camphausen K, Tofilon PJ. Enhanced cell killing induced by the combination of radiation and the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin: A multitarget approach to radiosensitization. Clin Cancer Res 2003; 9: 3749–3755
- Bull EE, Dote H, Brady KJ, Burgan WE, Carter DJ, Cerra MA, et al. Enhanced tumor cell radiosensitivity and abrogation of G2 and S phase arrest by the Hsp90 inhibitor 17 -(dimethylaminoethylamino)-17-demethoxygeldanamycin. Clin Cancer Res 2004; 10: 8077–8084
- Bisht KS, Bradbury CM, Mattson D, Kaushal A, Sowers A, Markovina S, et al. Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res 2003; 63: 8984–8995
- Enmon R, Yang WH, Ballangrud AM, Solit DB, Heller G, Rosen N, et al. Combination treatment with 17 -N-allylamino-17-demethoxy geldanamycin and acute irradiation produces supra-additive growth suppression in human prostate carcinoma spheroids. Cancer Res 2003; 63: 8393–8399
- Harashima K, Akimoto T, Nonaka T, Tsuzuki K, Mitsuhashi N, Nakano T. Heat shock protein 90 (Hsp90) chaperone complex inhibitor, radicicol, potentiated radiation21 induced cell killing in a hormone-sensitive prostate cancer cell line through degradation of the androgen receptor. Int J Radiat Biol 2005; 81: 63–76
- Machida H, Matsumoto Y, Shirai M, Kubota N. Geldanamycin, an inhibitor of Hsp90, sensitizes human tumour cells to radiation. Int J Radiat Biol 2003; 79: 973–980
- Stingl L, Stuhmer T, Chatterjee M, Jensen MR, Flentje M, Djuzenova CS. Novel Hsp90 inhibitors, NVP-AUY922 and NVP-BEP800, radiosensitise tumour cells through cellcycle impairment, increased DNA damage and repair protraction. Br J Cancer 2010; 102: 1578–1591
- Dote H, Burgan WE, Camphausen K, Tofilon PJ. Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res 2006; 66: 9211–9220
- Ha K, Fiskus W, Rao R, Balusu R, Venkannagari S, Nalabothula NR, et al. Hsp90 inhibitor-mediated disruption of chaperone association of ATR with Hsp90 sensitizes cancer cells to DNA damage. Mol Cancer Ther 2011; 10: 1194–1206
- Oda T, Hayano T, Miyaso H, Takahashi N, Yamashita T. Hsp90 regulates the Fanconi anemia DNA damage response pathway. Blood 2007; 109: 5016–5026
- Koll TT, Feis SS, Wright MH, Teniola MM, Richardson MM, Robles AI, et al. Hsp90 inhibitor, DMAG, synergizes with radiation of lung cancer cells by interfering with base excision and ATM-mediated DNA repair. Mol Cancer Ther 2008; 7: 1985–1992
- Noguchi M, Yu D, Hirayama R, Ninomiya Y, Sekine E, Kubota N, et al. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem Biophys Res Commun 2006; 351: 658–663
- Dungey FA, Caldecott KW, Chalmers AJ. Enhanced radiosensitization of human glioma cells by combining inhibition of poly(ADP-ribose) polymerase with inhibition of heat shock protein 90. Mol Cancer Ther 2009; 8: 2243–2254
- Ito A, Saito H, Mitobe K, Minamiya Y, Takahashi N, Maruyama K, et al. Inhibition of heat shock protein 90 sensitizes melanoma cells to thermosensitive ferromagnetic particle-mediated hyperthermia with low Curie temperature. Cancer Sci 2009; 100: 558–564
- van der Zee J, van Rhoon GC. Cervical cancer: Radiotherapy and hyperthermia. Int J Hyperthermia 2006; 22: 229–234
- Calderwood SK, Asea A. Targeting Hsp70-induced thermotolerance for design of thermal sensitizers. Int J Hyperthermia 2002; 18: 597–608
- Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother Pharmacol 2010; 66: 535–545
- Gortzak E, Azzarelli A, Buesa J, Bramwell VH, van Coevorden F, van Geel AN, et al. A randomised phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft tissue sarcoma. Eur J Cancer 2001; 37: 1096–1103
- Weiss C, Engehausen DG, Krause FS, Papadopoulos T, Dunst J, Sauer R, et al. Radiochemotherapy with cisplatin and 5-fluorouracil after transurethral surgery in patients with bladder cancer. Int J Radiat Oncol Biol Phys 2007; 68: 1072–1080
- Franckena M, Fatehi D, de Bruijne M, Canters RA, van Norden Y, Mens JW, et al. Hyperthermia dose–effect relationship in 420 patients with cervical cancer treated with combined radiotherapy and hyperthermia. Eur J Cancer 2009; 45: 1969–1978
- Ott OJ, Rodel C, Weiss C, Wittlinger M, St Krause F, Dunst J, et al. Radiochemotherapy for bladder cancer. Clin Oncol (R Coll Radiol) 2009; 21: 557–565
- Issels RD, Lindner LH, Verweij J, Wust P, Reichardt P, Schem BC, et al. Neoadjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft tissue sarcoma: A randomised phase 3 multicentre study. Lancet Oncol 2010; 11: 561–570
- Krause FS, Walter B, Ott OJ, Haberle L, Weiss C, Rodel C, et al. 15-year survival rates after transurethral resection and radiochemotherapy or radiation in bladder cancer treatment. Anticancer Res 2011; 31: 985–990
- Issels RD. Hyperthermia adds to chemotherapy. Eur J Cancer 2008; 44: 2546–2554
- Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, et al. Hyperthermia in combined treatment of cancer. Lancet Oncol 2002; 3: 487–497
- Horsman MR, Overgaard J. Hyperthermia: A potent enhancer of radiotherapy. Clin Oncol (R Coll Radiol) 2007; 19: 418–426
- Treszezamsky AD, Kachnic LA, Feng Z, Zhang J, Tokadjian C, Powell SN. BRCA1-and BRCA2-deficient cells are sensitive to etoposide-induced DNA double-strand breaks via topoisomerase II. Cancer Res 2007; 67: 7078–7081
- Tsaryk R, Fabian K, Thacker J, Kaina B. Xrcc2 deficiency sensitizes cells to apoptosis by MNNG and the alkylating anticancer drugs temozolomide, fotemustine and mafosfamide. Cancer Lett 2006; 239: 305–313
- Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, Yoshimura M, et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res 2005; 65: 11704–11711
- Sekine-Suzuki E, Yu D, Kubota N, Okayasu R, Anzai K. Sulforaphane induces DNA double strand breaks predominantly repaired by homologous recombination pathway in human cancer cells. Biochem Biophys Res Commun 2008; 377: 341–345