
Abstract
Purpose: Hepatocellular carcinoma (HCC) suffers high tumour recurrence rate after thermal ablation. Heat shock protein 90 (Hsp90) induced post-ablation is critical for tumour survival and progression. A combination therapy of thermal ablation and polymer conjugated Hsp90 chemotherapy was designed and evaluated for complete tumour eradication of HCC. Materials and methods: A thermo-responsive, elastin-like polypeptide (ELP)-based tri-block biopolymer was developed and conjugated with a potent Hsp90 inhibitor, geldanamycin (GA). The anti-cancer efficacy of conjugates was evaluated in HCC cell cultures with and without hyperthermia (43 °C). The conjugates were also administered twice weekly in a murine HCC model as a single treatment or in combination with single electrocautery as the ablation method. Results: ELP-GA conjugates displayed enhanced cytotoxicity in vitro and effective heat shock inhibition under hyperthermia. The conjugates alone significantly slowed the tumour growth without systemic toxicity. Four doses of thermo-responsive ELP-GA conjugates with concomitant simple electrocautery accomplished significant Hsp90 inhibition and sustained tumour suppression. Conclusion: Hsp90 inhibition plays a key role in preventing the recurrence of HCC, and the combination of ablation with targeted therapy holds great potential to improve prognosis and survival of HCC patients.
Introduction
Thermal ablation is widely utilised in clinics to treat unresectable primary liver tumours such as hepatocellular carcinoma (HCC). Although ablative technology has been improved over the years with increased coagulation efficiency and lowered complication rates, a range of high tumour recurrence rates for HCC (4–84%) [Citation1–3] remain among the current ablation modalities. Recent reports of 5-year tumour recurrence rates for HCC include radiofrequency ablation (RFA) 26–84% [Citation4], cryoablation (CA) 31–55% [Citation5,Citation6], high intensity focused ultrasound (HIFU) 18–58% [Citation7,Citation8], and microwave thermal ablation (MWA) 31–65% [Citation9,Citation10].
Multiple factors contribute to incomplete tumour ablation such as irregular, non-uniform tumour size and shape [Citation11,Citation12], high tumour tissue heterogeneity, liver cirrhosis, heat sink effects caused by proximate capillary drainage [Citation13–15], and the extreme difficulty of 3-D and real-time visualisation of all viable tumour tissues [Citation16] particularly at the polyclonal tumour margins (TMs) [Citation17–19]. The TMs are of particular interest, having been identified as one of the major risks of local tumour recurrence and patient mortality [Citation20,Citation21]. In addition, reports have shown that thermal ablation alters the tumour microenvironment by modulation of molecular signalling pathways involving growth factors, kinases, and cytokines [Citation22,Citation23]. Cellular growth kinetics, including those of hepatic stellate cells, macrophages, and cancer-associated fibroblasts, have also been increased post-ablation [Citation24]. Stroma cells, in particular, have been shown to recruit tumour cells to the margins in preparation for neoplastic growth [Citation25]. All these factors together with activated tumour stromal interactions present dynamic and daunting barriers to tumour remission. To combat local tumour recurrence following incomplete thermal ablation, complementary chemo- and/or radiotherapies have been combined for additive or synergistic benefit. For example, adjuvant chemotherapy with MWA increased recurrence-free survival 7-fold in large-HCC patients compared to MWA alone [Citation26]. RFA has been applied to animal tumour models with concomitant liposomal doxorubicin [Citation27], paclitaxel [Citation28], and quercetin [Citation29] achieving a 1.2–1.5-fold increase of apoptosis compared to each treatment alone. An ongoing phase II clinical trial combines sorafenib, a Raf kinase inhibitor, given orally twice daily with RFA to treat medium-sized HCC (clinical trial no. NCT00813293). The benefit of neoadjuvant sorafenib to RFA efficiency remains to be seen.
Heat shock proteins (HSPs) are induced under thermal stress caused by hypo- or hyperthermic ablation, which may lead to thermo-tolerance [Citation6,Citation30,Citation31]. Two major HSPs, Hsp70 and Hsp90, have been characterised in several animal tumour models. For example, in a renal tumour model these two HSPs are up-regulated at the ablated margins. Hsp70 displays a 3–8% increase up to 7 days after both CA and RFA, whereas Hsp90 shows an even higher elevation up to 15% in 3 days, but only with RFA [Citation30]. However, significant suppression of Hsp70 or Hsp90 has yet to be achieved to prevent post-ablation recurrence. Our group is particularly interested in Hsp90, an endogenous protein accounting for 1–2% of total cytosolic protein [Citation32] and elevated 2–4-fold in viable, thermal stressed cells [Citation33]. In human HCC tissue samples, Hsp90 has shown a 7-fold up-regulation compared to basal hepatocyte levels [Citation34]. More importantly, Hsp90 chaperones over 200 oncogenic proteins such as receptors for all the growth factors mentioned above, and various kinases including mTOR, Akt, and HIF-1 [Citation35] are critical for tumour angiogenesis [Citation36], invasion, and progression [Citation37]. Although it remains to be revealed how Hsp90 is directly involved in the ablation-altered tumour microenvironment, compounds for Hsp90 suppression hold potential to further increase ablation-induced tissue coagulation and, most importantly, eradicate the thermally stressed residual viable cells at the HCC tumour margins.
In this study, geldanamycin (GA), a potent Hsp90 inhibitor, was used as an adjuvant chemotherapeutic in combination with thermal ablation, to induce apoptosis by destabilising client proteins critical for tumour survival [Citation38–40]. Past studies with GA have been plagued with solubilisation issues and acute systemic toxicity. To solubilise this extremely hydrophobic agent (∼20–50 μM in water) and lower systemic toxicity, we proposed established, thermo-responsive, elastin-based biopolymers to carry high concentrations of GA for focal, locoregional drug targeting to hyperthermic tumour margins (41–45 °C). Briefly, elastin-like polypeptides (ELPs) are pentapeptide repeats of (VPGXG)n where guest residue ‘X’ may be any amino acid except proline [Citation41]. Native ELP homopolymers, genetically engineered ELP polymers, and ELP-GA conjugates all boast unique thermo-transitive properties revolving around a distinct inverse phase transition temperature (Tt) [Citation42–44]. At T < Tt ELP-GA bioconjugates are water soluble, linear, random coils; conversely, at T > Tt the bioconjugates undergo a rapid intra-/intermolecular hydrophobic collapse to be exploited as a targeted macromolecular delivery system at hyperthermic tissue sites.
Here we present the production of a new generation of ELP-based triblock biopolymers comprising 15 (D) aspartic acids on the C-terminal end to which GA is conjugated via pH-sensitive hydrazone linkers. This system is designed for site-specific targeting of hyperthermic tumour margins and pH-sensitive GA release in the acidic tumour microenvironment. Hsp90 inhibition levels and anti-tumour growth levels were evaluated as a function of ELP-GA concentration with concomitant hyperthermia. Clinically established electrocauterisation was the thermal ablation method of choice for tumour elimination in an HCC murine model. The results confidently demonstrated that ELP-GA bioconjugates were more efficient than free drug, 17-AAG (GA analogue) in preventing tumour recurrence. In addition, combined thermal ablation with ELP-GA chemotherapy proved superior to either treatment alone, demonstrating great potential in preventing HCC recurrence and improving patient survival.
Materials and methods
Nomenclature
ELPVAn stands for ELPs with different repeats of ELPVA monomer. ELPVA has a guest residue ratio of valines to alanines (V:A) of 1:10, e.g. ELPVA20 is a dimer of ELPVA with 22 amino acids: two valines and 20 alanines. ELPVA40 has four repeats of ELPVA with four valines and 40 alanines.
ELP1 library has a guest residue ratio of valines to glycine to alanines (V:G:A) = 5:3:2, e.g. ELP1-60 stands for six repeats of V5G3A2, 60 amino acids in total. ELP diblock copolymer (no spacer): ELP1-60-D15. ELP triblock copolymer: ELPVAn-60-(DADAV)7D. The ELP-GA conjugates, unless otherwise stated, stand for ELPVA40-60-(Asp)8GA7. ELP-fluorescein conjugates: ELP40-60-(Asp)6Fl9.
Cloning, expression, and purification of triblock ELP biopolymers
Previously we constructed a diblock ELP copolymer (ELP1-60-D15) and achieved rapid transition kinetics [Citation44]. However, the Tt of this copolymer is about 38 °C, which is not high enough to avoid aggregation upon systemic injection. Based on the physicochemical properties of ELP biopolymers, Tt can be raised to hyperthermic temperatures (41–45 °C) by constructing a new biopolymer with a more hydrophilic guest residue, or by making shorter ELPs with smaller molecular weight, and/or decreasing the polymer concentration or the ionic strength of the surrounding medium. We chose to add a hydrophilic block upstream of the ELP1-60 since decreased biopolymer concentration or molecular weight will hinder the acuteness of phase transition. Additionally, there are no charged residues on ELPs and the ionic strength of the blood is fixed. In order to have more control of the Tt with efficient cloning process, we inserted multiple hydrophilic ELPVAs at N-terminus of ELP1-60 (ELPVAn-ELP1-60). One valine was designed for cloning feasibility, and repeats of alanine, although not as hydrophilic as glycine, retained robust expression achieving high yields (>50 mg/L culture), as observed by other groups [Citation45]. We also inserted alanines between the aspartic acids (DADAV) to avoid steric hindrance to drug conjugation. Therefore, the sequence of our final construct from N to C-terminus was [ELPVAn-60-(DADAV)7D].
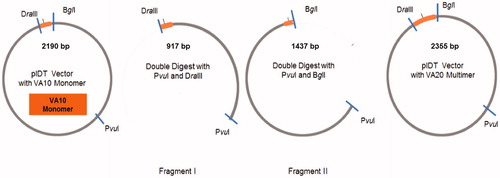
To construct an ELPVA library, three type II restriction enzymes were used to seamlessly link two VA10 monomers. Briefly, a cDNA of ELPVA monomer containing VA10 and a PvuI restriction site was synthesised by the core facility at the University of Utah. Four oligos comprising VA10 monomer were separately synthesised. Oligo sequences are provided in supporting information. Annealing was completed by 5 min of heating at 95 °C, followed by a slow cool down to 25 °C over 3 h. The cloning vector (pIDT) was first double digested with PvuI and DraIII to yield vector fragment I. Fragment II was simultaneously obtained by PvuII and BglI digestion. CIP was added to each digestion to prevent self-ligation. After digestion for 1 h at 37 °C, the bands were separated on agarose gel (1.5%, 90 V, 1 h). Each fragment with the expected size was cut and DNA was extracted from the gel using a QIAGEN extraction kit. The compatible ends of BglI and DraIII were recognised by ligase and the two fragments, each carrying one VA10 monomer, were ligated to obtain a new vector having VA10 dimers (VA20) (). Additional ELPVA repeats (e.g. ELPVA30, ELPVA40) were obtained by the same method. The ELPVAn was then inserted at the N-terminus of ELP1-60 using the DraIII recognition site. The block for drug conjugation – (DADAV)7D – was placed at the C terminus of ELP1-60 using two oligos (sequences in supporting information) synthesised by the Core facility at University of Utah.
Figure 1. Cloning scheme for building up ELPVAn library. The pIDT vector with VA monomer was first double digested with PvuI and DraIII to yield vector fragment I. Fragment II was simultaneously obtained by PvuI and BglI digestion. The compatible ends of BglI and DraIII were recognised by ligase and the two fragments, each carrying one VA10 monomer, were ligated to obtain a new vector having VA10 dimers (VA20)(I). Fragment II was simultaneously obtained by PvuI and BglI digestion. The compatible ends of BglI and DraIII were recognised by ligase and the two fragments, each carrying one VA10 monomer, were ligated to obtain a new vector having VA10 dimers (VA20).
The annealing and insertion of (DADAV)7D was performed the same as D15 (without spacers) in our previous construct [Citation44]. The final DNA with both ELPVAn (N-terminal) and (DADAV)7D (C-terminal) was verified by both a diagnostic digest and DNA sequencing. The completed construct was then digested with NdeI and HindIII, transferred to a pET25b expression plasmid, and transformed into BLR cells for expression. A single colony on the Petri dish was inoculated in 5 mL Circlegrow (Q-BIOgene, Carlsbad, CA) media containing 60 mg/mL carbenicillin at 240 rpm and 37 °C. After overnight shaking, the cell culture was centrifuged at 4000 rpm at 4 °C for 10 min to separate dead cells in the supernatant. The resultant pellet was resuspended in fresh media, and transferred to 4-L culture flasks and shaken under the same conditions for another 24 h for large-scale production. Typical yields of the triblock ELP biopolymer were 75–100 mg/L culture.
The purification of the ELP was realised by three rounds of hot/cold spin cycles (inverse transition cycling) as previously reported [Citation44]. The purity and precision of ELP triblock copolymers were characterised by both SDS-PAGE and mass spectrometry.
Synthesis and characterisation of triblock ELP-GA conjugates
Aldehyde-modified geldanamycin (GA-CHO) was conjugated to carbohydrazide-modified ELP triblock copolymer to yield a pH-sensitive (hydrazone) bond [Citation44]. The conjugation ratio (GAs per biopolymer) was calculated by both ultraviolet (UV) spectrometry (Agilent, Santa Clara, CA) and electrospray ionisation mass spectrometry. Particle size is an extremely delicate parameter; therefore, samples were solubilised in molecular biology grade water and measured by dynamic light scattering at 25 °C with a laser goniometer (Brookhaven, Holtsville, NY). The surface charge densities of both free biopolymer and biopolymer-GA conjugates were acquired on a Zetasizer Nano ZS (Malvern, Westborough, MA). To ensure thermo-targeting of hyperthermic tumour margins (∼41–45 °C), rapid thermal kinetics within this temperature range were essential. The thermal transition behaviours of the biopolymer alone and the biopolymer-GA conjugates were monitored using UV-Vis spectrophotometry as previously described [Citation44]. Briefly, 25 µM triblock ELP or ELP-GA equivalent conjugates were solubilised in phosphate buffered saline (PBS) and heated at a speed of 1 °C/min from 25–85 °C, and absorption at 650 nm was recorded as a function of temperature. To avoid false-positive absorption from GA 650 nm was chosen to monitor the change of optical density of ELP. The thermal Tt was calculated as the temperature at the maximum derivative of turbidity at 650 nm. All measurements were conducted in triplicate and expressed as mean ± standard error.
Drug release profile of GA-ELP biopolymer conjugates
The pH release kinetics at both acidic and neutral pH were conducted on an Alliance (Waters, Milford, MA) RP-HPLC column equipped with an absorbance reader. Aliquots of 25 μM ELP conjugates (concentration based on the ELP) were prepared in 1 mL of PBS at pH 5.0 or pH 7.4 and shaken at 37° or 43 °C at 180 rpm. Samples of 100 μL (cooled on ice first) were loaded at 0.5, 2, 6, 12, 24, 48, and 72 h with a 75 μL injection volume. ELP-GA conjugates and free GA-CHO were separated on a diphenyl column (Waters, Milford, MA) using an H2O/80% dichloromethane (DCM) gradient with 0.1% v/v trifluoroacetic acid. Initially, the binary gradient was 80:20 for 10 min at which point the 80% DCM concentration was raised to 100% over 35 min followed by a static 15 min period. The flow rate was 0.5 mL/min. Percentage of GA release as a function of time was based upon GA peak area under the curve normalised to the 0 h time point (n = 3).
Cytotoxicity
The Cell Counting Kit-8 (CCK-8, Dojindo, Rockville, MD) was used to evaluate cell viability through assessing mitochondrial dehydrogenase activity of cells treated with 17-AAG, the clinically tested GA analogue, GA-CHO, free ELP triblock biopolymer, and ELP-GA conjugates at both 37° and 43 °C. Briefly, HepG2 (in Dulbecco’s modified Eagle’s medium (DMEM)) and Hep3B (in MEM) cells were seeded onto 96-well culture plates (5000/well, n = 3) until 70% confluence. Thermo-treated groups were incubated at 43 °C for 30 min while the normothermia groups were incubated at standard 37 °C. After 10 min of recovery, each well was treated with 100 µL drug-containing solution. Specifically, 17-AAG and GA-CHO were freshly solubilised in 10 µL DMSO while ELP and conjugates were solubilised in 10 µL PBS; 90 µL media was added to each solution to reach ELP-normalised concentrations ranging from 10 nM to 10 µM. Cells were further incubated at 37 °C for another 48-h period before the CCK-8 assay. Here, 30 µL of diluted CCK-8 agent were added to each well and further incubated at 37 °C for 1 h. As per the vendor’s instructions, absorption at 450 nm was measured using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA). The cytotoxicity is presented by IC50 values defined as the drug concentrations eliciting a 50% decrease in absorbance of untreated control wells.
Cell uptake
Conjugation of fluorophore to biopolymers
Carbohydrazide modified ELP was conjugated with Atto 488 N-hydroxy-succinimidyl-esters (NHS ester) (Sigma Aldrich, St Louis, MO) to study the cell uptake after hyperthermia and normothermia. Briefly, Atto 488 NHS was dissolved in DMSO at a concentration of 10 mg/mL; 150 µL of dye solution was then mixed with 25 mg ELP in 2 mL Na2HCO3 buffer (0.1 M, pH 8.3 ± 0.2) and stirred in the dark at 25 °C for 1 h. Free dye was removed by filtration through a PD-10 column (GE Healthcare, Little Chalfont, Buckinghamshire, UK) and dialysis overnight against PBS. The final product was lyophilised for long-term storage. The concentration of loaded dye was measured using a Nanodrop spectrometer (Thermoscientific, Tewksbury, MA), which has built-in software for widely used fluorescein. The loading capacity was calculated by concentration after lyophilisation and divided by total concentration in the start solution.
Flow cytometry and microscopy
Cellular uptake was monitored using flow cytometry. Hep3B cancer cells were grown on a 6-well plate until 70–80% confluent. Endocytosis inhibitors (amiloride (0.08 mg/mL), mβCD (2.0 mg/mL), and chlorpromazine (10 µg/mL) inhibiting macropinocytosis, caveolae, and clathrin-mediated endocytosis, respectively) were administered, and cells were incubated at 37 °C for 30 min. Hyperthermia was performed after the endocytosis inhibition for 30 min at 43 °C. Free dye in 1% DMSO, ELPVA40-60-Fl (1 µM equivalent dye) in PBS was administered to the cells and incubated for an additional 4 h and 48 h. Cells were thoroughly washed in PBS three times, followed by trypsinisation and resuspension in fresh PBS before cell sorting.
For nucleus staining by fluorescence, each treated well was stained with one drop of Hoechst stain (Invitrogen, Carlsbad, CA) for nucleus visualisation. Excessive dye in each well was washed away with PBS five times before fluorescence microscopy (Olympus IX71Ftware, PA). All acquired images were processed by Image J software (National Institutes of Health, Baltimore, MD).
Co-localisation of polymer-conjugates with lysosomes
The co-localisation of fluorescence and lysosomes was monitored using confocal microscopy (Nikon A1R, Carlsbad, CA) at 60× magnification. Hep3B cells were cultured on a Lab-Tek 8-chamber cover-glass (Thermo Scientific, Tewksbury, MA) system at a density of ∼10 000 cells/chamber. After cells reached 70% in confluence, 2 µL of LysoTracker RFP (Invitrogen) was administered for lysosome staining as a result of transfection. After overnight incubation, the cover glass system was placed on a Nikon A1R confocal microscope and heated at 43 °C for 30 min (5% CO2 maintained). The temperature was then returned to 37 °C and 1 µM equivalent Atto 488 (Fl) or conjugated ELPVA40-60-Fl was administered to the labelled cells. Before imaging, cell nucleuses were labelled with Hoechst stain (50×). Images were taken at 60× with fixed positions every hour up to 12 h, using the following excitation/emission wavelengths: for Hoechst 350/461 nm, for Atto-488 fluorescein 498/520 nm, for lysosome-RFP 555/584 nm. The level of co-localisation was calculated using Image J software by computing the overlap of individual pixels from two fluorescence channels. The effect of endocytosis on co-localisation was evaluated using the same endocytosis inhibitors as in the cell uptake study.
Western blotting of Hsp90
To evaluate the inhibition of each drug, 20 000 cells were cultured in 6-well plates and grown until 70–80% confluent. Cells in each well were lysed with 150 µL sodium dodecyl sulphate (SDS) buffer (0.1 M Tris HCl, pH 8.3, 1% protease inhibitor). Total protein concentration was determined by BCA assay as per vendor instructions (Thermoscientific); 10–20 µg total protein was separated on a 4–12% Bis-Tris SDS gel (Invitrogen) and transferred to a polyvinylidene difluoride membrane for electroblotting. Primary antibodies against rabbit Hsp90 (1:1000) and β-actin (1:2000) were incubated at 4 °C overnight, followed by a 1-h incubation of rabbit anti-mouse IgG secondary antibody. The membranes were washed three times with Tris-buffered saline and Tween 20 buffer, incubated with a chemiluminescent reagent (ISC Bioexpress, Kaysville, UT), and visualised on a FluoroChem FC2 camera (Alpha Innotech, San Leandro, CA).
Tumour implantation
HepG2 cells were cultured in DMEM media in a 175 cm2 tissue flask until 70–80% confluent. Cells were washed with PBS, trypsinised, and centrifuged at 1000 rpm for 3 min. Cells were reconstituted in cold DMEM-Matrigel (BD Biosciences, San Jose, CA) solution (50:50, v/v). The right flank of each nude mouse was sterilised with isopropyl alcohol prior to injecting a 200 µL suspension (2–3 × 106 cells) under general anaesthesia. Animal weight and tumour size were recorded twice weekly according to the approved US Institutional Animal Care and Use Committee protocol.
Drug treatment and in vivo assessment of anticancer efficacy of ELP-GA biopolymer-drug conjugates
Tumours reaching 5 mm in all dimensions were subsequently intravenously injected with 200 µL of PBS, 17-AAG, or ELP-GA conjugates (3.7 mg/kg equivalent GA dose) twice weekly (for a total of seven doses) through the tail vein. Animal weight and tumour sizes were monitored up to 21 days after treatment. Tumour volume was calculated by 0.5 × (length of tumour) × (width of tumour)2.
Electrocauterised thermal ablation
Each animal was anaesthetised via inhalation of 5% isoflurane with a nose cone and subsequently placed on a sterile electrical grounding pad. A heating lamp was placed beside the animal to keep it warm during anaesthesia. The tumour area was sterilised with an alcohol swab and a 1-cm small incision was made to expose the tumour. Cauterisation was accomplished with an ESU-30 system (Olympus, Miami, FL) at 25 W with coagulation mode with an electrical pencil (Covidien, Mansfield, MA) placed in the centre of the tumour. To ensure that we could achieve hyperthermic margins, we first conducted a thermometry study where a temperature probe (Fluke, 80PK-25, Everett, WA) was placed consecutively at three positions (∼5 mm away from the pencil tip) and temperatures were recorded after demarcation appeared. The electrical surgery unit is able to achieve an ablated margin around 42 °C (supporting information). For tumour treatment, the duration of cauterisation was dependent on the tumour size and a 3–5-mm peripheral margin was left unablated. After the ablation 200 µL Rimadyl (50 mg/mL) was subcutaneously injected and the incision was closed with a surgical staple (3 M Health Care, St Paul, MN).
Combination therapy of electrocauterisation and thermosensitive biopolymer-GA conjugates
To evaluate additive and/or synergistic effects of combination therapy, each sample (PBS, 17-AAG, ELP-GA) was initially normalised to 3.7 mg/kg GA equivalents and intravenously injected. We kept the same dose in the combination while the flank HCC tumours on the mice were first cauterised and the tumour core coagulated. The injection of therapeutics was then performed immediately after the cauterisation and wound closure. Three days following the first dose a second dose was administered, with two more doses injected twice weekly. Physical activity and eating/drinking habits were monitored daily. The animal weight and tumour size were recorded twice weekly up to 70 days.
Histology and Hsp90 immunohistochemistry
Animals were euthanised when a tumour reached 2 cm in any dimension, or >20% loss of body weight was observed, or one day after the last treatment by CO2 inhalation, and the tissue samples were collected promptly. Briefly, a 0.5 cm incision was performed on the flank skin to expose the tumour. The fascia was removed and the tumour was washed three times with PBS. Two pieces from the ablated tumour core and tumour margins (tissues next to the demarcation, 3–5 mm long, and 100–200 mg), were excised from each animal. One piece of sample was immediately put into 10% neutral buffered formalin for 36–40 h. Fixed tissues were then transferred to 70% alcohol for storage prior to paraffin embedding. Tissue samples were dehydrated through graded alcohol and xylene, embedded in paraffin and sectioned at a thickness of 5 µm with a microtome. Ten slices were obtained per sample. Each slide was either stained with haematoxylin and eosin (H&E) for gross histopathological analysis or processed for Hsp90 immunochemistry (ARUP Laboratories, Salt Lake City, UT). Briefly, slides were pre-treated with monoclonal rabbit anti-Hsp90 (Cell Signaling Technology, Danvers, MA) for 60 min, followed by a 20-min incubation of mouse IgG blocker (BioCare Medical, Concord, CA) to block endogenous mouse IgG. The primary antibody was then applied at a dilution of 1:50 for 2 h at 37 °C. A secondary antibody (mouse F(ab)2) (Dako, Carpinteria, CA) was applied for 4 min at a dilution of 1:100. The slides were imaged with an IVIEW DAB detection kit (Ventana Medical Systems; Tucson, AZ) and visualised under optical microscopy with 10× and 40× magnification (assisted by Theodore Pysher, Division Chief of Pediatric Pathology, University of Utah). The relative Hsp90 expression was analysed using ImageJ. For each slide three random positions were selected at the tumour margins with the same areas of pixels to calculate Hsp90 intensity relative to untreated control.
The second section (100–200 mg) from the same tumour sample was flash frozen in liquid nitrogen and homogenised for western blot. Tumour tissues were homogenised in lysis buffer (100 mg tissue/mL) for 20 s on ice and immediately centrifuged at 12 000 rpm for 10 min at 4 °C. Protein concentrations were determined by BCA assay and samples loaded onto an SDS protein gel followed by electroblotting and processed as described above.
Statistical analysis
One-way analysis of variance (one-way ANOVA) with the Bonferroni post hoc analysis was used to compare three or more groups. A Student’s t-test was used for two groups; p < 0.05 indicated statistically significant differences between the groups.
Results
Characterisation of triblock ELP biopolymers and conjugates
To avoid potential aggregation upon systemic injection, we introduced a hydrophilic ELPVA library to the diblock polymer (ELP1-60-D15) to increase the Tt to the hyperthermic range (41–45 °C). The insertion of more VA repeats, as we expected, increased hydrophilicity (determined as solubilisation and no precipitation) of the polymer and resulted in elevated Tt. Briefly, the smallest VA polymer, VA20, has a Tt at 39 °C and the Tt rises to 43 °C for larger and more hydrophilic polymer VA40, as shown in . Thus, we chose the VA40 construct for drug conjugation and future experiments.
Figure 2. (A) Physicochemical properties of a new breed of ELP-VA library. The insertion of VA increases the molecular weight and hydrophilicity of ELP triblock polymers. All ELP-triblock copolymers were evaluated at 25 µM in PBS (pH 7.4, 147 mM). (B) Purity and molecular weight of the triblock polymer were displayed in a SDS gel. The thermo-responsiveness behaviors were evaluated in PBS and 10% serum after conjugation. The conjugates maintain the acute responsiveness within the desired hyperthermia range (41–45 °C) for combination therapy in the presence of serum. ELP triblock copolymer and conjugates were evaluated at 25 µM (ELP equivalent) in PBS (pH 7.4, 147 mM). (C) Size distribution of ELP-GA conjugates monitored by DLS. Conjugates having 25 μM equivalent ELP were solubilised in PBS (pH 7.4, 147 mM) and measured at 25 °C, n = 3.
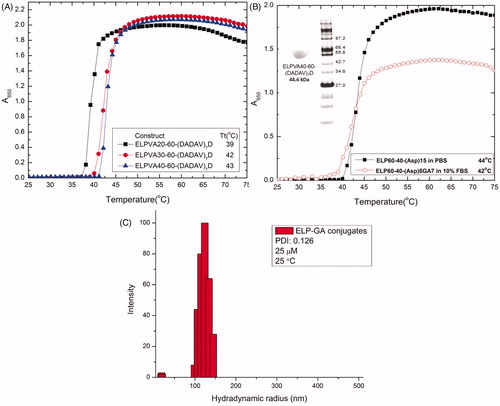
The insertion of alanine spacers between aspartic repeats [(DADAV)7D] reduces the steric hindrance for drug conjugation and allows more aldehyde modified GAs to access and react with the primary amines conjugated on aspartic acids. Therefore, the conjugation ratios (47–53%), although not dramatically increased, were more consistent and reproducible than our previous di-block and no-spacer construct (ELP1-60-D15). The size (hydrodynamic radius) and surface charges (zeta potential) of all ELP polymers and GA conjugates were summarised in . The increasing number of VA repeats showed gradual reduction of the hydrodynamic radius despite the increase of molecular weight. The conjugates displayed an approximate size of 116 nm and neutral surface charge, which may pass into the tumour through its leaky vasculature to exploit the enhanced permeation and retention (EPR) effect [Citation46]. The attachment of hydrophobic GA also results in the assembly of nanoparticles as shown in .
Table 1. Physicochemical characterizations of ELP homopolymers, triblock copolymers and ELP-GA conjugates.
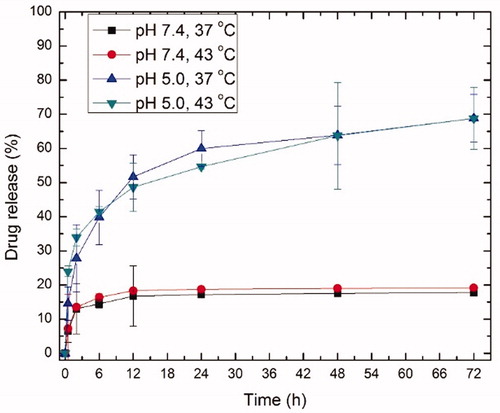
The pH sensitivity of the conjugates was characterised in both neutral and acidic buffers. A pH of 5.0 was chosen to mimic endosomal/lysosomal pH [Citation47] where the drug is cleaved. The drug release was significantly higher (∼70%) and faster in the acidic environment compared to normal physiological pH (∼15%) within 72 h ().
Figure 3. Release profile from ELP-GA conjugates at neutral and acidic pH under both normothermia (37 °C) and hyperthermia (43 °C). The concentration of GA and ELP free polymer were derived from the standard curves at 337 and 220 nm, respectively. Using the binary gradient, ELPVA40-60-(Asp)15 copolymer (Rt = 22 min) and free GA-CHO (Rt = 27 min) can be separated according to high performance liquid chromatography. We calculated the percentage of GA release as a function of time based upon a drop in the conjugate peak area percentage, normalised to the 0 h time point. These runs were repeated three times for each ELP-GA conjugate.
Cytotoxicity and Hsp90 inhibition of ELP-GA conjugates
To evaluate the cytotoxicity of all drugs, we used the CCK-8 assay to measure the level of formazan, which can be extrapolated to represent the percentage of viable cells. The IC50 value was defined as the concentration causing 50% cell death of the non-treated controls. Aldehyde modified GA (GA-CHO) showed slightly better cytotoxicity than the clinically tested GA analogue, 17-AAG, with the lowest IC50 around 160 nM. The triblock copolymer ELP was not toxic within the tested concentrations, but the resultant conjugate maintained strong cytotoxicity with IC50 below µM range, as shown in .
Table 2. Cytotoxicity of free drug, free ELP polymer, and ELP-GA conjugates to HCC cell lines.
Hyperthermia further enhances the cytotoxicity of GA and conjugates by 1.3–3.0-fold. GA has shown elevated toxicity upon hypothermia in various cancer cell lines including breast cancer [Citation48], melanoma [Citation49], and prostate cancer cells [Citation50]. Here, we achieved ubiquitous toxicity enhancement in both HCC cell lines. The increased toxicity can be attributed to the following reasons. First, hyperthermia is known to increase cell membrane mobility and permeability [Citation51], which allow more efficient cellular uptake for large molecules [Citation52]. At the same time, thermo-sensitive ELP conjugates form aggregates, which can be internalised more easily into the cells as a result of increased hydrophobicity, and function as a drug depot to kill the cells around aggregate domains. Those cells further increase the sensitivity of adjacent cells to chemotherapy (bystander effect) to cause vast and efficient cell depletion. Finally, the hyperthermia synergistically enhances the cellular ATPase activity which directly links to the favoured mechanism of Hsp90 and increases the rate of GA binding to Hsp90 to elicit the cytotoxic effect.
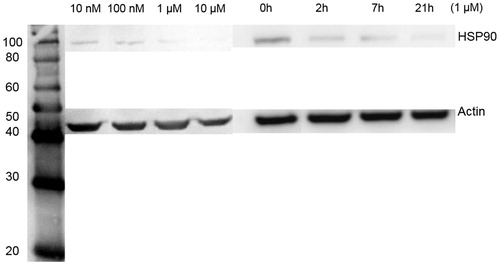
Hsp90 level was measured in the cells treated with the same conditions in the cytotoxicity studies. shows a dose- and time-dependent inhibition of Hsp90 by ELP-GA conjugates. The cellular Hsp90 was depleted at 21 h around the IC50 concentration, further substantiating that effective Hsp90 suppression leads to efficient cell death.
Figure 4. Quantitative western blotting of Hsp90 expression in HepG2 cells after treatment of ELP-GA conjugates as a function of dose and time. (A) Samples were taken at 48 h after each concentration (as in the cytotoxicity studies). Hsp90 was inhibited at a dose of 1 µm GA equivalent. (B) Hsp90 level as a function of time, treated by conjugates with a 1 µm GA equivalent concentration.
Characterisation and cellular uptake of ELP-fluorescent (ELP-Fl) conjugates
Since Hsp90 is mostly present in the cytosol we confirmed that conjugates are encapsulated inside the cells and released GA in the cytoplasm. To visualise the ELP-based polymer, Atto 488, a stable and reliable imaging agent, was linked to the ELP triblock copolymer backbone as a mimic of the ELP-GA conjugates to probe the extent and mechanism of cellular uptake. The same conjugation chemistry and purification was used for the ELP-Fl conjugate, which yielded a conjugate similar to GA’s, as shown in .
Table 3. Physicochemical characterization of ELP-Fl conjugates.
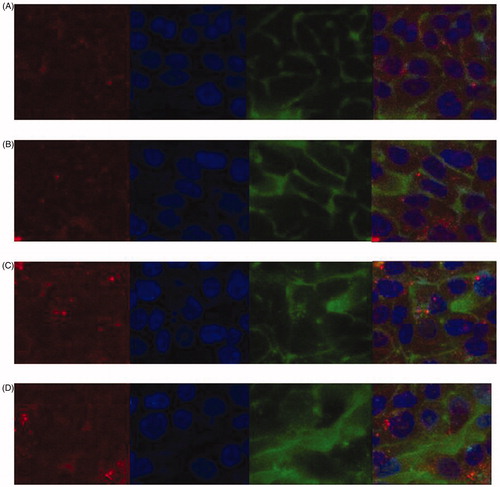
Both confocal microscopy and flow cytometry were conducted to examine the level of internalisation to the cytosol and lysosome. shows the cellular uptake by both free and conjugate after a 30 min treatment at 43 °C. The conjugate showed enhanced uptake at both 4 h and 48 h, during which 60% of the drug was still bound to the ELP backbone (confirmed by release studies using high-performance liquid chromatography). At the same time, lysosome-RFP was used to label the lysosome and monitor the co-localisation level of fluorescence and lysosome up to 48 h. Peak localisation (20%, calculated by ImageJ) was observed for conjugates while only trace levels were detected for the free fluorescent dye.
Figure 5. Confocal microscopy images of cell uptake of fluorescein only (Fl, panels A and B) and ELP-fluorescein (ELPVA40-60-(Asp)6Fl9 conjugates, panels C and D). Images were taken after 4 h (panels A and C) and 48 h (panels B and D) incubation. For each panel, images from left to right showing lysosome (red, lysosomes-RFP); cell nuclei (blue, Hoechst); fluorescein (green, Fl or ELP-Fl); merge (yellow and orange). scale bar: 20 μm.
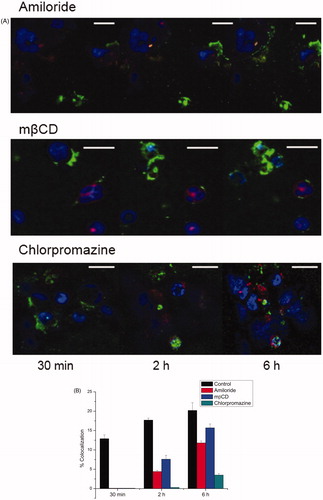
Localisation is important due to the fact that low pH of the lysosome provides cleavage of GA from the ELP backbone for therapeutic effect. To examine the molecular mechanisms of internalisation, three inhibitors: amiloride, mβCD, and chlorpromazine, suppressing three major pathways of endocytosis, were applied to the cell culture. All inhibitors decreased the level of co-localisation as shown in , suggesting that mixed and dynamic internalisation pathways exist for ELP-based biopolymers.
Figure 6. Co-localisation studies monitored using confocal microscopy and analysed by Image J. (A) Representative confocal images of live cells taken at 30 min, 2 h, and 6 h after endocytosis inhibition. Colour identification: lysosome (red, lysosomes-RFP); cell nuclei (blue, Hoechst); fluorescein (green, Fl or ELP-Fl); merge (white). (Scale bar, 20 μm). (B) Level of co-localisation with lysosomes as calculated from over-lapping pixels of lysosome-RFP and Fl divided by total Fl pixels.
Temperature also plays a major role in cellular uptake. As observed by other researchers, hyperthermia increases the fluidity of the cellular membrane [Citation53], which facilitates the dynamic interaction of ELP with the cell membrane. A higher temperature also contributes to increased cellular uptake [Citation54,Citation55] allowing targeted and efficient cancer cell abrogation.
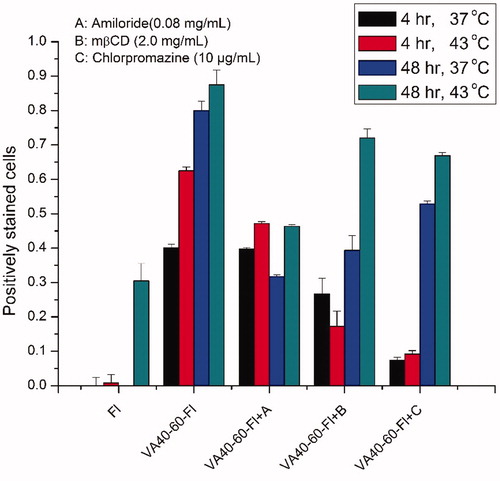
Hence, cellular uptake was confirmed using flow cytometry at the same dose under normo- and hyperthermia. Cells were analysed at both 4 h and 48 h post-treatment. Again, the free dye signal was sparse after extensive washing, while the conjugates maintained strong signals up to 48 h. The same endocytosis inhibitors were used to examine the mechanism of internalisation. We found that all inhibitors had some effect on endocytosis as shown in . Chlorpromazine, which inhibits the clathrin-mediated endocytosis pathway [Citation56], displayed the most suppression of cellular uptake, similar to the findings in confocal microscopy studies.
Figure 7. Fluorescent dye uptake of Hep3B cells after 30 min of endocytotic inhibition. The percentage of gated fluorescing cells was quantified by flow cytometry at 4 h and 48 h after treatment of conjugates and equivalent doses of fluorescein (1 µm). The results are shown as mean ± SD, n = 3.
Anti-tumour efficacy and histological evaluation of heat shock inhibition
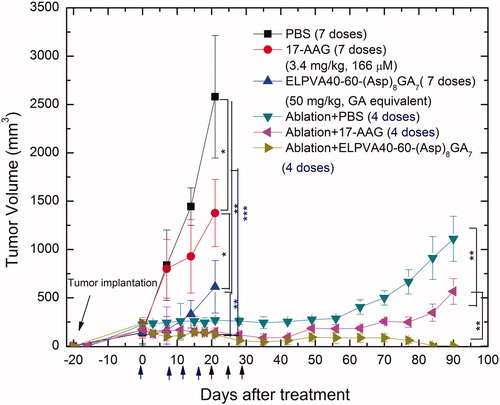
Tumour growth rate for each treatment is summarised in . Due to the aggressive nature of HCC, most animals in the PBS group were quickly (15–20 days) subject to euthanasia as the tumour size reached 2 cm in one dimension. The conjugates and free drug displayed limited efficacy in tumour stasis. However, the ELP-GA achieved slower tumour growth than the free drug (p < 0.05), which is in accordance with the level of Hsp90 inhibition.
Figure 8. Tumour growth following treatment with intravenous free 17-AAG, ELP-GA conjugates, and/or ablation (electrocautery). With a total of seven doses given twice weekly, the conjugates alone exhibited slower tumour growth compared to PBS or 17-AAG. Combination of ablation and four doses of adjuvant therapy (either with 17-AAG or ELP-GA or PBS) resulted in tumour regression but only the ELP-GA conjugates achieved no tumour recurrence up to 90 days (The results are shown as mean ± SD, n = 4. *p < 0.05, **p < 0.01, ***p < 0.001).
All mice tolerated the drug throughout the course of the treatment since the doses were given at 50% of the maximum tolerated dose (MTD). No severe distress, weight loss or deaths were observed. There are some side effects for the combination therapy. First, the mice experienced 3–5% weight loss the first day after treatment. The activity level was restored with time, but they displayed slower weight gain compared to untreated controls. The mice also showed some reduced physical strength in clawing and declined response to other stimuli.
Before the combination therapy, we did a thermometry study to confirm that the electrocautery was effective in killing the tumours. As shown in , the power and duration of the cauterisation achieved >50% tissue necrosis; there are distinct regions where clusters of cells remained viable. Indeed, one of the major limitations for all ablation modalities is that the heat was not evenly distributed and was significantly affected by the tumour size, location, cell type, and adjacency to large blood vessels. As a result, Hsp90 immunohistochemistry showed variable intensity of staining. The levels of Hsp90 expression throughout the whole tumour were confirmed by the western blot in where ELP-GA conjugates displayed the most inhibition of Hsp90 compared to other treatments. No tumour growth was observed over 80 days after the reduction of tumour achieved in the first 10 days of treatment. However, slow tumour growths (compared to non-treated control) at the ablated tumour site were observed in ablation (electrocautery) alone and with the 17-AAG group. Therefore, the conjugate played a critical role in preventing tumour recurrence by superior Hsp90 inhibition in tumours in general (), as well as in the marginal cells (viable cells adjacent to necrotic cells), as shown in .
Figure 9. (A) Histology and Hsp90 immunohistochemistry of tumour margin samples taken after combination therapy. Square on low magnification is the region of high magnification image. Original magnification ×25. (B) Hsp90 expression levels after each combination treatment (normalised to ablation-only controls). The results are shown as mean ± SD, n = 3 (**p < 0.01). (C) Quantitative western blotting of Hsp90 in tumour samples excised after ablation only (tumour core, margin, 4 h post-ablation), and from tumour margins after combined treatment (electrocautery with PBS, 17-AAG, and ELP-GA) following the last dose. (D) Hsp90 expression levels from margin cells (viable cells adjacent to necrotic cells) after combination treatment. The results are shown as mean ± SD, n = 3 (**p < 0.01). Bars in panels B and D represent quantitative analysis from immunohistochemistry studies.

Discussion
Primary HCC accounts for more than 90% of liver cancers and the 5-year survival rate remains as low as 12% [Citation57]. Despite the higher response rates and longer survival afforded by current treatment modalities, the vast majority of patients die due to recurrent tumours or distant metastasis. Thermal ablation treatment is widely used in HCC patients who are not eligible for surgery or transplantation. However, prevention of post-ablation recurrence remains underexplored and requires targeted molecular intervention for better prognosis.
Thermal ablation is effective in killing cells at hyperthermic conditions [Citation58]. At temperatures > 60 °C, almost all cells undergo coagulation necrosis with massive protein denaturation and DNA damage [Citation59]. With advanced mathematical simulation and technical developments, faster hyperthermic kinetics, better probe design, and larger coagulation zones have been achieved in major ablation modalities [Citation60–62]. Paradoxically, tumour thermal ablation has also been shown to enhance tumour progression and recurrence, regardless of the extent of ablation [Citation30]. Not all tumour cells can be ablated. It has been shown that the location of the tumour has been one of the risk factors of incomplete tumour ablation and recurrence [Citation54]. Specifically, tumour cells adjacent to large blood vessels not only have better nutrient supplies, but also receive less heating, either due to the position of the probe (usually away from major vessels to avoid bleeding) or to the heat sink effect, which is more pronounced in liver cancer due to its abundant vasculature. Limited clinical efficacy also persists among large tumours (>3 cm) [Citation63,Citation64], which cannot be overcome even by multiple ablations. More importantly, the cells further from the ablation tip, especially those at the periphery of ablated tumours, will not experience coagulation necrosis, but rather sub-lethal hyperthermia or mild heat stress. In such cases, protein denaturation and DNA impairment occur at a much lower level [Citation65]. The cells are able to express and secrete multiple cellular factors to minimise the impact of thermal stress for survival and serve as new niches for neoplastic growth [Citation25].
In this study we used electrocauterisation, a simplified but widely used clinical ablation device, to ablate tumours in a HCC murine model. Up to 50% tumour relapse was observed after a single ablation treatment, despite strong evidence of cell killing by histology studies. In order to increase ablation efficiency and decrease tumour recurrence, several groups have combined thermal ablation with either chemotherapy or radiation [Citation66–68]. These adjuvant therapies can target tumour cells at the ablated margins by inducing cell apoptosis and enhancing cytotoxicity. Chemotherapies can also fill the gaps inside the high temperature area as observed by other groups [Citation69] and confirmed in this study. Specifically, Lindner et al. used RFA combined with Thermo-Dox to deliver the doxorubicin in breast cancer cells [Citation70,Citation71]. This combination has displayed superior efficacy over either ablation or chemotherapy alone and the study is currently moving into clinical trials (no. NCT00617981). The formulation of Thermo-Dox has been optimised such that the therapeutic payload is preferentially released at hyperthermic temperatures. Further studies have been encouraging as it achieved a 2-fold increase of cell apoptosis compared with free drug ablation [Citation28]. Using similar formulations, Yang et al. further delivered both paclitaxel and Hsp70 inhibitor quercetin to a rat breast cancer model with increased tumour abrogation [Citation29].
To explore more significant clinical benefits of combined chemotherapy and ablation, a logical step forward is to exploit the synergy between ablation and adjuvant chemotherapies to obtain optimal tumour cell injury. Several groups have probed the molecular mechanisms of tumour recurrence in the context of ablation. Our study reconciles these observations, identifying that Hsp90 was up-regulated after tumour ablation. The viable cells post-ablation showed stronger Hsp90 staining than non-ablated tumour cells, without nuclear or cytoplasmic degeneration observed by light microscopy. This observation highlights the protection role of Hsp90 in stressed tumour cells which was further supported by the fact that 50% of tumours were relapsed in murine models receiving ablation only. Therefore, we proposed a synchronous thermal ablation with systemic Hsp90 inhibition through thermo-responsive ELP-GA conjugated to not only kill the tumour cells around the ablation core but also to eradicate the cells at the ablated margins where viable cells express more Hsp90.
Hsp90 inhibitors have been developed over three decades and continuous effort has been made to develop more potent agents as more understanding of the heat shock pathway in cancer is revealed. Numerous oncogenic proteins remain targets as client proteins of Hsp90. Thus, inhibition of Hsp90 can lead to suppression of multiple targets with reduced drug resistance, drug tolerance, or drug–drug interaction. We chose geldanamycin (GA) for its high potency and improved cytotoxicity when combined with hyperthermia. However, GA has limited clinical use due to its hepatotoxicity, so requiring an advanced formulation. We demonstrated that thermo-responsive ELP-based biopolymers not only increase GA’s water solubility but also achieve lower systemic toxicity through conjugation with a pH-sensitive hydrozone linkage. We achieved a 2–3-fold increase of cell eradication when exposed to 30 min of hyperthermia in vitro with strong suppression of Hsp90.
The safety of conjugates was further confirmed by toxicity studies including blood chemistry and endotoxin testing (results not shown). ELP-GA is biocompatible in vivo and the maximum tolerated dose (MTD, 100 mg/kg) is 200 times that of IC50 in vitro. We arbitrarily chose half of the MTD and administered GA twice weekly in an aggressive HCC murine model and showed that down-regulation of Hsp90 contributed significantly to slower tumour progression. However, although the conjugates alone displayed superior efficacy to the free drug (17-AAG) and controls, they are still limited for tumour regression. Various studies using ELP-based polymer conjugates have been published with significant anti-cancer effect [Citation72,Citation73]. However, similar to many other macromolecular systems, sufficient drug delivery into the tumour has been a daunting task, where multiple barriers including high interstitial pressure, limited EPR effect, and tumour cell heterogeneity are yet to be overcome. Impressively, when ELP-GA conjugates were combined with thermal ablation (electrocautery), we achieved >90% cell eradication, greater Hsp90 inhibition, and no tumour recurrence up to 90 days. Ongoing studies are comparing the concentration of GA in both the tumour core and tumour margins after single or combined treatment. This information will help probe the mechanism of two-way synergy between ablation and macromolecular drug delivery. Given the fact that blood circulation of mice is 5–6 s, immediate injection after ablation is likely to get some benefit from hyperthermia as a result of ablation. However, the therapeutic potential of ELP phase transition can be further illustrated by a control study where ELP conjugates are injected long after ablation to rule out the effect of hyperthermia. In addition, we initially designed the ELPVA80 polymer to be a thermal-insensitive control but the yields were too poor for animal studies due to its higher Tt. Nevertheless, a separate animal study to quantify ELP concentrations as a function of ablation is warranted. This information will help to improve the design of ELP-based polymer conjugates to rapidly aggregate at the ablated site to achieve maximum accumulation and distribution into tumour tissues. Continued effort is also needed to optimise the dosing regimens and improve the ablation method to further exploit thermal targeting of ELP conjugates. For a translational point of view, an orthotropic liver cancer model which has a more comprehensive tumour microenvironment and similarity to human HCCs should be further pursued and optimised for clinical development of chemotherapy combined with thermal ablation. Our study paves the way for combining other chemotherapies, especially those with Hsp90-sensitive clients (e.g. HER2) to achieve better clinical outcomes.
In conclusion, the current study suggests that a combination of thermal ablation and adjuvant Hsp90 inhibition via thermo-responsive ELP-GA conjugates can exert a superior anti-tumour effect in an HCC murine model. The vast tumour eradication provides compelling rationale for further exploring the post-ablation kinetics of Hsp90 expression for optimal dosing to achieve progression-free and long-term survival of HCC patients.
Acknowledgements
The authors thank Sheryl Tripp for the technical assistance of the histological study of the manuscript and Amanda Brooks for providing the cloning vector for protein engineering.
Declaration of interest
This work was supported by the US National Institutes of Health (1R21NS064541-01A1) and University of Utah start up-funds. The authors alone are responsible for the content and writing of the paper.
Supplementary material (supporting information) available online
Supplementary Material
Download PDF (150.2 KB)References
- Lu MD, Xu HX, Xie XY, Yin XY, Chen JW, Kuang M, et al. Percutaneous microwave and radiofrequency ablation for hepatocellular carcinoma: A retrospective comparative study. J Gastroenterol 2005;40:1054–60
- Lu DS, Yu NC, Raman SS, Limanond P, Lassman C, Murray K, et al. Radiofrequency ablation of hepatocellular carcinoma: Treatment success as defined by histologic examination of the explanted liver. Radiology 2005;234:954–60
- Park W, Chung YH, Kim JA, Jin YJ, Lee D, Shim JH, et al. Recurrences of hepatocellular carcinoma following complete remission by transarterial chemoembolization or radiofrequency therapy: Focused on the recurrence patterns. Hepatol Res 2013;43:1304–12
- Waki K, Aikata H, Katamura Y, Kawaoka T, Takaki S, Hiramatsu A, et al. Percutaneous radiofrequency ablation as first-line treatment for small hepatocellular carcinoma: Results and prognostic factors on long-term follow up. J Gastroenterol Hepatol 2010;25:597–604
- Yang Y, Wang C, Lu Y, Bai W, An L, Qu J, et al. Outcomes of ultrasound-guided percutaneous argon-helium cryoablation of hepatocellular carcinoma. J Hepatobiliary Pancreat Sci 2012;19:674–84
- Liu Q, Zhai B, Yang W, Yu LX, Dong W, He YQ, et al. Abrogation of local cancer recurrence after radiofrequency ablation by dendritic cell-based hyperthermic tumor vaccine. Mol Ther 2009;17:2049–57
- Zhang Y, Zhao J, Guo D, Zhong W, Ran L. Evaluation of short-term response of high intensity focused ultrasound ablation for primary hepatic carcinoma: Utility of contrast-enhanced MRI and diffusion-weighted imaging. Eur J Radiol 2011;79:347–52
- Cheung TT, Fan ST, Chu FS, Jenkins CR, Chok KS, Tsang SH, et al. Survival analysis of high-intensity focused ultrasound ablation in patients with small hepatocellular carcinoma. HPB 2013;15:567–73
- Itoh S, Ikeda Y, Kawanaka H, Okuyama T, Kawasaki K, Eguchi D, et al. Efficacy of surgical microwave therapy in patients with unresectable hepatocellular carcinoma. Ann Surg Oncol 2011;18:3650–56
- Liu Y, Zheng Y, Li S, Li B, Zhang Y, Yuan Y. Percutaneous microwave ablation of larger hepatocellular carcinoma. Clin Radiol 2013;68:21–6
- Veltri A, Guarnieri T, Gazzera C, Busso M, Solitro F, Fora G, et al. Long-term outcome of radiofrequency thermal ablation (RFA) of liver metastases from colorectal cancer (CRC): Size as the leading prognostic factor for survival. Radiol Med 2012;117:1139–51
- Yamakado K, Nakatsuka A, Ohmori S, Shiraki K, Nakano T, Ikoma J, et al. Radiofrequency ablation combined with chemoembolization in hepatocellular carcinoma: Treatment response based on tumor size and morphology. J Vasc Interv Radiol 2002;13:1225–32
- Mulier S, Ni Y, Jamart J, Ruers T, Marchal G, Michel L. Local recurrence after hepatic radiofrequency coagulation: Multivariate meta-analysis and review of contributing factors. Ann Surg 2005;242:158–71
- Lin SM, Lin CJ, Lin CC, Hsu CW, Chen YC. Randomised controlled trial comparing percutaneous radiofrequency thermal ablation, percutaneous ethanol injection, and percutaneous acetic acid injection to treat hepatocellular carcinoma of 3 cm or less. Gut 2005;54:1151–6
- Wiggermann P, Puls R, Vasilj A, Sieron D, Schreyer AG, Jung EM, et al. Thermal ablation of unresectable liver tumors: Factors associated with partial ablation and the impact on long-term survival. Med Sci Monit 2012;18:CR88–92
- Schumann C, Rieder C, Bieberstein J, Weihusen A, Zidowitz S, Moltz JH, et al. State of the art in computer-assisted planning, intervention, and assessment of liver-tumor ablation. Crit Rev Biomed Eng 2010;38:31–52
- Goldberg SN, Solbiati L. Tumor dissemination after radiofrequency ablation of hepatocellular carcinoma. Hepatology 2001;34:609–11
- Kuszyk BS, Boitnott JK, Choti MA, Bluemke DA, Sheth S, Magee CA, et al. Local tumor recurrence following hepatic cryoablation: Radiologic-histopathologic correlation in a rabbit model. Radiology 2000;217:477–86
- Supic G, Kozomara R, Jovic N, Zeljic K, Magic Z. Prognostic significance of tumor-related genes hypermethylation detected in cancer-free surgical margins of oral squamous cell carcinomas. Oral Oncol 2011;47:702–8
- Kim YS, Rhim H, Cho OK, Koh BH, Kim Y. Intrahepatic recurrence after percutaneous radiofrequency ablation of hepatocellular carcinoma: Analysis of the pattern and risk factors. Eur J Radiol 2006;59:432–41
- Berber E, Siperstein A. Local recurrence after laparoscopic radiofrequency ablation of liver tumors: An analysis of 1032 tumors. Ann Surg Oncol 2008;15:2757–64
- Yoshida S, Kornek M, Ikenaga N, Schmelzle M, Masuzaki R, Csizmadia E, et al. Sublethal heat treatment promotes epithelial-mesenchymal transition and enhances the malignant potential of hepatocellular carcinoma. Hepatology 2013;58:1667–80
- Burks SR, Ziadloo A, Hancock HA, Chaudhry A, Dean DD, Lewis BK, et al. Investigation of cellular and molecular responses to pulsed focused ultrasound in a mouse model. PLoS One 2011;6:e24730
- Yang JD, Nakamura I, Roberts LR. The tumor microenvironment in hepatocellular carcinoma: Current status and therapeutic targets. Semin Cancer Biol 2011;21:35–43
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423–37
- Groeschl RT, Wong RK, Quebbeman EJ, Tsai S, Turaga KK, Pappas SG, et al. Recurrence after microwave ablation of liver malignancies: A single institution experience. HPB 2013;15:365–71
- Goldberg SN, Kamel IR, Kruskal JB, Reynolds K, Monsky WL, Stuart KE, et al. Radiofrequency ablation of hepatic tumors: Increased tumor destruction with adjuvant liposomal doxorubicin therapy. Am J Roentgenol 2002;179:93–101
- Yang W, Ahmed M, Elian M, Hady el-SA, Levchenko TS, Sawant RR, et al. Do liposomal apoptotic enhancers increase tumor coagulation and end-point survival in percutaneous radiofrequency ablation of tumors in a rat tumor model? Radiology 2010;257:685–96
- Yang W, Ahmed M, Tasawwar B, Levchenko T, Sawant RR, Collins M, et al. Radiofrequency ablation combined with liposomal quercetin to increase tumour destruction by modulation of heat shock protein production in a small animal model. Int J Hyperthermia 2011;27:527–38
- Kroeze SGC, Van Melick HHE, Nijkamp MW, Kruse FK, Kruijssen LWJ, Van Diest PJ, et al. Incomplete thermal ablation stimulates proliferation of residual renal carcinoma cells in a translational murine model. BJU Int 2012;110:E281–6
- Sajjadi AY, Mitra K, Grace M. Expression of heat shock proteins 70 and 47 in tissues following short-pulse laser irradiation: Assessment of thermal damage and healing. Med Eng Phys 2013;35:1406–14
- Parsell DA, Lindquist S. The function of heat-shock proteins in stress tolerance: Degradation and reactivation of damaged proteins. Annu Rev Genet 1993;27:437–96
- Yavelsky V, Vais O, Piura B, Wolfson M, Rabinovich A, Fraifeld V. The role of Hsp90 in cell response to hyperthermia. J Therm Biol 2004;29:509–14
- Sun Y, Zang Z, Xu X, Zhang Z, Zhong L, Zan W, et al. Differential proteomics identification of Hsp90 as potential serum biomarker in hepatocellular carcinoma by two-dimensional electrophoresis and mass spectrometry. Int J Mol Sci 2010;11:1423–33
- Koga F, Kihara K, Neckers L. Inhibition of cancer invasion and metastasis by targeting the molecular chaperone heat-shock protein 90. Anticancer Res 2009;29:797–807
- Bohonowych JE, Gopal U, Isaacs JS. Hsp90 as a gatekeeper of tumor angiogenesis: Clinical promise and potential pitfalls. J Oncol 2010;2010:412985
- Teng Y, Ngoka L, Mei Y, Lesoon L, Cowell JK. Hsp90 and Hsp70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J Bio Chem 2012;287:10051–9
- Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 1999;42:260–6
- Blagosklonny MV, Toretsky J, Neckers L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene 1995;11:93393–9
- Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol 1995;36:305–15
- Chilkoti A, Dreher MR, Meyer DE. Design of thermally responsive, recombinant polypeptide carriers for targeted drug delivery. Adv Drug Deliv Rev 2002;54:1093–111
- Urry DW, Trapane TL, Prasad KU. Phase-structure transitions of the elastin polypentapeptide-water system within the framework of composition-temperature studies. Biopolymers 1985;24:2345–56
- Furgeson DY, Dreher MR, Chilkoti A. Structural optimization of a ‘smart’ doxorubicin-polypeptide conjugate for thermally targeted delivery to solid tumors. J Control Release 2006;110:362–9
- Chen Y, Youn P, Furgeson DY. Thermo-targeted drug delivery of geldanamycin to hyperthermic tumor margins with diblock elastin-based biopolymers. J Control Release 2011;155:175–83
- Fujita Y, Mie M, Kobatake E. Construction of nanoscale protein particle using temperature-sensitive elastin-like peptide and polyaspartic acid chain. Biomaterials 2009;30:3450–7
- Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J Control Release 2000;65:271–84
- Szmacinski H, Lakowicz JR. Optical measurements of pH using fluorescence lifetimes and phase-modulation fluorometry. Ann Chem 1993;65:1668–74
- Bae Y, Buresh RA, Williamson TP, Chen THH, Furgeson DY. Intelligent biosynthetic nanobiomaterials for hyperthermic combination chemotherapy and thermal drug targeting of Hsp90 inhibitor geldanamycin. J Control Release 2007;122:16–23
- Ito A, Saito H, Mitobe K, Minamiya Y, Takahashi N, Maruyama K, et al. Inhibition of heat shock protein 90 sensitizes melanoma cells to thermosensitive ferromagnetic particle-mediated hyperthermia with low Curie temperature. Cancer Sci 2009;100:558–64
- Larson N, Gormley A, Frazier N, Ghandehari H. Synergistic enhancement of cancer therapy using a combination of heat shock protein targeted HPMA copolymer–drug conjugates and gold nanorod induced hyperthermia. J Control Release 2013;170:41–50
- Song CW, Kang MS, Rhee JG, Levitt SH. The effect of hyperthermia on vascular function, pH, and cell survival. Radiology 1980;137:795–803
- Kong G, Anyarambhatla G, Petros WP, Braun RD, Colvin OM, Needham D, et al. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: Importance of triggered drug release. Cancer Res 2000;60:6950–7
- Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, et al. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol 2002;43:33–56
- Huang HW. Influence of blood vessel on the thermal lesion formation during radiofrequency ablation for liver tumors. Med Phys 2013;40:073303
- Raucher D, Chilkoti A. Enhanced uptake of a thermally responsive polypeptide by tumor cells in response to its hyperthermia-mediated phase transition. Cancer Res 2001;61:7163–70
- Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol 1993;123:1107–17
- Szklaruk J, Silverman PM, Charnsangavej C. Imaging in the diagnosis, staging, treatment, and surveillance of hepatocellular carcinoma. Am J Roentgenol 2003;180:441–54
- Nikfarjam M, Muralidharan V, Christophi C. Mechanisms of focal heat destruction of liver tumors. J Surg Res 2005;127:208–23
- Chu KF, Dupuy DE. Thermal ablation of tumours: Biological mechanisms and advances in therapy. Nat Rev Cancer 2014;14:199–208
- Wright AS, Lee FT, Jr Mahvi DM. Hepatic microwave ablation with multiple antennae results in synergistically larger zones of coagulation necrosis. Ann Surg Oncol 2003;10:275–83
- Dong BW, Zhang J, Liang P, Yu XL, Su L, Yu DJ, et al. Sequential pathological and immunologic analysis of percutaneous microwave coagulation therapy of hepatocellular carcinoma. Int J Hyperthermia 2003;19:119–33
- Mala T. Cryoablation of liver tumours – A review of mechanisms, techniques and clinical outcome. Minim Invasive Ther Allied Technol 2006;15:9–17
- de Baere T, Elias D, Dromain C, Din MG, Kuoch V, Ducreux M, et al. Radiofrequency ablation of 100 hepatic metastases with a mean follow-up of more than 1 year. Am J Roentgenol 2000;175:1619–25
- Wood TF, Rose DM, Chung M, Allegra DP, Foshag LJ, Bilchik AJ. Radiofrequency ablation of 231 unresectable hepatic tumors: indications, limitations, and complications. Ann Surg Oncol 2000;7:593–600
- Fajardo LF, Egbert B, Marmor J, Hahn GM. Effects of hyperthermia in a malignant tumor. Cancer 1980;45:613–23
- Grieco CA, Simon CJ, Mayo-Smith WW, DiPetrillo TA, Ready NE, Dupuy DE. Percutaneous image-guided thermal ablation and radiation therapy: Outcomes of combined treatment for 41 patients with inoperable stage I/II non-small-cell lung cancer. J Vasc Interv Radiol 2006;17:1117–24
- Ahmed M, Goldberg SN. Combination radiofrequency thermal ablation and adjuvant IV liposomal doxorubicin increases tissue coagulation and intratumoural drug accumulation. Int J Hyperthermia 2004;20:781–802
- Horkan C, Dalal K, Coderre JA, Kiger JL, Dupuy DE, Signoretti S, et al. Reduced tumor growth with combined radiofrequency ablation and radiation therapy in a rat breast tumor model. Radiology 2005;235:81–8
- Barnes KD, Shafirstein G, Webber JS, Koonce NA, Harris Z, Griffin RJ. Hyperthermia-enhanced indocyanine green delivery for laser-induced thermal ablation of carcinomas. Int J Hyperthermia 2013;29:474–9
- Goldberg SN, Girnan GD, Lukyanov AN, Ahmed M, Monsky WL, Gazelle GS, et al. Percutaneous tumor ablation: increased necrosis with combined radio-frequency ablation and intravenous liposomal doxorubicin in a rat breast tumor model. Radiology 2002;222:797–804
- Lindner LH, Eichhorn ME, Eibl H, Teichert N, Schmitt-Sody M, Issels RD, et al. Novel temperature-sensitive liposomes with prolonged circulation time. Clin Cancer Res 2004;10:2168–78
- MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat Mater 2009;8:993–9
- McDaniel JR, Macewan SR, Li X, Radford DC, Landon CD, Dewhirst M, et al. Rational design of ‘heat seeking’ drug loaded polypeptide nanoparticles that thermally target solid tumors. Nano Lett 2014;14:2890–5