
Abstract
Purpose: There were two primary objectives of this study: (1) to determine whether treatment of a tumour site with systemically administered thermally sensitive liposomes and local hyperthermia (HT) for triggered release would have dual anti-tumour effect on the primary heated tumour as well as an unheated secondary tumour in a distant site, and (2) to determine the ability of non-invasive optical spectroscopy to predict treatment outcome. The optical end points studied included drug levels, metabolic markers flavin adenine dinucleotide (FAD), nicotinamide adenine dinucleotide phosphate (NAD(P)H), and physiological markers (total haemoglobin (Hb) and Hb oxygen saturation) before and after treatment.
Materials and methods: Mice were inoculated with SKOV3 human ovarian carcinoma in both hind legs. One tumour was selected for local hyperthermia and subsequent systemic treatment. There were four treatment groups: control, DOXIL® (non-thermally sensitive liposomes containing doxorubicin), and two different thermally sensitive liposome formulations containing doxorubicin. Optical spectroscopy was performed prior to therapy, immediately after treatment, and 6, 12, and 24 h post therapy.
Results: Tumour growth delay was seen with DOXIL and the thermally sensitive liposomes in the tumours that were heated, similar to previous studies. Tumour growth delay was also seen in the opposing tumour in the thermally sensitive liposome-treated groups. Optical spectroscopy demonstrated correlation between growth delay, doxorubicin (DOX) levels, and changes of NAD(P)H from baseline levels. Hb and Hb saturation were not correlated with growth delay.
Discussion: The study demonstrated that thermally sensitive liposomes affect the primary heated tumour as well as systemic efficacy. Non-invasive optical spectroscopy methods were shown to be useful in predicting efficacy at early time points post-treatment.
Introduction
Successful cancer chemotherapy requires the efficient delivery of therapeutic drug to the tumour at an efficacious concentration. It is well known that the physiology of tumours presents a significant impediment to drug delivery because the microvasculature is inefficient for transport and high interstitial fluid pressure in the tumour can impede delivery, particularly for protein-bound drugs or macromolecules [Citation1]. One approach to overcome this resistance to drug delivery is the use of drug vehicles such as liposomes in combination with local/regional hyperthermia (HT), which increases perfusion [Citation2–4] and endothelial pore sizes [Citation5], promoting greater extravasation into tumour interstitium. An additional improvement in drug delivery mediated by hyperthermia can be achieved by using a thermally sensitive liposome formulation, which releases drug from the liposome intravascularly [Citation6]. This drives the drug into the tumour by Fickian diffusion following its concentration gradient. The increase in drug penetration achieved by this effect makes the drug more bioavailable for tumour cell killing. This concept was pioneered by Yatvin [Citation7,Citation8], but his formulation was not thermally sensitive in the temperature range that is easily achievable clinically (40–43 °C) where the benefits of relatively non-cytotoxic heating are found. Furthermore, the formulation exhibited very slow drug release when heated, which was not ideal for taking advantage of Fickian diffusion, since the liposomes would exit from the tumour vasculature before releasing their contents. Needham et al. [Citation9] was the first study to report on a low temperature-sensitive liposome (LTSL) that contained doxorubicin (LTSL-DOX). LTSL-DOX releases its contents within seconds when the local tissue temperature is ∼40–42 °C. Lindner has reported on a thermally sensitive formulation with prolonged circulation time [Citation10], which also shows very rapid DOX release when heated. When LTSL-DOX has been combined with hyperthermia in vivo, drug delivery to the tumour has been increased by as much as 20–30-fold compared with free drug, and 4–5-fold over that achievable with non-thermally sensitive liposomes [Citation11,Citation12]. Drug penetration distances from the nearest blood vessel are twice that of Doxil® or free drug [Citation6].
Several preclinical studies [Citation9,Citation11,Citation13] have shown that heat-mediated localised LTSL-DOX delivery yields improved anti-tumour effects. A phase I trial in dogs with spontaneous tumours showed that LTSL-DOX was safe and that it exhibited anti-tumour activity [Citation14]. A human phase I trial reported that LTSL-DOX was safe when combined with thermal ablation for hepatocellular carcinoma, and that substantial anti-tumour effects were seen [Citation15]. LTSL-DOX was also shown to be safe after administration of multiple courses in patients with chest wall recurrences of breast cancer [Citation16].
Despite the potential promise of LTSL-DOX to improve local anti-tumour effects, there are concerns about using an agent that focuses therapy on the primary tumour as a first line agent when there is a risk for presence of unknown systemic metastases. The risk of using a locally delivered chemotherapy is that the metastases may not be treated. We recently completed a phase I clinical trial of patients with chest wall recurrences of breast cancer that were treated with LTSL-DOX with hyperthermia. The overall response rate was over 45% in this heavily pre-treated set of patients; however, we also observed some tumour responses that were outside of the heated volume, suggesting that this drug may have systemic effects. This could occur if some of the DOX that was released from liposomes remained in circulation and entered tumours at distant sites. We have shown previously in several tumour models that LTSL-DOX does not have anti-tumour properties in the absence of local heating, including in SKOV3, which is used in this current study [Citation13].Therefore, it is most likely that the systemic anti-tumour effects seen with local heating would have to be the result of recirculating released drug.
Optical spectroscopy is a non-invasive means of characterising the tissue absorption, scattering, and fluorescence properties. Using Monte Carlo-based analysis methods it is possible to quantify these optical properties in tissue and relate them to the underlying physiological parameters that influence tissue optical properties, including: (1) absorbers oxy- and deoxyhaemoglobin, (2) scattering properties, which are determined by tissue morphology including extracellular matrix, and cellular density, and (3) fluorescence properties, in this case NAD(P)H, FAD and doxorubicin [Citation17]. In the current work we have focused on haemoglobin concentration, which is indicative of changes in blood volume, and haemoglobin oxygen saturation, which is indicative of tissue hypoxia. In addition, doxorubicin is fluorescent, and we have previously shown that doxorubicin concentration in tissue can be quantified using optical methods [Citation18]. Finally, NAD(P)H and FAD are indicative of oxidative metabolism, and have been used as indicators of the redox status of tumours and other tissues [Citation17].
The purpose of this study was to determine whether the combination of LTSL-DOX and local HT has a systemic anti-tumour effect. To test this we created a distant pseudo-metastasis by transplanting two tumours in opposing flanks and heating only one in conjunction with administration of LTSL-DOX.
Methods
All animal procedures were approved by the Duke University Institutional Care and Use Committee. The goal was to explore distant efficacy of using the standard clinical formulation with 3.8–4% polyethylene glycol (PEG) lipid (used in Hauck et al. [Citation14] and Viglianti et al. [Citation2]) along with a second formulation with a higher PEG concentration (5.2%) since molecular modelling suggested that at the lower concentration of 3.8–4% PEG there was lack of complete steric coverage of the liposome surface. We previously reported that PEGylation increases vascular permeability to liposomes. Increasing PEG coverage may improve the enhanced permeability and retention (EPR) effect in the unheated tumour if this is important for any anti-tumour effect in that site [Citation19]. Further, others have reported that circulation half-life is increased by full PEG coverage. The extended circulation time could also improve liposomal uptake at distant non-heated sites (similar to DOXIL). Thus, we examined the question of whether accumulation of DOX in the distant unheated tumour would explain any abscopal effect on tumour growth delay.
Liposomes
Chemicals
1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC), 1-stearoyl-2-hydroxy-sn-glycero-3-phosphatidylcholine (MSPC), and 1,2-distearoyl-sn-glycero-3-phosphatidyl ethanol amine-N-[methoxy(polyethylene glycol)-2000] – ammonium salt (DSPE-PEG 2000) were obtained from Avanti Polar Lipids (Alabaster, AL, USA) and used without further purification. Doxorubicin-HCl, citric acid, Sephadex G-50, HEPES sodium salt, sodium carbonate anhydrous, and other chemicals (chloroform, methanol and hydrochloric acid) were obtained from Sigma-Aldrich (St Louis, MO). All chemicals involved were standard reagent grade and used without further purification.
Preparation of liposome
To study the effect of PEG2000 on tumour regression, two different lipid compositions with varied PEG content were used for LTSL. LTSL1 contained 3.8 mol% PEG2000 (DPPC:MSPC:DSPE-PEG2000 at 86.5:9.7:3.8 mol%) and LTSL2 contained 5.2 mol% PEG-2000 (DPPC:MSPC:DSPE-PEG2000 at 85.0:9.8:5.2 mol%). The lipids were dissolved together in chloroform and methanol (4:1 volume ratio) within a small round-bottom flask, and the solvent was removed by a roto-evaporator under a vacuum. The lipid film was then dried overnight under a vacuum. The dried lipids were hydrated at 55 °C for 30 min with citrate buffer (300 mM, pH 4) containing 1 μM of dissolved MSPC to a lipid concentration of 100 mg/mL. The hydrated lipid vesicles were extruded twice through a polycarbonate membrane of 200-nm pore size and then eight times through 100-nm pores at 55 °C and 400 psi nitrogen pressure by using a water-jacketed extruder (Northern Lipids, Vancouver, BC, Canada). This procedure resulted in the formation of unilamellar liposomes averaging about 116 nm ± 3 nm in diameter. Once the extrusion procedure was completed, the extruded liposomes were immediately placed into 4 °C until further loading with DOX.
DOX loading procedure
DOX is loaded into the liposome by the remote pH gradient method [Citation20]. The extruded liposome formulation was pH adjusted to 7.4 through the addition of sodium carbonate buffer (0.5 M, pH 11). DOX (5 mg/mL) and the extruded liposome formulation were incubated for 5 min individually at 35 °C, then combined to a ratio of 10 mg lipid:0.5 mg DOX and further incubated for 30 min, with gentle agitation every 5 min. Following loading, the liposomes were immediately transferred to a pre-cooled glass tube on ice, left to rest for 10 min on ice followed by 5 min at room temperature. The remaining unencapsulated DOX was removed from the liposome samples by the gel filtration column using Sephadex G-50. Liposomes were added to Sephadex G-50 columns and centrifuged. Sephadex column separation was repeated twice. Liposome-encapsulated DOX was estimated by lysing the vesicles with Triton X-100 (5 uL in 1 mL HEPES added to the sample) and measuring emission (λex 480 nm, λem 550 nm) with a spectrofluorophotometer. The hydrodynamic radius of DOX-loaded liposomes was measured using a particle size analyser (Brookhaven Instruments, Holtsville, NY).
Animals and tumours
Six-week-old female homozygous nu/nu 100 mice (3 did not have tumors take) were purchased from the National Cancer Institute and housed five per cage with appropriate food and water. Animals were anaesthetised with isofluorane and an AVID MUSICC RFID chip was subcutaneously implanted via syringe in the dorsal skin fold so that each mouse could be easily identified. One week later, two SKOV3 tumours were established by subcutaneous injection of 3 × 106 cells in 100 μL suspension into both the left and right flanks. Subsequently, mouse weight and tumour volume were measured every three days using the formula volume = (length × width × width × π)/6. Animals were randomised into one of four groups: saline control, DOXIL™ (commercial thermally stable liposomal DOX), LTSL1 with 3.8% PEG, and LTSL2 with 5.2% PEG, having n = 19, 20, 28, and 26 animals, respectively. Two additional groups, LTSL1 and LTSL2 at a higher treatment dose, were also added with 5 animals each to see if a higher dose provided an additional survival benefit/effect on tumour metabolism/physiology. Treatment commenced when one of the tumours reached a volume ≥ 200 mm3. The larger of the tumours (defined as the flank tumour to first reach a volume >) was chosen to be the primary, ‘treatment side’ for two reasons. First, we ideally wanted both sides to achieve 5× treatment volume. If the larger side was not treated, it was possible that the tumour on that side would rapidly reach Institutional Animal Care and Use Committees’ humane end points necessitating sacrifice prior to study termination point of 5× treatment volume. Allowing the smaller tumour to be the untreated, secondary site increased the overall time to sacrifice. Furthermore, deeming the larger tumour to be the primary, treated side creates a pseudometastatic model where there is a large ‘primary’ tumour in one flank and a smaller ‘metastatic’ tumour in the other. For the final survival analysis, results were reported with both 3× and 5× initial treatment time. This was done since the growth rates were different between the treated and control sides. This could result in censoring of one side, if the animal reached the target volume on the other side. The 3× treatment volume end point virtually eliminated the need for data censoring of either group, however the 5× data (with censoring present) was included given that is the standard used for tumour growth delay studies.
DOX was administered at either the free DOX MTD of (5 mg/kg) or at 7.5 mg/kg for the two high dose LTSL-DOX groups. Animals were anaesthetised with 85 mg/kg pentobarbital during the HT procedure. All treatment occurred via tail vein injection. Hyperthermia was applied for 1 h, starting immediately after drug was administered. Tumour-bearing limbs were placed into a 44 °C water bath, which yields an intratumoural temperature of 41–42 °C, as previously described. A fan was used to cool the rest of the body and thus maintain core body temperature at 38 °C [Citation11].
Optical spectroscopy
An optical spectrometer consisting of a Triax 320 imaging spectrograph and Gemini 180 monochromator with a 450 W xenon light source (JY Horiba, Irvine, CA) was coupled through a fibre-optic probe, consisting of separate columns of 42 illumination and 42 collection fibres divided by a 2-mm wide dark spacer against the tumour. Diffuse reflectance spectra were acquired using zero-order (white light) illumination and collecting the remitted spectrum from 340–612 nm. Fluorescence spectra were acquired at 350, 400, 440, 460, and 490 nm excitation. Optical spectra were then modelled using a Monte Carlo-based model of diffuse reflectance [Citation21] and fluorescence [Citation22] to extract the absorption, scattering, and intrinsic fluorescence properties of the tissue (fluorescence corrected for tissue absorption and scattering). Briefly, diffuse reflectance spectra were modelled by assuming that the tissue absorption was described by the summed contribution of oxy- and deoxyhaemoglobin modulated by a pigment packing correction [Citation23] as well as a tissue baseline absorption spectrum as previously described [Citation18]. Scattering was fitted to a scatter power model: , where
is the reduced scattering coefficient, A is the scattering amplitude, b is the scatter power, and λ is the wavelength in nm. Normalisation to 400 nm reduces the large dependence of the magnitude of the scattering coefficient on b. A non-linear least squares fitting algorithm was applied to fit the measured data to the modelled data for each measurement. Modelling the diffuse reflectance thus provides the absorption and scattering coefficients as well as the haemoglobin concentration and oxygen saturation.
For extraction of relative DOX levels from these spectra, these optical properties were then applied to a Monte Carlo-based model of fluorescence spectra in turbid media [Citation22]. This allows for correction of the measured fluorescence spectra for the effects of absorption and scattering, and allows for extraction of the intrinsic fluorescence, which is proportional to the fluorophore concentration, its excitation and emission spectra, and its fluorescence quantum yield, summed for all constituent fluorophores. Fluorescence spectra of NAD(P)H and flavins FAD/FMN were not corrected for absorption and scattering, since in the case of NAD(P)H, UV excitation is used, which introduces additional absorbers that would complicate the model, and this was also not applied to FAD for consistency. Thus, these spectra may reflect changes in absorption and scattering, as well as intrinsic changes in the fluorescence properties. For the purposes of quantification, the DOX fluorescence ratio was taken at 490 nm excitation as the ratio of the average emission between 580–610 (DOX fluorescence signal), normalised to the tissue autofluorescence taken as the average value between 510–540 nm (background autofluorescence signal). This ratio was found in a previous study to have a linear relationship to the DOX concentration [Citation18], although this relationship was not calibrated in the present study and so is reported in relative units. NAD(P)H fluorescence was taken as the mean fluorescence signal acquired at 350 nm excitation, 390–634 nm emission, while FAD fluorescence was taken as the mean signal acquired at 460 nm excitation, 516–558 nm emission. Optical spectra and associated end points were acquired from each animal at pretreatment, immediately after treatment, and at 6 and 24 h post-treatment.
Data analysis and statistics
The GraphPad Prism 5 program (GraphPad Software, San Diego, CA) was used to perform all statistical analyses. Growth delay statistics were analysed using the log-rank (Mantel-Cox) test. Relative DOX fluorescence data versus growth delay was plotted as mean and standard error of mean (SEM) of DOX fluorescence (SEM of growth delay was not shown for clarity). Relative DOX fluorescence versus time was plotted as mean treatment group value (again SEM was not given for clarity given that there was significant overlap in the secondary side values). Metabolic data (NADP(H)) was averaged for each group and time point. Data was then plotted as mean percentage change from the pretreatment value. Metabolic data at 6 h (percentage change from baseline) versus tumour growth delay was plotted and linear regression with 95% confidence interval was performed.
Results
Times to reach three and five times treatment volumes were calculated for both the HT treated tumour (primary) and the non-HT treated tumour (secondary). DOXIL treatment and LTSL treatments (independent of PEG concentration) demonstrated statistically significant prolongation of tumour growth delay time at both 3× and 5× the treatment volume on the primary side () compared to controls. The hazard ratios were 2.1 for DOXIL, 2.5 for LTSL1, and 3.6 for LTSL2 compared with saline control (). The LTSL at 3.8% for the 3× time point and the LTSL at 5.2% for the 5× time point were statistically different from control (p < 0.05). The hazard ratios for these groups were 3.2 and 2.6 respectively (). Time to 3× with the 5.2% PEG TSL had p = 0.09.
Table 1. Survival analysis for the different treatment groups.
These efficacies are related to the intratumoural drug concentration. In , the time to reach 5× treatment volume versus DOX fluorescence levels at 6, 12, and 24 h for the primary and secondary side is shown. Growth times are positively correlated with the amount of drug delivered, as assessed spectroscopically, in both the heated and non-heated tumours. The kinetics of intratumoural drug level for the different treatment groups are shown in for the primary and secondary side.
Figure 1. Growth delay of the primary and secondary side as a function of relative intratumoural DOX measured with optical spectral imaging at 6 h (A), 12 h (B), and 24 h (C) after treatment. Data is given as mean and SEM for each group (n = 19, 20, 28, 26, 5, and 5 animals for control, DOXIL, LTSL1 with 3.8% PEG, LTSL2 with 5.2% PEG, LTSL1 (7.5 mg/kg) with 3.8% PEG, LTSL2 (7.5 mg/kg) with 5.2% PEG, respectively).
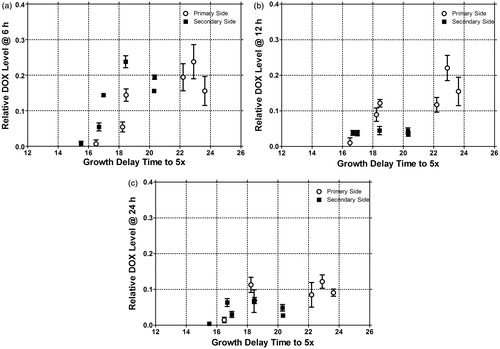
Figure 2. Primary treated side relative DOX level versus time (a). Secondary treated side relative DOX level versus time (b). Data is given as mean for each group (n = 19, 20, 28, 26, 5, and 5 animals for control, DOXIL, LTSL1 with 3.8% PEG, LTSL2 with 5.2% PEG, LTSL1 (7.5 mg/kg) with 3.8% PEG, LTSL2 (7.5 mg/kg) with 5.2% PEG, respectively).
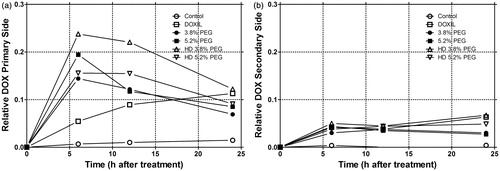
From it is clear that the combination of local heat and thermally sensitive liposomes results in rapid increase in primary tumour drug levels peaking at 6 h. In contrast, the DOXIL liposome exhibits a continuous gradual increase in DOX fluorescence levels up to 24 h. This kinetic accumulation pattern fits the expected enhanced permeability and retention effect, which would be manifested by steady extravasation. Additional increase in fluorescence could come from slow leakage of the encapsulated DOX (fluorescence is quenched at high concentrations when drug is encapsulated inside the liposome) [Citation24]. The secondary side also shows an increase in DOX fluorescence slowly over the course of 24 h (). Here LTSLs behave similarly to the DOXIL liposomes. gives the area under the curve for each treatment group and tumour side. LTSL-DOX + HT delivered DOX at a faster rate, to reach a peak value at an earlier time point compared with DOXIL. By 24 h, however, the relative concentrations of DOX were similar for both methods. Growth time correlated with peak DOX concentration (which ever time point it occurred at) and with the area under the curve (data not shown).
Table 2. DOX accumulation results. Normalised area under the curve (AUC) and peak concentration for each treatment group and tumour side (Primary had local HT).
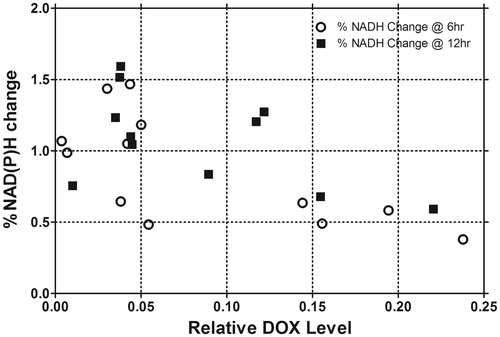
Optical spectroscopic measurements of Hb concentration, Hb oxygen saturation, FAD, and NAD(P)H, were also performed prior to therapy and 6, 12, and 24 h post-therapy. Baseline normalised changes of these metabolic markers, as a function of time from treatment, were compared with tumour growth time. NAD(P)H decreased in the primary tumour side after treatment with a maximal change at 6 h. NAD(P)H then recovered and then increased toward baseline in the groups that received HT; in contrast the untreated LTSL-DOX groups exceeded baseline levels by 50%. The secondary side exhibited no significant time dependence in any of the groups ( and ). Additionally, normalised NAD(P)H change from baseline had an inverse relationship with growth time; NAD(P)H values showed decreased normalised values as growth delay time increased. This was observed at both 6 and 12 h (). The results were significant at 6 h with a slope that was not equal to 0 (p = 0.05) based on linear regression. A similar trend was seen at 12 h (p = 0.097). The results for FAD were similar, but less dramatic (data not shown). Since there was a correlation between DOX fluorescence and growth delay along with NAD(P)H and growth delay, NAD(P)H percentage change from baseline (at 6 and 12 h) versus DOX fluorescence level was compared in . A significant relationship between DOX fluorescence intensity and NAD(P)H was observed using linear regression at 6 h (p = 0.0135). A trend was observed at 12 h (p = 0.08).
Figure 3. a and b demonstrate the mean temporal relationship for each treatment group of NAD(P)H after therapy for the primary side (a) and the secondary side (b). c and d demonstrate the mean percentage change of the NAD(P)H of each treatment group versus tumour growth delay of both the primary and secondary side at 6 h (c) and 12 h (d) with linear regression and 95% confidence bounds. At 6 h the results were statistically significant (n = 19, 20, 28, 26, 5, and 5 animals for control, DOXIL, LTSL1 with 3.8% PEG, LTSL2 with 5.2% PEG, LTSL1 (7.5 mg/kg) with 3.8% PEG, LTSL2 (7.5 mg/kg) with 5.2% PEG, respectively).
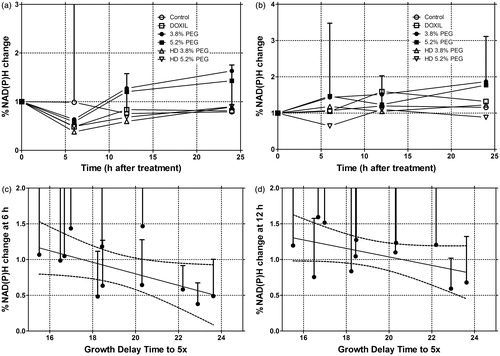
Figure 4. The mean NAD(P)H percentage change for each treatment group versus mean relative DOX concentration for each group at 6 and 12 h combining both treatment sides. Linear regression was then performed demonstrating a significant slope not equal to unity with 95% confidence intervals at 6 h. Note, linear regression overlay was not included, for clarity (n = 19, 20, 28, 26, 5, and 5 animals for control, DOXIL, LTSL1 with 3.8% PEG, LTSL2 with 5.2% PEG, LTSL1 (7.5 mg/kg) with 3.8% PEG, LTSL2 (7.5 mg/kg) with 5.2% PEG, respectively).
Discussion
Delay in tumour growth is known to occur with local HT in combination with non-thermally sensitive liposomes (DOXIL) and thermally sensitive liposomes [Citation11,Citation25,Citation26]. The data generated here demonstrates significant growth delay with the DOXIL and two LTSL treatment groups on the primary tumour side. The data are also consistent with our previous work showing that growth time correlates with intratumoural drug levels [Citation11,Citation18] and that optical spectroscopy can be used to quantify intratumoural DOX levels non-invasively [Citation18].
The main goal of this study was to determine whether treatment of a primary tumour site with local HT and systemic LTSLs would have both local and systemic anti-tumour effects. It was demonstrated that a systemic anti-tumour effect on the secondary tumour following treatment with LTSLs and local HT therapy at the primary tumour site did occur.
The mechanism of the abscopal effect of LTSL-DOX is most likely due to recirculation of intravascularly released drug. Compared with DOXIL, both LTSL formulations have increased membrane permeability to DOX at physiological temperatures [Citation11,Citation27]. Although there is this increased membrane permeability, prior studies have demonstrated that in the absence of local HT, LTSLs have similar efficacy to DOXIL [Citation11,Citation18]. In the case of SKOV3, however, LTSL has no anti-tumour effect in the absence of hyperthermia. Further, hyperthermia itself has no anti-tumour effect in this model [Citation13]. While a DOXIL without heat experimental arm was not performed directly, there was no efficacy on the secondary side in the DOXIL group (a pseudo similar experiment). We previously reported that this tumour line is relatively sensitive to DOX, in vitro, with an IC50 of 128 nM [Citation13]. If adequate concentrations of DOX reach the tumour, there should be an anti-tumour effect. However, we previously demonstrated that vascular permeability of the SKOV3 tumour is very low in the absence of heat, but that it is greatly enhanced by heating [Citation12]. Without adequate extravasation into the unheated tumour there would be little anti-tumour effect expected from DOXIL.
There was improved tumour growth delay in the secondary tumour with the LTSL formulations when combined with HT. The mechanism for this effect may relate to intravascular release of drug from LTSL-DOX when it enters the heated tumour-bearing leg of the mouse. Intravascular drug release would enable some of the released drug to enter the systemic circulation along with unreleased LTSL-encapsulated drug. It is also possible that there is extravasation of partially loaded LTSL into the secondary tumour. However, we believe this is very unlikely because of the low vascular permeability of SKOV3 to nanoparticles under normothermic conditions [Citation12]. Further, the half-life of LTSL-DOX in vivo is very short, compared with DOXIL, which would greatly lessen the possibility for an EPR effect. In dogs and rats, the half-life of LTSL-DOX is 1 and 2 h, respectively. In humans, however, it is shorter, in the order of 35–40 min [Citation14–16,Citation28,Citation29]. Given the short circulation time of LTSL, it is not likely that enhanced extravasation of the liposomes plays a role in anti-tumour effects at distant sites. Another potential mechanism of enhanced effect could come from heat-mediated liposome-released lysolipids, which have been shown to be cytotoxic [Citation30]. However, materials characterisation of these liposomes reveals that little if any lysolipid is desorbed upon heating [Citation31].
The hazard ratios for growth delay were consistently larger for the 5.2% versus the 3.8% PEG lipid concentration. This means that the anti-tumour effect of the 5.2% PEG lipid formulation was relatively greater than the 3.8% PEG lipid formulation. There are two potential reasons for this difference. The higher PEG concentration may have yielded a longer circulation time. Alternatively, the vascular permeability of the 5.2% PEG lipid formulation may have been larger than the 3.8% PEG lipid formulation. However, these were separate experiments, so a direct head-to-head comparison of the two formulations was not made. Additional studies examining potential differences in pharmacokinetics and/or vascular permeability may yield an explanation for differences if seen in a direct comparison study.
Optical spectroscopy was also performed to look at Hb concentration and oxygen saturation along with NAD(P)H and FAD fluorescence. It is known that HT alone increases tumour oxygenation and perfusion [Citation32] and that this effect is independent of whether chemotherapy is used. In this case, however, there was not a consistent change in Hb or Hb saturation after treatment. However, the results demonstrated that NAD(P)H decreased in all treatment groups for the primary tumour, reaching a maximal change at 6 h and then slowly recovering. Similar but less dramatic results occurred in the secondary tumour site. Decreases in NAD(P)H were correlated with larger growth delay. Similarly, the change in NAD(P)H was also associated with levels of DOX within the tumour. Considering that NAD(P)H levels monitor cellular metabolism, the results are consistent with DOX causing cell death and/or increasing oxidative stress either directly or by shutting down blood flow [Citation33], or by decreasing aerobic metabolic activity, resulting in lowered NAD(P)H. The levels subsequently recovered as the tumour DOX levels decreased. A larger NAD(P)H drop may thus reflect greater tumour growth delay due to a larger effect on cell stress/death from chemotherapy.
DOX fluorescence levels showed a positive correlation with treatment response on the primary side, which is consistent with prior work [Citation11,Citation18,Citation26], and demonstrates the importance of measuring (ideally non-invasively) tumour drug levels as a method to predict treatment response. In addition, the optical metabolic measurements demonstrated promise for predicting treatment response and/or monitoring response; however, further work is needed to validate the technique and its robustness.
Conclusion
In summary, this study demonstrates that there are systemic anti-tumour effects of LTSL-DOX when it is combined with local heating of a primary tumour. The most likely contribution to the anti-tumour effect is recirculated free drug, which occurs as a result of intravascular drug release in the heated volume. We demonstrate the value of optical spectroscopy as a method to predict extent of anti-tumour effect. The natural fluorescence of DOX provides a convenient optical biomarker of drug concentration. However, changes in NAD(P)H fluorescence intensity, which reflect cellular metabolic state, may also prove valuable in the prediction of response.
Declaration of interest
G.M.P. and Duke University have financial interest in Zenalux Biomedical, Inc., which is commercializing optical spectroscopy technologies.
References
- Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. II. Role of heterogeneous perfusion and lymphatics. Microvasc Res 1990;40:246–63
- Viglianti BL, Lora-Michiels M, Poulson JM, Lan L, Yu DH, Sanders L, et al. Dynamic contrast-enhanced magnetic resonance imaging as a predictor of clinical outcome in canine spontaneous soft tissue sarcomas treated with thermoradiotherapy. Clin Cancer Res 2009;15:4993–5001
- Griffin RJ, Dings RP, Jamshidi-Parsian A, Song CW. Mild temperature hyperthermia and radiation therapy: Role of tumour vascular thermotolerance and relevant physiological factors. Int J Hyperthermia 2010;26:256–63
- Song CW, Park H, Griffin RJ. Improvement of tumor oxygenation by mild hyperthermia. Radiat Res 2001;155:515–28
- Kong G, Braun RD, Dewhirst MW. Hyperthermia enables tumor-specific nanoparticle delivery: Effect of particle size. Cancer Res 2000;60:4440–5
- Manzoor AA, Lindner LH, Landon CD, Park JY, Simnick AJ, Dreher MR, et al. Overcoming limitations in nanoparticle drug delivery: Triggered, intravascular release to improve drug penetration into tumors. Cancer Res 2012;72:5566–75
- Weinstein JN, Magin RL, Yatvin MB, Zaharko DS. Liposomes and local hyperthermia: Selective delivery of methotrexate to heated tumors. Science 1979;204:188–91
- Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. Design of liposomes for enhanced local release of drugs by hyperthermia. Science 1978;202:1290–3
- Needham D, Anyarambhatla G, Kong G, Dewhirst MW. A new temperature-sensitive liposome for use with mild hyperthermia: Characterization and testing in a human tumor xenograft model. Cancer Res 2000;60:1197–201
- Lindner LH, Eichhorn ME, Eibl H, Teichert N, Schmitt-Sody M, Issels RD, et al. Novel temperature-sensitive liposomes with prolonged circulation time. Clin Cancer Res 2004;10:2168–78
- Kong G, Anyarambhatla G, Petros WP, Braun RD, Colvin OM, Needham D, et al. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: Importance of triggered drug release. Cancer Res 2000;60:6950–7
- Kong G, Braun RD, Dewhirst MW. Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature. Cancer Res 2001;61:3027–32
- Yarmolenko PS, Zhao Y, Landon C, Spasojevic I, Yuan F, Needham D, et al. Comparative effects of thermosensitive doxorubicin-containing liposomes and hyperthermia in human and murine tumours. Int J Hyperthermia 2010;26:485–98
- Hauck ML, LaRue SM, Petros WP, Poulson JM, Yu D, Spasojevic I, et al. Phase I trial of doxorubicin-containing low temperature sensitive liposomes in spontaneous canine tumors. Clin Cancer Res 2006;12:4004–10
- Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: An adjuvant to increase the cure rate of radiofrequency ablation in liver cancer. Future Oncol 2011;7:937–45
- Zagar T VZ, Formenti S, Rugo H, Muggia F, O'Connor B, Myerson R, et al. Two phase I dose escalation/pharmacokinetics studies of low temperature liposomal doxorubicin (LTLD) and mild local hyperthermia in heavily pretreated patients with local regionally recurrent breast cancer. Int J Hyperthermia 2014 in press
- Brown JQ, Vishwanath K, Palmer GM, Ramanujam N. Advances in quantitative UV-visible spectroscopy for clinical and pre-clinical application in cancer. Curr Opin Biotechnol. 2009;20:119–31
- Palmer GM, Boruta RJ, Viglianti BL, Lan L, Spasojevic I, Dewhirst MW. Non-invasive monitoring of intra-tumor drug concentration and therapeutic response using optical spectroscopy. J Control Release 2010;142:457–64
- Wu NZ, Da D, Rudoll TL, Needham D, Whorton AR, Dewhirst MW. Increased microvascular permeability contributes to preferential accumulation of stealth liposomes in tumor tissue. Cancer Res 1993;53:3765–70
- Mayer LD, Tai LC, Bally MB, Mitilenes GN, Ginsberg RS, Cullis PR. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim Biophys Acta 1990;1025:143–51
- Palmer G, Ramanujam N. Monte Carlo-based inverse model for calculating tissue optical properties. Part I: Theory and validation on synthetic phantoms. Appl Opt 2006;45:1062–71
- Palmer GM, Ramanujam N. Monte-Carlo-based model for the extraction of intrinsic fluorescence from turbid media. J Biomed Opt 2008;13:024017
- van Veen RL, Verkruysse W, Sterenborg HJ. Diffuse-reflectance spectroscopy from 500 to 1060 nm by correction for inhomogeneously distributed absorbers. Opt Lett 2002;27:246–8
- Wu NZ, Braun RD, Gaber MH, Lin GM, Ong ET, Shan S, et al. Simultaneous measurement of liposome extravasation and content release in tumors. Microcirculation 1997;4:83–101
- Kong G, Dewhirst MW. Hyperthermia and liposomes. Int J Hyperthermia 1999;15:345–70
- Ponce AM, Viglianti BL, Yu D, Yarmolenko PS, Michelich CR, Woo J, et al. Magnetic resonance imaging of temperature-sensitive liposome release: Drug dose painting and antitumor effects. J Natl Cancer Inst 2007;99:53–63
- Viglianti BL, Abraham SA, Michelich CR, Yarmolenko PS, MacFall JR, Bally MB, et al. In vivo monitoring of tissue pharmacokinetics of liposome/drug using MRI: Illustration of targeted delivery. Magn Reson Med 2004;51:1153–62
- Abdel-Wahab OI, Grubbs E, Viglianti BL, Cheng TY, Ueno T, Ko S, et al. The role of hyperthermia in regional alkylating agent chemotherapy. Clin Cancer Res 2004;10:5919–29
- Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: A novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin Pharmacother 2009;10:333–43
- Jantscheff P, Schlesinger M, Fritzsche J, Taylor LA, Graeser R, Kirfel G, et al. Lysophosphatidylcholine pretreatment reduces VLA-4 and P-selectin-mediated b16.f10 melanoma cell adhesion in vitro and inhibits metastasis-like lung invasion in vivo. Mol Cancer Therapeut 2011;10:186–97
- Mills JK, Needham D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim Biophys Acta 2005;1716:77–96
- Hong WK, Bast RC, Halt WN, Kufe DW, Pollock RE. Cancer Medicine, 8th ed. Shelton, CN: People’s Medical, 2010, pp xxv, 2,021
- Chen Q, Tong S, Dewhirst MW, Yuan F. Targeting tumor microvessels using doxorubicin encapsulated in a novel thermosensitive liposome. Mol Cancer Therapeut 2004;3:1311–17