
Abstract
Purpose: From the role of double strand DNA dependent protein kinase (DNA-PKcs) activity of non-homologous end joining (NHEJ) repair for DNA double strand breaks (DSBs), we aim to define possible associations between thermo-sensitisation and the enzyme activities in X-ray irradiated cells. Materials and methods: DNA-PKcs deficient mouse, Chinese hamster and human cultured cells were compared to the parental wild-type cells. The radiosensitivities, the number of DSBs and DNA-PKcs activities after heat-treatment were measured. Results: Both DNA-PKcs deficient cells and the wild-type cells showed increased radiosensitivities after heat-treatment. The wild-type cells have two repair processes; fast repair and slow repair. In contrast, DNA-PKcs deficient cells have only the slow repair process. The fast repair component apparently disappeared by heat-treatment in the wild-type cells. In both cell types, additional heat exposure enhanced radiosensitivities. Although DNA-PKcs activity was depressed by heat, the inactivated DNA-PKcs activity recovered during an incubation at 37 °C. DSB repair efficiency was dependent on the reactivation of DNA-PKcs activity. Conclusion: It was suggested that NHEJ is the major process used to repair X-ray-induced DSBs and utilises DNA-PKcs activity, but homologous recombination repair provides additional secondary levels of DSB repair. The thermo-sensitisation in X-ray-irradiated cells depends on the inhibition of NHEJ repair through the depression of DNA-PKcs activities.
Introduction
Ionizing radiation produces major lesions in DNA, such as DSBs, which subsequently lead to numerous biological effects, including cell death and mutations [Citation1,Citation2]. In eukaryotic cells, DSBs are repaired mainly through the two processes of non-homologous end joining (NHEJ) [Citation3,Citation4] and homologous recombination repair (HR) [Citation5]. NHEJ is one of the major repair pathways in mammalian cells. Double strand DNA dependent protein kinase (DNA-PK) plays an important role in NHEJ during all stages of the cell cycle [Citation6]. DNA-PK is composed of DNA-PK catalytic subunit (DNA-PKcs) and a double-stranded DNA end binding subunit (the Ku70/80 heterodimer). DNA-PKcs combines with the Ku70/Ku80 heterodimer and phosphorylates a variety of proteins, including XRCC4 and Artemis [Citation6]. The severe combined immunodeficiency (scid) mouse was identified as being an immuno-deficient mutant due to the lack of V(D)J recombination activity during lymphocyte development [Citation7]. Cells isolated from scid mice are defective in rejoining DSBs and are hypersensitive to ionizing radiation when compared to the wild-type mouse cells [Citation8]. The mutation in the scid mouse was mapped to mouse chromosome 16 [Citation9]. If human chromosome 8 (8q11), which includes the DNA-PKcs structural gene, is incorporated into scid mouse cells, a stable hybrid cell line is established. These hybrid cells exhibit normal DNA-PKcs activity and normal radiation sensitivity when compared to the parental wild-type cells [Citation10]. Scid cells lack a DNA-PKcs gene, but carry the Ku70 and Ku80 genes (XRCC6 and XRCC5). The xrs-5 and sxi-3 cell mutants are defective in the Ku80 gene and DNA-PKcs activity is also defective, and these cells are hypersensitive to ionizing radiation [Citation11,Citation12]. Kurimasa et al. reported that full length cDNA of human DNA-PKcs can complement DSB repair in a radiation sensitive mutant cell line [Citation13]. These results indicate that the protein kinase activity of DNA-PK is essential for the repair of DSBs induced by ionizing radiation.
Heat exposures inactivate DSB repair activity [Citation14]. In a previous report [Citation15], DNA-PKcs activity was shown to be highly heat sensitive, and was inactivated by heat exposure of 44 °C for 15 min. However, many reports have concluded that the HR pathway is also involved in thermo-sensitisation in the cells irradiated with radiation [Citation16–21]. Not all of the targets affected by heat exposure are completely known, even now. In the present paper, DSBs were examined, and DNA-PKcs activity was investigated in relation to the loss and recovery of heat-inactivated repair enzymes.
Materials and methods
Cells
Mouse scid cells (SC3VA2) and hybrid cells containing a fragment of human chromosome 8 in scid mouse cells (RD13B2) [Citation10] were used in this study. Other mouse cells (PK33N and CB17), Chinese hamster cells (V3 and CHO-K1) and human (M059J and M059K) cells were used to compare between DNA-PKcs deficient type cells and the parental wild-type cells, respectively among different mammalian cultured cells. They were cultured in Dulbecco’s modified Eagle’s minimal essential medium (DMEM, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (Equitech-Bio, Kerrville, TX).
Heat-treatments
Cells (1 × 106) were inoculated into a culture dish (100 mm in diameter) and were grown at 37 °C for 24 h before exposure to heat. Cells were exposed to heat for different periods by immersing dishes sealed with parafilm into a water bath maintained at 44 °C. Following the heat exposures, cells were incubated at 37 °C for the indicated times.
X-ray irradiation
Cells were irradiated with 200 kV, 15 mA X-ray (by a Toshiba X-ray machine model EXS300-5; Tokyo, Japan) with 0.5 mm aluminium and 0.5 mm copper filters at a dose rate of 0.731 Gy/min at room temperature. To measure colony formation, exponentially growing cells were trypsinised and irradiated at room temperature. Cells were then plated on to 100 mm diameter culture dishes and incubated at 37 °C for two weeks. The number of cells plated per dish was chosen to ensure that about 50 colonies would survive. For measurements of DSB repair, heat-treated cells were irradiated with 10 Gy. To clearly detect DSBs, non-heat-treated cells were irradiated with 20 Gy.
Assay of DNA-PKcs activity
DNA-PKcs activity was assayed as described previously [Citation15]. Briefly, the frozen cells were suspended in 20 μL of low salt buffer (10 mM HEPES-KOH, 25 mM KCl, 10 mM NaCl, 1.1 mM MgCl2, 0.1 mM EDTA, 0.5 mM PMSF, pH 7.2), frozen, and then thawed. After centrifugation at 14 000 rpm for 5 min at 4 °C, the supernatant was collected and used as a crude cell extract. The crude cell extract (10 μg protein) was mixed with oligopeptide as a substrate, double-stranded DNA and (γ-32P) ATP. The mixture was then incubated at 30 °C for 30 min. The mixture was then transferred to a phosphocellulose disc. The discs were washed with 1% acetic acid and distilled water twice. DNA-PKcs activity was evaluated from the radioactivity bound to the discs.
Measurement of DSBs
The number of DSBs was calculated from the density of the bands separated from the applied wells after pulsed-field gel electrophoresis (PFGE) (CHEF DRII Bio-Rad Laboratories Hercules, CA) as described previously [Citation22]. Briefly, heat-treated and the control cells were irradiated and then washed with PBS (80 mM Na2HPO4, 20 mM NaH2PO4, 100 mM NaCl), harvested using a cell scraper, suspended in 1.5 mL of PBS and transferred to microcentrifuge tubes. After counting the number of cells with a Coulter counter (Industrial D, Coulter Electronics, Bedfordshire, UK), cells were sedimented in a microcentrifuge at 6000 rpm for 5 min at 4 °C. The cells were resuspended in PBS to a final concentration of 2×107 cells/mL. An equal volume of 1% low melting point (LMP) agarose (Beckman Coulter, Brea, CA) in 0.25 M EDTA (pH 9.0) was added to the cells and thoroughly mixed. Aliquots (100 μL) of the resulting cell suspension were transferred to a plug mounter (Beckman Coulter) and allowed to cool. The plugs were incubated in ESP buffer (3 mg/mL of proteinase K and 0.3% lauryl sarcosine in 0.5 M EDTA (pH 9.0)) at 50 °C overnight. The plugs were placed in 1.2% agarose in 0.5× TBE buffer (44.6 mM Tris, 44.5 mM boric acid, 1 mM EDTA), and pulsed-field gel electrophoresis was performed at 200 V with a 120-s pulse time for 24 h at 4 °C. After electrophoresis, gels were stained with ethidium bromide (0.5 μg/mL) for 1 h and de-stained in 0.5× TBE buffer for 1 h. The stained gels were photographed with Polaroid 665 film under ultraviolet light. The density of the DNA fragment bands which migrated from the plug into the agarose gel was determined with densitometry (Photometrics, Tucson, AZ) and analysed with IP Lab Spectrum and IP Lab Gel software (Scanalytics Inc., Milwaukee, WI). The number of DSBs was calculated from the band densities.
Western blot analysis
Cells were treated at 44 °C for 15 min as described above. RIPA (50 mM Tris-HCl (pH 7.5), 1% NP40, 0.1 mM sodium deoxycholate, 50 mM NaCl, 1 mM EDTA) buffer was used to prepare cellular extract. Cellular extracts were applied to sodium dodecyl sulphate polyacrylamide gel electrophoresis. Then the proteins were transferred to nitrocellulose membrane (Hybond ECL, GE Healthcare, Amersham, UK). They were visualised by enhanced chemiluminescence system (ECL, GE Healthcare). Antibodies used were anti-Ku70, anti-Ku80 (Santa Cruz Biothchnology, Santa Cruz, CA), anti-ATM (Abcam, Cambridge, UK), anti-BRCA1 (Acris Antibodies, San Diego, CA), anti-53BP1 (Bethyl Laboratories, Montgomery, TX), anti-Rad51 (Bioacademia, Ibaraki, Osaka, Japan) and anti-actin (Sigma-Aldrich, St Louis, MO).
Results
Radiosensitivities between DNA-PKcs deficient type cells and the wild-type cells among different mammalian cultured cells
Radiosensitivities of DNA-PKcs defective cells and the parental wild-type cells from mouse, Chinese hamster and human were measured by colony forming assays (). The doses at D10 values of DNA-PKcs defective cells were 2.44, 1.88, 1.65 and 1.31 Gy for scid, PK33N, V3 and M059J cells, respectively. The doses at D10 values of their wild-type cells were 5.51, 6.36, 5.45 and 4.26 Gy for hybrid, CB17, CHO-K1 and M059K cells, respectively. These results indicate that all DNA-PKcs defective cells were more sensitive than their wild-type cells.
Figure 1. Heat-induced radiosensitisation. Cells were heated at 44°C for 15 min (open symbols) or not heated (closed symbols), and then irradiated by X-ray at the indicated doses on the abscissa. Triangles, wild-type cells; circles, DNA-PKcs deficient cells. (A) mouse SC3VA2 (scid, DNA-PKcs−/−) and RD13B2 (hybrid, wild); (B) mouse PK33N (DNA-PKcs) and CB17 (wild); (C) Chinese hamster V3 (DNA-PKcs) and CHO-K1 (wild); (D) human M059J (DNA-PKcs) and M059K (wild).
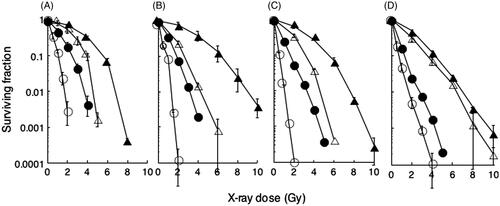
Enhancement of radiosensitivity with heat exposure before X-ray-irradiation
Radiosensitivities after heat exposure were measured by colony forming assays (). D10 values were 5.51, 3.92, 2.44 and 1.08 Gy for hybrid cells, heat-treated hybrid cells, scid cells and heat-treated scid cells, respectively. When D10 values were compared, scid cells were found to be about 2.3 times more sensitive to X-ray than the hybrid cells. When the hybrid cells were heated for 15 min at 44 °C before X-ray exposure, the cells were about 1.4 times more sensitive when compared to non-heat-treated cells. When the scid cells were heated for 15 min at 44 °C before X-irradiation however, the cells were 2.3 times more sensitive than the non-heat-treated cells. The doses at D10 values in the heat-treated wild-type cells were 3.92, 2.67, 3.13 and 3.52 Gy in hybrid, CB17, CHO-K1 and M059K cells, respectively. The doses at D10 values of heat-treated DNA-PKcs defective cells were 1.08, 0.91, 0.51 and 0.74 Gy for scid, PK33N, V3 and M059J cells, respectively. When D10 doses were compared among DNA-PKcs defective cells, they were found to be about 2.3, 3.4, 3.3 and 3.3 times more sensitive to X-ray than the wild-type cells of hybrid mouse, mouse, Chinese hamster and human cells, respectively. When the wild-type cells were heated for 15 min at 44 °C before X-ray exposure the cells were about 1.4, 2.4, 1.7 and 1.2 times more sensitive when compared to non-heat-treated cells in hybrid mouse, mouse, Chinese hamster and human cells, respectively. When DNA-PKcs defective cells were heated for 15 min at 44 °C before X-irradiation, however, the cells were 2.3, 2.0, 3.2 and 1.8 times more sensitive than the non-heat-treated cells in hybrid mouse, mouse, Chinese hamster and human cells, respectively. These results show that a heat exposure before radiation exposure enhanced radiosensitivities in both types of cells, whether they contained the DNA-PKcs gene or not. The pattern of survival curve of human cells was different from other rodent cells.
Repair kinetics for radiation-induced DSBs in scid cells and hybrid cells
The number of radiation-induced DSBs was measured by PFGE analysis. These data exhibit the repair kinetics for DSBs during an incubation at 37 °C after irradiation with X-ray (). The number (N) of DSBs after an incubation of t h after an irradiation was N = N0e−at where N0 is the number of DSBs immediately after irradiation. The repair kinetics for DSBs in the hybrid cells show two exponential components, for fast repair and slow repair, which are indicated by the lines labelled I and II in a semi-logarithmic plot, respectively (Figure 2A). From the values of the relative number of DSBs shown on the y-axis for lines I and II extrapolated to 0 h, it appeared that about 59% of the DSBs were repaired by a fast repair process, and about 41% of the breaks were repaired by a slow process. The mean half times for DSB repair for the fast repair and slow repair components were calculated to be 40 min and 11.4 h, respectively. Fast repair was completely finished within 3 h after irradiation. On the other hand, the number of DSBs in scid cells decreased linearly on a semi-logarithmic plot, even without heat-treatment (). These data show the presence of only a slow repair process, and no evidence for a fast repair process in scid cells. The mean half time for repair was about 8 h in these cells.
Figure 2. Effect of heat exposures prior to X-ray irradiation on DSB repair. (A) hybrid cells (closed symbols). (B) scid cells (open symbols). Cells were exposed to heat at 44 °C and then irradiated with 20 Gy of X-ray. Circles: non-heat-exposed cells; squares: heat exposure for 15 min; triangles: heat exposure for 30 min; reverse triangles: heat exposure for 45 min. These data are averages of four independent experiments.
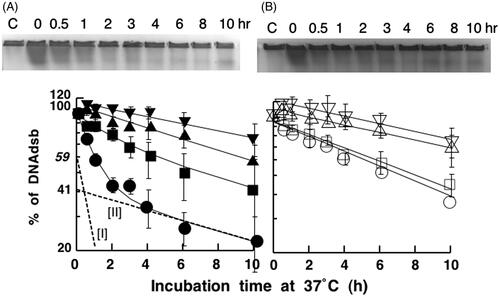
Repair of radiation-induced DSBs in scid cells and hybrid cells exposed to heat
also shows the repair kinetics for DSBs when cells were exposed to heat at 44 °C for 15, 30, and 45 min prior to X-ray irradiation. In , when hybrid cells were heated at 44 °C for 15 min before X-ray irradiation, the fast repair process was apparently reduced when compared to repair in non-heated cells. In addition, the decrease in the extent of DSB repair in the X-ray-irradiated cells was dependent on the incubation period in the presence of heat. When the hybrid cells were exposed to heat at 44 °C for 15, 30 or 45 min, the mean half times for slow repair, mostly without fast repair, were 12.5, 10.7 and 16.6 h, respectively. On the other hand, only slow repair was observed in scid cells. When scid cells were heated at 44 °C for 15, 30 or 45 min, the mean half time for the repair of breaks was 8.8, 15.2 or 15.1 h, respectively (). The heat exposure before irradiation depressed the decrease in the number of DSBs. This depression occurred in a manner which was dependent on the length of the incubation period with heat prior to X-ray irradiation.
As shown in , more than 100% of the initial number of DSBs detected (about 120%) were sometimes observed after a 30-min to 2-h incubation period following heat exposure, and this was observed in both hybrid cells and scid cells. This was observed when both types of cells were heated at 44 °C for 45 min prior to X-ray irradiation (). However, the number of DSBs decreased during the incubation periods at 37 °C.
Recovery of heat-treated DNA-PKcs activity and its correlation with DSB repair
In hybrid cells, DNA-PKcs activity was undetectable after heat exposure at 44 °C for 15 min (closed circles) at 0 h-incubation in the un-irradiated cells (). The similar values were detected at 0 h-incubation after heat-treatment at 44°C for 30 min (closed triangles) and 45 min (reverse closed triangles) in the un-irradiated cells. The recovery rates of DNA-PKcs activities in the heat-treated cells for these three different periods were 52%, 19% and 2% after a 10-h incubation at 37°C, respectively. Almost no recovery was found in the heat-treated cells at 44°C for 45 min. As shown in , the repair of DSBs was dependent primarily on fast repair up to 3 h after X-irradiation in unheated hybrid cells, and was then dependent on slow repair.
Figure 3. DNA-PK reactivation after a heat exposure in hybrid cells. (A) Hybrid cells were heat-treated at 44 °C for 15 (•), 30 (▴) and 45 (▾) min without irradiation and then cultured at 37 °C for the indicated time on the abscissa. The percentage of DNA-PKcs activity is shown on the vertical axis. (B) The fast repair fraction is indicated on the vertical axis from . The relative DNA-PKcs activity during an incubation from 0 h to 3 h after heat exposure at 44 °C for 15 min (▵) or 30 min (▿) was calculated from results of compared with the non-heated case (○). The abscissa is indicated with a logarithmic scale. Other details are described in the text.
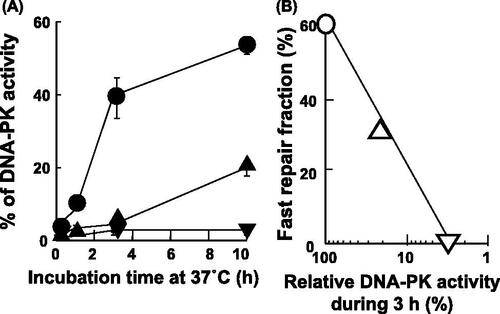
In Figure 3B, a linear relationship was observed between fast repair fraction and relative DNA-PK activity during an incubation for 3 h after heat-treatment. As the control, the relative DNA-PK activity in the non-heat-treated cells represents as 100%. In this case, about 59% of the fast repair fraction are shown (○), because the value was obtained from Figure 2A line [I]. About 27% of fast repair fraction and 18% of recovered DNA-PK activity were detected during 3-h incubation at 37°C in the heat-treated cells for 15 min at 44°C (▵). Almost 0% of fast repair fraction was observed in the heat-treated cells for 30 min at 44°C, when DNA-PK activity was recovered to 3% during 3-h incubation at 37°C (▿).
Heat sensitivity of repair enzymes
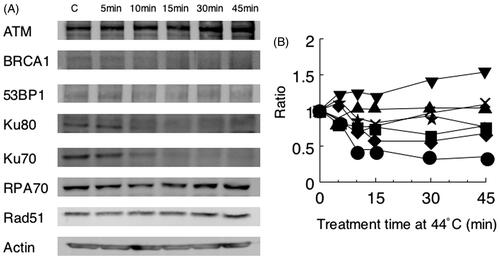
To explain the heat-sensitisation for radiosensitivity, we investigated the heat sensitivity of repair proteins by Western blot (). The cellular contents of Rad51 and RPA70 were almost unchanged even after heat-treatment at 44 °C for 15 min. However, the amounts of Ku70, Ku80, BRCA1 and 53BP1 were decreased to 44%, 56%, 76% and 83% by the heat-treatment, respectively. RPA70 was decreased to 69% at 10 min, and then increased to 108% at 45 min. On the other hand, the ATM protein was apparently increased after heat-treatment.
Figure 4. Heat sensitivity of repair proteins from hybrid cells. Results of Western blot (A) and the analysis by densitometry (B) are shown. Key: ▾ ATM; ▪ BRCA1; ★ 53BP1; • Ku70; ♦ Ku80; × RPA70; ▴ Rad51.
Discussion
The focus in many kinds of DNA damage has been on DSBs, because the repair of DSBs is mostly dependent on the cell lethality. There are essentially two repair processes of NHEJ and HR in mammalian cells. In the present paper, we compared DNA-PKcs deficient cells and the wild-type cells (). The gene is one of the major repair pathways in NHEJ during all stages of the cell cycle. Radiosensitivities of the wild-type cells differed among mammalian cells used here. The doses (Gy) at D10 values for wild-type cells were in the range of 6.36 to 4.26. Those of mouse and Chinese hamster cells were similar at levels, whereas the most sensitive human M059K cells was 1.5-fold more sensitive to radiation than the most resistant mouse CB17 cells. These results suggest repair capacity of human cells is lower than mouse cells. The doses (Gy) at D10 values of DNA-PKcs defective cells were in the range of 2.44 to 1.31. The D10 values of DNA-PKcs defective cells were decreased 2.26-, 3.38-, 3.30- and 3.25-fold compared with their wild-type cells in mouse scid and PK33N, Chinese hamster V3 and human M059J cells, respectively. These results support that DNA-PKcs protein plays the important role in DNA repair. The most sensitive human M059J cells was 1.9 fold sensitive than the most resistant mouse scid cells. This indicates that the human cells have low activity in other than NHEJ, namely HR. These results suggest that repair capacities of at least HR is low in human cells as compared with mouse and Chinese hamster cells.
The radiosensitivities of the wild-type cells were enhanced by a heat exposure prior to the irradiation. When we compared them at D10 values, heat-treatment enhanced the radiosensitivity 1.41-, 2.38-, 1.74- and 1.21-fold for hybrid, CB17, CHO-K1 and M059K cells, respectively. The enhancement ratio in human cells was the lowest among all of them. It suggested that radiosensitivity has little capacity for DSB repair in human cells here, too. In the case of DNA-PKcs defective cells, the radiosensitivity after heat-treatment was enhanced in all of them. The enhancement ratios were 2.26-, 2.07-, 3.24- and 1.77-fold by heat-treatment for scid, PK33N, V3 and M059J cells, respectively. In this case, too, the enhancement ratio in human M059J cells was the smallest. These results suggest that the capacity of NHEJ repair may the lowest among them. Other species might possess higher DNA repair capacity for NHEJ.
From the DSB repair analysis shown in , it is clear that DSBs induced by ionizing radiation are repaired in a biphasic manner consisting of a fast repair component (indicated by line I) and a slow repair component (indicated by line II) in hybrid cells. These repair components are indicated by the dotted straight lines on the bottom curve in .
As shown in , the repair process for DSBs is composed of two types of repair, designated as fast and slow repair, and which compose about 60% and 40% of the DSB repair process, respectively. These results suggest that the major repair process for DSB is NHEJ repair which is dependent on DNA-PKcs activity. Some previous reports have estimated the contribution of NHEJ toward DSB repairs to be over a wide range of values from 40–90% (with an average of 66.3%) using various methods, but most of the reported values are close to the results reported here of about 60% () [Citation23–29]. The number of DSBs in the hybrid cells decreased through both repair processes in a linear manner on a semi-logarithmic scale (). The mean half times for the fast repair and slow repair processes were shown to be 40 min and 11.5 h, respectively. The fast repair process was almost complete at 3 h in hybrid cells exposed to X-ray. Núñez et al. reported that the surviving fraction after an exposure to ionizing radiation correlated strongly with the half time of the fast repair process [Citation23]. It was also reported that the fast repair process disappeared after heat-treatment at 45 °C for 20 min in Chinese hamster ovary (CHO)-1A cells [Citation27], and at 45.5 °C for 15 min in CHO cells [Citation25].
On the other hand, only the slow repair process was found in scid cells which are defective in DNA-PKcs activity. The mean half time for repair was about 8 h. This value is similar to the mean half time value of the slow repair process in hybrid cells (11.5 h, ) in contrast to the mean value of half time for fast repair which was about 40 min. These results suggested that the fast repair and slow repair processes were associated with the NHEJ and HR repair systems, respectively. Since DNA-PKcs activity plays a major role in the NHEJ repair process, it is consistent with the fact that the fast repair component was not found in scid cells (). These results are consistent with previous reports indicating that the mean half time for repair of DSBs was 12 h and 18.4 h in DNA-PKcs defective M059J human glioblastoma cells [Citation30] and in Ku70-knockout DT40 chicken cells [Citation31], respectively. At the same time, this suggests that NHEJ repair, rather than HR repair, preferentially acts to repair DSBs. This is also supported by reports that the induction of thermo-sensitisation in the X-ray-irradiated cells occurs through dysfunction or depression in NHEJ activity [Citation15]. The reports indicated that one or more factors involved in NHEJ are highly heat sensitive. In this report, the heat sensitivity of DNA-PKcs activity was observed to correlate with the major component (about 60%) of DSB repair (). These results are consistent with previous reports showing that Ku70 and/or Ku80 are heat-sensitive components of the DNA-PK complex. Burgman et al. [Citation32] and Matsumoto et al. [Citation33] reported that heat-treatment inactivated Ku subunits. Heat exposure at 44 °C for 15 min reduced the fast repair component in hybrid cells (). Longer heat exposures of 30 and 45 min at 44 °C resulted in only the slow repair fraction of DSB repair being observed in the same cells.
Heat exposure of 30 and 45 min, but not of 15 min, at 44 °C depressed DSB repair processes. These results suggest that heat exposure prior to X-ray irradiation also inhibits the HR repair process which is distinct from the NHEJ repair process. Umeda et al. reported that heat sensitivity of DNA dependent protein kinase activity are different among mouse, hamster and human cells [Citation34]. We analysed heat sensitivities of repair enzymes from mouse hybrid cells. As shown in , protein bands of Ku70, Ku80 and BRCA1 disappeared with heat-treatment at 44 °C for 15 min. 53BP1 may also be heat sensitive. Because Ku70 and Ku80 participate NHEJ and BRCA1 and 53BP1 work to HR, these results indicate that not only NHEJ but also HR is heat sensitive. On the other hand, the cellular contents of ATM were increased by heat-treatment. These results indicate ATM is activated by heat-treatment. Similarly to our study here, other reports have indicated that the HR repair process also involves heat-sensitive factors [Citation16–21]. To confirm heat sensitivity in the role of HR repair, we examined Rad51 foci after radiation in heat-treated or non-heat-treated cells. Average numbers of Rad51 foci in hybrid cells were 4.2, 13.1 and 6.7 in the non-heat-treated control cells, after radiation and after heat-treatment, respectively. Average numbers of Rad51 foci in scid cells were 3.0, 19.7 and 10.8 (data not shown). Because NHEJ was not working in scid cells, HR may be enhanced. Therefore, these results suggest that HR might also be heat sensitive. Genet et al. also demonstrated that radiation-induced Rad51 protein foci at the DSB site was dissociated in response to the temperature and time of hyperthermia exposure [Citation35]. These results indicate that heat could prevent HR. Recently, the inhibition of one or more DSB repair pathway was developed for clinical cancer treatment by synthetic lethality. Bergs et al. reported that in the temporary inhibition of HR by hyperthermia, DSB repair was shifted to NHEJ in human cells [Citation36]. Krawczyk et al. demonstrated that mild hyperthermia induced BRCA2 degradation [Citation37]. They showed the combination of hyperthermia and PARP1 inhibitor was well tolerated. These results indicate a strong role of HR in heat-induced radiosensitisation. In addition, Ma et al. reported that BRCA1, a major contributor of HR was also inactivated by heat-treatment [Citation38]. The BRCA1 protein decreased to about 50% at 60 min after heat-treatment at 42 °C and disappeared completely at 80 min. Although the treatment temperature is different from our experiment, we considered almost the same time course for inactivation. Eppink et al. showed that hyperthermia might induce the depression of DNA repair from the results of the inhibition of the cellular heat shock response [Citation39]. These results support the development of novel anti-cancer therapy by the combination of hyperthermia and drugs to inhibit repair systems other than HR and/or heat-shock response.
Iliakis et al. reported that alternative pathways of NHEJ also contributed to heat-induced radiosensitisation [Citation14]. Although heat-sensitive components are not known at present, alternative pathways of NHEJ must be considered to contribute to the heat-induced radiosensitisation. Alternative pathways of NHEJ especially will be important in NHEJ or HR deficient cells. As another target of thermo-sensitisation in the X-ray-irradiated cells, Takahashi et al. reported that DNA polymerase-β was also heat sensitive [Citation40].
When both cell types were exposed to heat at 44 °C for more than 30 min, and specifically for 45 min, about 120% of the initial number of DSBs was observed at 30 min (). These results mean that heat exposures for long periods at 44 °C can induce DSBs. However, the appearance of heat-induced DSBs requires an incubation time of more than 30 min. It is also possible that DSBs are produced indirectly from heat through the action of radicals such as oxygen and/or nitrogen oxide [Citation41] during the incubation at 44 °C, and such DSBs would require more time to become apparent. It was previously reported that heat exposure at high temperature induced DSBs, and this was confirmed by using the γH2AX-positive focus assay, which is the most sensitive assay available in cultured mammalian cells [Citation42,Citation43]. In addition, heat-induced DSBs may enhance radiosensitivity in both cell lines through depressing the two DSB repair processes, NHEJ and HR (). Okamoto et al. described that heat sensitivity was increased when NSB1 function was down-regulated by siRNA in human anaplastic thyroid carcinoma cells [Citation44]. It was suggested that it was possible for DSBs to be accumulated by heat-treatment.
Heat-treatment at 44 °C for different periods was utilised to depress DNA-PKcs activity in hybrid cells, and a recovery of DNA-PKcs activity was measured during subsequent incubation periods at 37 °C (). Almost half the activity of DNA-PK was recovered in the heat-treated cells at 44 °C for 15 min during post-incubation at 37 °C for 10 h. When the cells were heat-treated at 44 °C for 30 min and 45 min, the recovered activity was only about 20% and 5%, respectively. These results were easily understood from the heat-labile enzyme structure of DNA-PK proteins, especially Ku proteins, depending on high temperature. Thereafter, the mechanism responsible for the recovery of DNA-PKcs activity is likely de novo synthesis of new DNA-PKcs protein and Ku proteins. In particular, the recovery of the fast repair component corresponded closely with the recovery of total DNA-PKcs activity during a 3-h incubation period following heat exposure (). Alternatively, heat shock proteins might become available and provide chaperone activity to the denatured DNA-PKcs proteins and/or the Ku proteins, because many types of heat-shock proteins are induced by heat. In the present experiments we used a temperature of 44 °C to clarify the effect of heat on DSB repair. Clinical application mostly uses below 43 °C, because the critical temperature of hyperthermia is 42.5 °C in the efficacy of temperature-survival curves. Although higher outcome is observed above 43 °C, higher temperature is difficult in hyperthermic therapy for deep cancer of patients. It was also shown that fast repair acted preferentially during DSB repair in hybrid cells and was heat sensitive. The slow repair component was also depressed by heat for additional periods. A full understanding of these experimental findings will hopefully contribute to improvements in combination cancer therapies which utilise both hyperthermia and radiation.
Conclusions
An examination of radiosensitivity after heat exposure in the wild-type mammalian cultured cells for DSB repair led to the conclusion that heat depressed DNA-PKcs activities which play the role for fast repair, because the thermo-sensitisation in the X-ray-irradiated cells was mainly dependent on inhibiting the NHEJ repair process through the inhibition of DNA-PKcs activity. Since DNA-PKcs defective cells were also enhanced by heat-treatment, it was suggested that the HR process of slow repair for DSBs was depressed by heat-treatment.
Declaration of interest
This work was supported by Grants-in-Aid (21310040 and 25514007) for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Hakem R. DNA-damage repair: The good, the bad, and the ugly. EMBO J 2008;27:589–605
- Jeggo P, Lavin MF. Cellular radiosensitivity: How much better do we understand it? Int J Radiat Biol 2009;85:1061–81
- Lieber MR, Ma Y, Pannicke U, Schwarz K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair 2004;3:817–26
- Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J 2009;417:639–50
- Wyman C, Ristic D, Kanaar R. Homologous recombination-mediated double-strand break repair. DNA Repair 2004;3:827–33
- Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair 2004;3:909–18
- Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature 1983;301:527–30
- Fulop GM, Phillips RA. The scid mutation in mice causes a general defect in DNA repair. Nature 1990;347:479–82
- Bosma GC, Davisson MT, Ruetsch NR, Sweet HO, Shultz LD, Bosma MJ. The mouse mutation severe combined immune deficiency (scid) is on chromosome 16. Immunogenetics 1989;29:54–7
- Komatsu K, Ohta T, Jinno Y, Niikawa N, Okumura Y. Functional complementation in mouse–human radiation hybrids assigns the putative murine scid gene to the pericentric region of human chromosome 8. Hum Mol Genet 1993;2:1031–4
- Jeggo PA, Kemp LM. X-ray-sensitive mutants of Chinese hamster ovary cell line. Isolation and cross-sensitivity to other DNA-damaging agents. Mutat Res 1983;112:313–27
- He DM, Lee SE, Hendrickson EA. Restoration of X-ray and etoposide resistance, Ku-end binding activity and V(D)J recombination to the Chinese hamster sxi-3 mutant by a hamster Ku86 cDNA. Mutat Res 1996;363:43–56
- Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, et al. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol 1999;19:3877–84
- Iliakis G, Wu W, Wang M. DNA double strand break repair inhibition as a cause of heat radiosensitization: Re-evaluation considering backup pathways of NHEJ. Int J Hyperthermia 2008;24:17–29
- Ihara M, Suwa A, Komatsu K, Shimasaki T, Okaichi K, Hendrickson EA, et al. Heat sensitivity of double-stranded DNA-dependent protein kinase (DNA-PK) activity. Int J Radiat Biol 1999;75:253–8
- Dynlacht JR, Bittner ME, Bethel JA, Beck BD. The non-homologous end-joining pathway is not involved in the radiosensitization of mammalian cells by heat shock. J Cell Physiol 2003;196:557–64
- Woudstra EC, Konings AW, Jeggo PA, Kampinga HH. Role of DNA-PK Subunits in radiosensitization by hyperthermia. Radiat Res 1999;152:214–18
- Ostapenko VV, Wang X, Ohnishi K, Takahashi A, Yamamoto I, Tanaka Y, et al. Increased resistance of the radiosensitive M10 mutant cells of the L5178Y mouse lymphoma cell line to heat-induced apoptosis. Radiat Res 1999;152:321–7
- Zeng ZC, Jiang GL, Wang GM, Tang ZY, Curran WJ, Iliakis G. DNA-PKcs subunits in radiosensitization by hyperthermia on hepatocellular carcinoma hepG2 cell line. World J Gastroenterol 2002;8:797–803
- Raaphorst GP, Maude-Leblanc J, Li L. Evaluation of recombination repair pathways in thermal radiosensitization. Radiat Res 2004;161:215–18
- Yin HL, Suzuki Y, Matsumoto Y, Tomita M, Furusawa Y, Enomoto A, et al. Radiosensitization by hyperthermia in the chicken B-lymphocyte cell line DT40 and its derivatives lacking nonhomologous end joining and/or homologous recombination pathways of DNA double-strand break repair. Radiat Res 2004;162:433–41
- Shimasaki T, Ihara M, Furusawa Y, Okumura Y. Induction of DNA double strand breaks in scid cells by carbon ions. Radiat Prot Dosimetry 2002;99:155–7
- Núñez MI, Villalobos M, Olea N, Valenzuela MT, Pedraza V, McMillan TJ, et al. Radiation-induced DNA double-strand break rejoining in human tumour cells. Br J Cancer 1995;71:311–16
- Pinto M, Prise KM, Michael BD. Evidence for complexity at the nanometer scale of radiation-induced DNA DSBs as a determinant of rejoining kinetics. Radiat Res 2005;164:73–85
- Wong RS, Dynlacht JR, Cedervall B, Dewey WC. Analysis by pulsed-field gel electrophoresis of DNA double-strand breaks induced by heat and/or X-irradiation in bulk and replicating DNA of CHO cells. Int J Radiat Biol 1995;68:141–52
- van Waarde MAWH, van Assen AJ, Konings AWT, Kampinga HH. Feasibility of measuring radiation-induced DNA double strand breaks and their repair by pulsed field gel electrophoresis in freshly isolated cells from the mouse RIF-1 tumor. Int J Radiat Oncol Biol Phys 1996;36:125–34
- Dahm-Daphi J, Brammer I, Dikomey E. Heat effects on the repair of DNA double-strand breaks in CHO cells. Int J Radiat Biol 1997;72:171–9
- Dikomey E, Dahm-Daphi J, Brammer I, Martensen R, Kaina B. Correlation between cellular radiosensitivity and non-repaired double-strand breaks studied in nine mammalian cell lines. Int J Radiat Biol 1998;73:269–78
- Karlsson KH, Radulescu I, Rydberg B, Stenerlow B. Repair of radiation-induced heat-labile sites is independent of DNA-PKcs, XRCC1 and PARP. Radiat Res 2008;169:506–12
- DiBiase SJ, Zeng ZC, Chen R, Hyslop T, Curran WJ Jr, Iliakis G. DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res 2000;60:1245–53
- Wang H, Zeng ZC, Bui TA, Sonoda E, Takata M, Takeda S, et al. Efficient rejoining of radiation-induced DNA double-strand breaks in vertebrate cells deficient in genes of the RAD52 epistasis group. Oncogene 2001;20:2212–24
- Burgman P, Ouyang H, Peterson S, Chen DJ, Li GC. Heat inactivation of Ku autoantigen: Possible role in hyperthermic radiosensitization. Cancer Res 1997;57:2847–50
- Matsumoto Y, Suzuki N, Sakai K, Morimatsu A, Hirano K, Murofushi H. A possible mechanism for hyperthermic radiosensitization mediated through hyperthermic lability of Ku subunits in DNA-dependent protein kinase. Biochem Biophys Res Commun 1997;234:568–72
- Umeda N, Matsumoto Y, Yin HL, Tomita M, Enomoto A, Morita A, et al. Difference in the heat sensitivity of DNA-dependent protein kinase activity among mouse, hamster and human cells. Int J Radiat Biol 2003;79:671–80
- Genet SC, Fujii Y, Maeda J, Kaneko M, Genet MD, Miyagawa K, et al. Hyperthermia inhibits homologous recombination repair and sensitizes cells to ionizing radiation in a time- and temperature-dependent manner. J Cell Physiol 2013;228:1473–81
- Bergs JWJ, Krawczyk PM, Borovski T, Cate R, Rodermond HM, Stap J, et al. Inhibition of homologous recombination by hyperthermia shunts early double strand break repair to non-homologous end-joining. DNA Repair 2013;12:38–45
- Krawczyk PM, Eppink B, Essers J, Stap J, Rodermond H, Odijk H, et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc Natl Acad Sci USA 2011;108:9851–6
- Ma YX, Fan S, Xiong J, Yuan RQ, Meng Q, Gao M, et al. Role of BRCA1 in heat shock response. Oncogene 2003;22:10–27
- Eppink B, Krawczyk PM, Stap J, Kanaar R. Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. Int J Hyperthermia 2012;28:509–17
- Takahashi A, Yamakawa N, Mori E, Ohnishi K, Yokota S, Sugo N, et al. Development of thermotolerance requires interaction between polymerase-beta and heat shock protein. Cancer Sci 2008;99:973–8
- Takahashi A, Ohnishi T. Molecular mechanisms involved in adaptive responses to radiation, UV light, and heat. J Radiat Res 2009;50:385–93
- Takahashi A, Matsumoto H, Nagayama K, Kitano M, Hirose S, Tanaka H, et al. Evidence for the involvement of double-strand breaks in heat-induced cell killing. Cancer Res 2004;64:8839–45
- Takahashi A, Mori E, Somakos GI, Ohnishi K, Ohnishi T. Heat induces γH2AX foci formation in mammalian cells. Mutat Res/Genet Toxicol Environ Mutagen 2008;656:88–92
- Okamoto N, Takahashi A, Ota I, Ohnishi K, Mori E, Kondo N, et al. siRNA targeted for NBS1 enhances heat sensitivity in human anaplastic thyroid carcinoma cells. Int J Hyperthermia 2011;27:297–304