
Abstract
Background: Clinical efficacy of thrombolytic drugs is limited by lack of specific delivery and requires large therapeutic doses which increase toxicity. Encapsulating these drugs in temperature-sensitive liposomes and applying hyperthermia to deliver thrombolytic agents locally to thrombus might theoretically favourably alter the therapeutic window. The objectives of this study were to formulate liposomes encapsulating thrombolytics and assess thrombolytic activity following hyperthermia. Methods: Three liposome formulations were investigated: temperature-sensitive liposome (TSL, DPPC:DSPE-PEG2000 (mol% 95:5)), low temperature-sensitive liposome (LTSL, DPPC:MSPC:DSPE-PEG2000 (mol% 85.3:9.7:5)), and traditional temperature-sensitive liposome (TTSL, DPPC:HSPC:Chol:DSPE-PEG2000 (mol% 55:25:15:5)). To characterise temperature-dependent release of high molecular weight cargo from each formulation, fluorescein-conjugated dextrans (70 kDa) were loaded and release was quantified via spectrophotometry. Staphylokinase (SAK), urokinase, and tissue-type plasminogen activator were also loaded individually into each liposome formulation. Leakage at 37 °C and release at 38–44 °C were quantified via chromogenic enzymatic activity assay. Clot lysis was evaluated by measuring mass of blood clots before and after thrombolytic liposome treatment. Results: The LTSL formulation had optimal release characteristics with maximum release at 41.3 °C. Release of dextrans from LTSLs was observed to be 11.5 ± 1.5%, 79.7 ± 1.6%, and 93.6 ± 3.7% after 15 min in plasma at 37°, 39°, and 41.3 °C, respectively. The SAK LTSL had the highest release/leakage ratio and demonstrated greater clot lysis. Conclusions: The SAK LTSL achieves significant clot lysis in vitro. When combined with local hyperthermia, the SAK LTSL potentially produces sufficient thrombolysis while minimising systemic side effects.
Introduction
Thromboembolic disease is a leading cause of morbidity and mortality worldwide and is categorised into arterial thromboembolic diseases (e.g. myocardial infarction and stroke) and venous thromboembolic diseases (e.g. deep vein thrombosis (DVT) and pulmonary embolism (PE)) [Citation1]. Thromboembolic diseases are often treated with thrombolytics, which are serine proteases that activate the endogenous fibrinolytic system by cleaving the arginine 560-valine 561 bond in plasminogen [Citation2], producing active plasmin. Plasmin then degrades fibrin, leading to clot dissolution [Citation3]. The most widely used thrombolytic is alteplase and is indicated in the management of acute ischaemic strokes (within the first 3 h of onset) [Citation4,Citation5] and large PEs with haemodynamic compromise in combination with heparin [Citation6]. Thrombolytics are also commonly used to treat DVT [Citation7].
Thrombolytics have been shown to effectively dissolve thrombi and recanalise occluded blood vessels. However, the effectiveness of thrombolytics is limited clinically by the large therapeutic doses required, re-occlusion of vessels [Citation8], and an increased risk of intracranial haemorrhage in treated patients compared to untreated patients or those treated with anti-coagulation only [Citation9,Citation10]. Large doses are required because protein-based thrombolytics have short half-lives due to rapid inactivation upon injection into the bloodstream [Citation4]. For example, native tissue plasminogen activator (tPA) has a half-life of only 5 min [Citation11], and even the more stable recombinant tPAs such as alteplase and reteplase have half-lives of only 15–20 min [Citation11,Citation12].
Due to the narrow therapeutic index and short circulation times of thrombolytics, significant efforts have gone into developing improved thrombolytics that reduce systemic bleeding complications and have improved bioavailability [Citation13]. As an alternative to improving the thrombolytic agent itself, drug delivery systems (DDS) offer the potential to localise sufficient quantities of thrombolytic agents to the desired thrombus site while minimising systemic exposure and complications. Liposomes consisting of a bi-layer phospholipid membrane offer both an aqueous space and hydrophobic membrane for incorporation of thrombolytics which exhibit both hydrophobic and hydrophilic properties. Liposomes have primarily been investigated for encapsulation and delivery of small molecular agents, such as chemotherapeutics [Citation5], but they also have been used to deliver higher molecular weight (MW > 10 kDa) proteins [Citation14]. Several studies have highlighted the promise of liposomal thrombolytic formulations [Citation13,Citation15–17]. For example, Heeremans et al. [Citation18] demonstrated liposomal delivery of tPA improved thrombolytic efficacy approximately four-fold compared to free tPA in a rabbit jugular vein thrombosis model. Similarly, Nguyen et al. [Citation6] showed improved vessel patency by liposomal streptokinase delivery as compared to free streptokinase in a canine myocardial infarction model. Activate-able liposomal systems that liberate their cargo in response to an internal and/or external trigger represent a novel approach for thrombolytic delivery. For example, echogenic liposomes loaded with thrombolytics in combination with ultrasound demonstrate synergistic clot lysis through mechanical disruption (with or without ultrasound-induced cavitation) and activated release of thrombolytics. Laing et al. [Citation15] and Tiukinhoy-Laing et al. [Citation7] found that echogenic liposomes loaded with tPA produced enhanced clot lysis when combined with ultrasound.
Temperature-sensitive liposomes (TSLs) that release their contents in response to temperature elevations greater than body temperature have been used for over three decades combined with externally focused hyperthermia (T = 40–45 °C) to actively deliver small molecule therapeutics [Citation19], but such liposomes have not been extensively studied for the delivery of macromolecules. Recently, Zhang et al. [Citation14] demonstrated encapsulation and release of FITC-BSA (MW 66 kDa) in 145 nm lysolipid-based thermosensitive liposomes. Temperature-triggered liposomal formulations show phase transition temperature (Tm, solid/gel to liquid disordered phase change) in the range of 40–45 °C. These liposomes consist of dipalmitoylphosphatidylcholine (DPPC), which has a Tm of 41 °C, and may be modified to have increased rate of release (incorporation of lysolipid (monostearoyl-2-hydroxy-sn-glycero-3-phosphocholine (MPPC) or 1-myristoyl-2-stearoyl-sn-glycero-3-phosphocholine (MSPC), 1,2-dipalmitoyl-sn-glycero-3-phosphodiglycerol (DPPG2), or Brij® surfactant), greater stability (incorporation of polyethylene glycol (PEG)), or adjustable temperature for release (cholesterol, hydrogenated soy phosphatidylcholine (HSPC), and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)) [Citation20–23].
The objective of the present study was to investigate the release of high MW agents from TSLs, formulate and characterise thrombolytic TSLs, and determine the in vitro ability of TSLs to provide selective temperature-triggered thrombolysis. Release of fluorescein-conjugated dextran (70 kDa) was first investigated as a function of time, temperature, and buffer (4-(2-hydroxyethyl)piperazine-1-ethanesulphonic acid sodium salt (HEPES buffer, pH 7.4) or human plasma). The thrombolytics investigated were staphylokinase (SAK), urokinase (UK), and alteplase (a class of tPA). SAK is a 15.5 kDa microbial plasminogen activator isolated from Staphylococcus, UK is a 54 kDa plasminogen activator isolated from human urine, and alteplase is a 68 kDa recombinant protein that is similar to native tPA. UK and tPA directly cleave plasminogen to plasmin, whereas SAK forms a 1:1 complex with plasminogen. This SAK-plasminogen complex then cleaves other plasminogen molecules [Citation24]. These thrombolytics were then loaded into different temperature-sensitive liposome formulations and investigated for preferential release at a target temperature and their ability to effect clot lysis in vitro.
Materials and methods
Recombinant SAK, UK (ProSpec, Ness-Ziona, Israel) and tPA (Genentech, San Francisco, CA) were obtained as lyophilised powders and reconstituted in distilled water. Fluorescein isothiocyanate (FITC)-conjugated dextran (70 kDa, Sigma, St Louis, MO) was reconstituted in distilled water. To formulate liposomes, DPPC, MSPC, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (PEG)2000] (DSPE-PEG2000), HSPC, cholesterol (Avanti, Alabaster, AL) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD, Invitrogen, Eugene, OR) were dissolved in chloroform (Sigma) at a concentration of 10 mg/mL. A syringe extruder with a 100-nm pore size filter (Mini-Extruder; Avanti) was used to generate uniformly sized liposomes. Size-exclusion chromatography using a vertical column packed with Sepharose® G-4B (Sigma) was used to separate non-encapsulated dextran or protein. Temperature dependence and kinetics of dextran release from liposomes were quantified using a spectrofluorometer (Cary Eclipse, Agilent, Santa Clara, CA) equipped with multi-cell Peltier, temperature controller, and kinetic software. Enzymatic activity was assessed using chromogenic activity assays with substrates specific for each thrombolytic (Diapharma, West Chester, OH: S-2251 for SAK, S-2444 for UK, and S-2288 for tPA) and measured on a microplate reader (SpectraMax M5, Molecular Devices, Downingtown, PA) at 405 nm.
Preparation and purification of liposomes
Three liposome formulations were investigated: a standard temperature-sensitive liposome (TSL, DPPC:DSPE-PEG2000 (mol% 95:5)), a low temperature-sensitive liposome (LTSL, DPPC:MSPC:DSPE-PEG2000 (mol% 85.3:9.7:5)), and a traditional temperature-sensitive liposome (TTSL, DPPC:HSPC:Chol:DSPE-PEG2000 (mol% 55:25:15:5)). Lipid components were weighed and dissolved in chloroform in a round bottom flask. DiD (0.1% w/v; 4 µL/100 mg lipids) was added to the lipid solution for coloration. The chloroform was evaporated under vacuum at 45 °C to give a thin layer of film. The lipid thin film was further vacuum dried overnight in a desiccator to remove any traces of chloroform. The lipid film was then hydrated with a solution of FITC-dextran (25 mg/mL), SAK (500 µg/mL), UK (1 mg/mL), or tPA (2 mg/mL) in de-ionised water (Hydro water purification system, Rockville, MD) at a final lipid concentration of 20 mg/mL. The liposome suspension was incubated with gentle agitation in a 50 °C water bath for 1 h. After incubation, liposomes were extruded at 50 °C forty times to achieve uniform diameter (∼100 nm). Liposomes were separated from non-encapsulated dextrans or enzymes using size exclusion chromatography (Sepharose® G-4B, 34 × 2 cm column and PBS is used as an eluent) and then stored at 4 °C until use.
Characterisation of dextran-encapsulated liposomes
Liposomal release of FITC-conjugated dextrans (70 kDa) as a function of temperature was assessed via spectrophotometry. The liposome suspension (20 µL) was pipetted into 3 mL HEPES buffer or pooled normal plasma in a quartz cuvette. At 25 °C, liposomes exhibited low baseline fluorescence due to the self-quenching of fluorescein molecules within the liposomes. For release experiments in plasma, a blank plasma fluorescent reading was taken for background correction. Temperature-activated release was measured by monitoring fluorescence increase from 25–50 °C at a rate of 1 °C/min using a Cary Eclipse spectrofluorometer equipped with Eclipse multi-cell Peltier, temperature controller, and Eclipse Kinetic Software (Varian, Palo Alto, CA) at excitation and emission wavelengths of 494 and 518 nm, respectively. Temperature for release (TR) was empirically determined based on the inflection point from the signal intensity vs. temperature graph. The TM for each liposome formulation was not directly measured but is related to TR. Kinetics of dextran release were determined by measuring fluorescence at 25°, 37°, 39° and 42 °C for TSLs and LTSLs and 44 °C for TTSLs for 15 min. Measurements at 25 °C were to provide baseline fluorescence readings. Release with heating was compared to leakage at body temperature (37 °C). Percentage release was calculated by assuming 100% release with 2 µL aqueous solution of Triton® X-100 (5% v/v) and 0% release at 25 °C using the following formula:
where Io, It, and Imax are defined as signal intensity recorded at 25 °C, at various time t, and after adding Triton® X-100, respectively. Triton® X-100 was used as a standard method to induce complete dextran release.
Chromogenic activity assay for thrombolytic encapsulated liposomes
Enzyme release from liposomes with heating and baseline leakage at 37 °C were quantified using a chromogenic enzymatic activity assay. Chromogenic substrates were reconstituted at 5 mM in distilled water. The liposomes were incubated at 25°, 37°, 42° (TSL and LTSL), or 44 °C (TTSL) for 8 min. After incubation the liposome suspension (5 µL) was pipetted into a 96-well flat bottom plate in triplicate. HEPES buffer (75 µL) was added to wells containing liposomal UK or tPA. For liposomal SAK, 5 µL of liposome was added to 20 µL of human plasminogen (5 µM stock solution, Diapharma, West Chester, OH) and 55 µL of HEPES. The appropriate chromogenic substrate (20 µL) was then added to each well. Enzymatic activity was determined via the release of p-nitroaniline at 37 °C which was monitored with a spectrophotometer at an absorbance of 405 nm. Enzymatic activity was proportional to the initial rate of reaction (absorbance units/second over the first 5 min) and quantified using a standard curve generated from the reaction rates of known enzyme concentrations.
Whole blood clot lysis assay for thrombolytic liposomes
Whole blood was drawn into 50 mL polypropylene tubes via venipuncture from healthy male volunteers. All subjects had normal clotting and laboratory parameters and were not taking medications known to affect clotting. Whole blood (600 µL) was added to glass tubes containing compression springs 0.75 inches long with 0.12 inch outer diameter (SPR10-7, WM Berg, Cudahy, WI) with 4-0 suture attached. Tubes were covered in Parafilm (Neenah, WI) and incubated in a 37 °C water bath for 3 h allowing blood to clot within and around the spring. After incubation the springs were carefully removed and placed into 2 mL microcentrifuge tubes containing distilled water for 5 min. The springs with clots were gently blotted and weighed to determine pre-lysis clot weight. The clots on springs were temporarily placed into microcentrifuge tubes containing 0.9% normal saline. Subsequently the clots on springs were submerged in glass tubes containing 500 µL of the following solutions in replicates of 5: normal saline (negative control), SAK LTSL diluted in normal saline heated to either 37 °C or 42 °C for 8 min, SAK LTSL lysed with Triton® X-100 (2 µL, 5% v/v), and free enzyme diluted in normal saline (positive control). The free enzyme concentration used was equal to the effective enzyme concentration of the fully lysed liposome solution as determined by chromogenic assay (6–8 µg/mL). Clots were treated with saline, liposome, or free enzyme for 15 min at 37 °C. Clots were then removed from the water bath, blotted gently, and weighed to determine post-lysis weight. Percentage lysis was calculated as:
where Wp is the weight of the spring + clot after lysis, Ws is the weight of the empty spring, and Wc is the pre-lysis weight of the spring + clot. In order to compare percentage lysis across samples, values for liposome-treated clots were normalised to the controls from the respective samples, with the negative control (saline) serving as 0% lysis and the positive (free enzyme) control serving as 100% lysis.
Statistical analysis
All analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). Groups were compared for differences using an ANOVA and post-hoc test (Tukey or Neumann-Keuls). All p-values were two-sided, and a p value less than 0.05 indicated statistical significance. Values are reported as mean ± SEM unless otherwise indicated.
Results
Properties of temperature-sensitive liposomes
In order to model release of large macromolecules from liposomes, TSLs were loaded with FITC-conjugated dextrans (70 kDa), which approximated the molecular weights and Stokes radii of the thrombolytics investigated. The FITC-conjugated dextrans exhibited self-quenching inside the liposomes, resulting in low baseline fluorescence. Upon release from the liposome, fluorescein self-quenching ceased, and the observed fluorescence increased, allowing for calculation of percentage release. Loading TSLs with dextran enabled determination of the release kinetics and TR (a temperature where a sharp increase in release occurred) () for each liposome formulation in HEPES buffer or plasma. The TSL and LTSL exhibited similar TR, whereas the TTSL required heating to a slightly higher temperature in order to achieve release. Temperature of release for all liposomes was similar in HEPES or plasma. illustrates 70-kDa dextran–FITC release from LTSLs in physiological buffer (HEPES) and plasma, respectively, as the temperature is increased (1 °C/min). There was minimal to no release in HEPES until the transition temperature was reached (), yet release occurred slowly in human plasma before the transition temperature was reached; thereafter release was more rapid (). The influence of buffer on release characteristics is further emphasised when assessing the kinetics of release at a fixed temperature (). Minimal leakage of 70 kDa dextran–FITC was observed in HEPES but 12% leakage occurred in human plasma. Furthermore, a greater release of large cargo was observed in plasma at lower temperatures (e.g. 39 °C). In addition to the LTSL, all liposomes had greater leakage in plasma than HEPES. Dextrans 20–70 kDa in size had similar release kinetics in the LTSL formulation (data not shown). The passive loading efficiency was similar for dextrans with MW of 20–70 kDa, but larger dextrans (150 kDa) had a lower loading efficiency and were not evaluated further.
Figure 1. Time and temperature-dependence of a high MW cargo release (70 kDa dextran-FITC; 25 mg/mL) from LTSLs in HEPES buffer (A and C) and in human plasma (B and D). Data are mean ± SEM, n = 3.
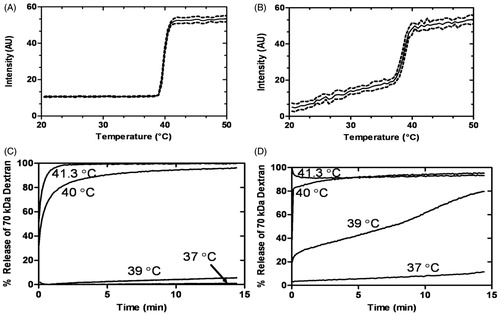
Table 1. TR (°C) in HEPES and plasma refers to a temperature at which a sharp increase in release of encapsulated 70-kDa dextran-FITC from the liposome was observed.
Release and leakage of thrombolytics from temperature-sensitive liposomes
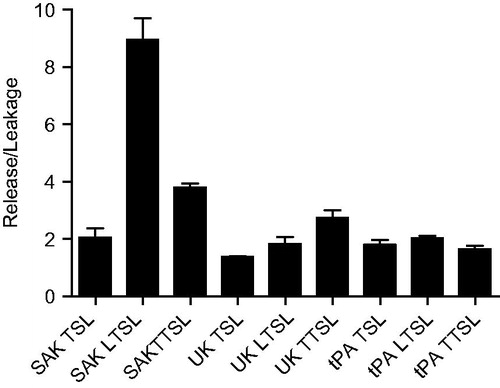
The three temperature-sensitive liposome formulations were loaded with SAK, UK, or tPA, yielding nine possible combinations. Release/leakage ratios were determined via chromogenic assay for each liposome–thrombolytic pairing (). The pairing with the highest release/leakage ratio was the SAK LTSL (8.9 ± 0.7, p < 0.0001) and thus was chosen for further evaluation.
Figure 2. Release/leakage ratios for nine thrombolytic-liposome pairings as determined by chromogenic assay. The SAK LTSL had the highest release/leakage ratio (8.9 ± 0.7). Data are mean ± SEM, n = 3, p < 0.0001, Tukey post-hoc test.
Whole blood clot lysis with thrombolytic liposomes
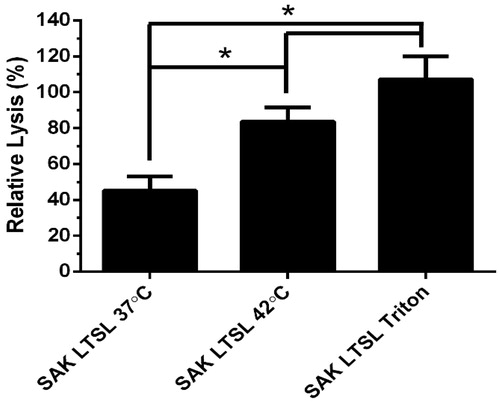
The SAK LTSL was investigated in terms of its ability to lyse whole blood clots because of its high release/leakage ratio. Whole blood clots appeared dark red and cylindrical on the spring scaffold. Heating the SAK LTSL at 42 °C for 8 min resulted in significantly greater clot lysis than unheated liposome (; 45.2 ± 7.9% and 83.7 ± 7.8% for 37 °C and at 42 °C, respectively; p < 0.05). Furthermore, there was no significant difference in clot lysis between heated LTSL, fully lysed LTSL (p > 0.05), and free enzyme positive controls.
Figure 3. Thrombolytic activity of the SAK LTSL as determined by the whole blood clot lysis assay. The SAK LTSL demonstrated significantly higher clot lysis with heating than without, but there was no significant difference in clot lysis between heated or fully lysed SAK LTSL and the free enzyme positive control. Data are mean ± SEM, n = 4, p < 0.05, Neuman-Keuls post-hoc test. *significant difference.
Discussion
The present study investigated the release of high MW agents from TSLs and formulated thrombolytic TSLs, and determined the in vitro ability of TSLs to provide selective temperature-triggered thrombolysis. The current management of thromboembolic diseases involves either systemic [Citation25,Citation26] or local catheter-mediated administration of thrombolytics [Citation27], which has some efficacy but also significant associated risks (e.g. cerebral haemorrhage) [Citation28]. The limited efficacy and risk of significant complications motivates the use of DDS to broaden the therapeutic window of thrombolytics. We investigated a temperature-activatable thrombolytic DDS by combining TSLs with thrombolytics. When combined with externally focused hyperthermia, this approach transforms an invasive procedure (e.g. insertion of a catheter into a clot and mechanical thrombectomy) into a non-invasive one. Alternatively, it is speculative although possible that an acutely inflamed thrombus or vulnerable plaque might have slightly elevated temperatures that may allow lipids to selectively deposit into the vascular plaque [Citation29,Citation30].
Although TSLs have been used for over three decades to deliver small molecules in combination with hyperthermia, there are limited reports for the release of large cargo [Citation14]. Herein, we demonstrated ultrafast release of dextrans with a hydrodynamic radius up to 6.49 nm (73 kDa) [Citation31]. Although hydrophilic dextrans are highly flexible, these data suggest the formation of water-filled pores in liposomes with diameters (perhaps ∼100 nm) sufficient for the transport of 70 kDa dextrans. It is clear from that plasma proteins increase the release rate of large cargo but also induce leakage at normal body temperature. Plasma proteins may destabilise the membrane by creating packing defects and/or transfer of lipids to and from the liposomes [Citation32–34].
Temperature triggered release of thrombolytics was evaluated with three liposome formulations (TTSL, TSL, and LTSL), each encapsulating individual thrombolytic agents (tPA, UK, or SAK) of different sizes and hydrophobic character. tPA is the most widely used thrombolytic, but because of its hydrophobic character, up to 70% may be associated with the liposomal membrane [Citation35]. This association may in part explain the low release/leakage ratio for tPA (). UK and SAK may be more readily encapsulated in the aqueous liposome interior, but the lower MW of SAK (15.5 vs. 54 kDa) may explain why its release/leakage ratio was highest for all thrombolytics evaluated. The 12% of SAK leakage from LTSLs in plasma after 15 min at 37 °C is not ideal, but a thrombolysis procedure may last approximately 30 min, suggesting that the stability may be sufficient for a clinical procedure. The SAK LTSL liposome formulation was further investigated in terms of its ability to lyse whole blood clots and yielded significantly more clot lysis with the application of hyperthermia than without (; p < 0.05).
The SAK LTSL offers several potential benefits beyond improved thrombolytic efficacy and reduced side effects. Even though liposome-mediated delivery of SAK should reduce systemic generation of plasmin, the fibrin-specific nature of this enzyme provides an additional layer of safety against undesired systemic plasmin activation. SAK has also been shown in clinical trials to be effective in the management of acute myocardial infarction. The Study of Tamoxifen and Raloxifene (STAR) trial was an open, randomised, multicentre trial that compared SAK to weight-adjusted alteplase in 100 patients with acute myocardial infarction. After 90 min, complete arterial recanalisation was observed in 58% of patients treated with alteplase and 62% of patients treated with SAK. Patients treated with SAK had unchanged levels of plasma fibrinogen, plasminogen, and α2-antiplasmin, whereas treatment with alteplase resulted in a 30% reduction in fibrinogen and a 60% reduction in plasminogen and α2-antiplasmin [Citation8]. These results showed that SAK is as effective as alteplase in the management of acute coronary syndrome but offers better fibrin specificity than alteplase.
A potential limitation of SAK is that it is immunogenic due to its bacterial origin. Clinical trials have shown that patients treated with SAK develop neutralising IgG antibodies after two weeks, limiting SAK to single-use [Citation36]. However, efforts are underway to develop SAK variants with reduced immunogenicity [Citation37–39]. Several studies have shown that simple amino acid substitutions produce SAK mutants that elicit fewer circulating antibodies in rabbits [Citation40] and baboons [Citation41] than the wild-type enzyme. A simple substitution of alanine for lysine in position 74 produces a variant with intact specific activity and thrombolytic potency but significantly reduced immunogenicity [Citation42]. More recently, Liu et al. [Citation43] used site-specific PEGylation to create a SAK variant with greatly reduced immunogenicity in guinea pig and rabbit models.
Despite the promising preliminary data for SAK LTSL, substantial hurdles and opportunities exist for optimisation of this temperature-active thrombolytic paradigm, including greater stability, more active thrombolytics, and clot-specific affinity targeting. Several options exist for targeting thrombolytic liposomes to sites of clot. Fibrin can be targeted using fibrin-specific monoclonal antibodies [Citation44] or inactivated tPA [Citation45]. Activated platelets can be targeted through the surface addition of RGD-based peptides [Citation46]. More recently, Peters et al. [Citation47] showed that clotted plasma proteins within atherosclerotic plaques can be targeted via the addition of the pentapeptides cysteine-arginine-glutamic acid-lysine-alanine (CREKA).
There are several hyperthermia applicators that may be suitable for use in temperature-activated thrombolysis. One potential applicator is high intensity focused ultrasound (HIFU). Conventional ultrasound, which may improve thrombolysis through acoustic cavitation, is potentially damaging to tissues [Citation48], limiting the practicality and safety of this technology. Damaging cavitation from HIFU may be detected, controlled and predicted, based in part upon selected HIFU parameters and tissue biology. Pulsed HIFU has been shown to cause acoustic radiation forces which can cause local mechanical displacement of tissue at the cellular level [Citation49]. Pulsed HIFU enhances thrombolysis in part by producing macroscopic alterations in clots which increase penetration or binding of tPA [Citation50,Citation51]. Alternatively, the EkoSonic Endovascular System (EKOS, Bothell, WA), an ultrasound-assisted catheter-based treatment strategy which emits low intensity high frequency ultrasound [Citation52], could also be paired with a thrombolytic liposome to mediate delivery of thrombolytic to the site of clot. Although the in vitro studies presented herein demonstrate the concept of heat-activated thrombolysis, additional in vivo studies are required to define any true potential of this approach.
Conclusion
In this study it was demonstrated that large cargo may be actively released from TSLs in the mild hyperthermic temperature range (40–45 °C). Plasma was found to increase the rate of dextran release. Nine TSL formulations were evaluated, and the SAK LTSL had the greatest release/leakage ratio. These liposomes lysed significantly more clott when heated to 42 °C. Targeted delivery with hyperthermia potentially offers increased bioavailability and reduced side effects by limiting systemic activity. The SAK LTSL has potential for local thrombolytic activity, but hurdles remain in terms of stability in plasma, immunogenicity, and selective or targeted delivery to clots. Future work could also clarify whether the trace hyperthermia present in native acute clots or whether local HIFU application could augment thrombolysis in vivo.
Declaration of interest
This research was supported by the Center for Interventional Oncology, the Intramural Research Program of the National Institutes of Health (NIH), the Howard Hughes Medical Institute NIH Research Scholars Program (V.S.), and the NIH Imaging Sciences Training Program (C.G.J.). NIH and Biocompatibles BTG have a cooperative research and development agreement. None of the authors have any forms of conflicts of interest and the mention of commercial products, their source, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the National Institutes of Health. The authors alone are responsible for the content and writing of the paper.
References
- Pierce T, Razzuk M, Razzuk L, Hoover S. A comprehensive review of the physiology of hemostasis and antithrombotic agents. BUMC Proc 1999;12:39–49
- Patel SC, Mody A. Cerebral hemorrhagic complications of thrombolytic therapy. Prog Cardiovasc Dis 1999;42:217–33
- Collen D, Lijnen HR. Basic and clinical aspects of fibrinolysis and thrombolysis. Blood 1991;78:3114–24
- Eppler S, Senn T, Gilkerson E, Modi NB. Pharmacokinetics and pharmacodynamics of recombinant tissue-type plasminogen activator following intravenous administration in rabbits: A comparison of three dosing regimens. Biopharm Drug Dispos 1998;19:31–8
- Martins S, Sarmento B, Ferreira DC, Souto EB. Lipid-based colloidal carriers for peptide and protein delivery – Liposomes versus lipid nanoparticles. Int J Nanomedicine 2007;2:595–607
- Nguyen PD, Orear EA, Johnson AE, Patterson E, Whitsett TL, Bhakta R. Accelerated thrombolysis and reperfusion in a canine model of myocardial-infarction by liposomal encapsulation of streptokinase. Circ Res 1990;66:875–8
- Tiukinhoy-Laing SD, Huang SL, Klegerman M, Holland CK, McPherson DD. Ultrasound-facilitated thrombolysis using tissue-plasminogen activator-loaded echogenic liposomes. Thromb Res 2007;119:777–84
- Vanderschueren S, Barrios L, Kerdsinchai P, Vandenheuvel P, Hermans L, Vrolix M, et al. A randomized trial of recombinant staphylokinase versus alteplase for coronary artery patency in acute myocardial infarction. Circulation 1995;92:2044–9
- Chatterjee S, Chakraborty A, Weinberg I, Kadakia M, Wilensky RL, Sardar P, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: A meta-analysis. JAMA 2014;311:2414–21
- Wardlaw JM, Murray V, Berge E, del Zoppo G, Sandercock P, Lindley RL, et al. Recombinant tissue plasminogen activator for acute ischaemic stroke: An updated systematic review and meta-analysis. Lancet 2012;379:2364–72
- Cohen A. Pharmacokinetics of the recombinant thrombolytic agents – What is the clinical significance of their different pharmacokinetic parameters? Biodrugs 1999;11:115–23
- Modi NB, Eppler S, Breed J, Cannon CP, Braunwald E, Love TW. Pharmacokinetics of a slower clearing tissue plasminogen activator variant, TNK-tPA, in patients with acute myocardial infarction. Thromb Haemostasis 1998;79:134–9
- Elbayoumi TA, Torchilin VP. Liposomes for targeted delivery of antithrombotic drugs. Expert Opin Drug Deliv 2008;5:1185–98
- Zhang X, Luckham PF, Hughes AD, Thom S, Xu XY. Development of lysolipid-based thermosensitive liposomes for delivery of high molecular weight proteins. Int J Pharmaceutics 2011;421:291–2
- Laing ST, Moody MR, Kim H, Smulevitz B, Huang SL, Holland CK, et al. Thrombolytic efficacy of tissue plasminogen activator-loaded echogenic liposomes in a rabbit thrombus model. Thromb Res 2012;130:629–35
- Tiukinhoy-Laing SD, Buchanan K, Parikh D, Huang SL, MacDonald RC, McPherson DD, et al. Fibrin targeting of tissue plasminogen activator-loaded echogenic liposomes. J Drug Target 2007;15:109–14
- Torchilin VP. Targeting of drugs and drug carriers within the cardiovascular system. Adv Drug Deliv Rev 1995;17:75–101
- Heeremans JLM, Prevost R, Bekkers MEA, Los P, Emeis JJ, Kluft C, et al. Thrombolytic treatment with tissue-type plasminogen-activator (t-PA)-containing liposomes in rabbits – A comparison with free t-PA. Thromb Haemostasis 1995;73:488–94
- May JP, Li SD. Hyperthermia-induced drug targeting. Expert Opin Drug Deliv 2013;10:511–27
- Lindner LH, Eichhorn ME, Eibl H, Teichert N, Schmitt-Sody M, Issels RD, et al. Novel temperature-sensitive liposomes with prolonged circulation time. Clin Cancer Res 2004;10:2168–78
- Lindner LH, Hossann M, Vogeser M, Teichert N, Wachholz K, Eibl H, et al. Dual role of hexadecylphosphocholine (miltefosine) in thermosensitive liposomes: Active ingredient and mediator of drug release. J Control Release 2008;125:112–20
- Needham D, Dewhirst MW. The development and testing of a new temperature-sensitive drug delivery system for the treatment of solid tumors. Adv Drug Deliv Rev 2001;53:285–305
- Li L, ten Hagen TLM, Schipper D, Wijnberg TM, van Rhoon GC, Eggermont AMM, et al. Triggered content release from optimized stealth thermosensitive liposomes using mild hyperthermia. J Control Release. 2010;143:274–9
- Lijnen HR, Vanhoef B, Decock F, Okada K, Ueshima S, Matsuo O, et al. On the mechanism of fibrin-specific plasminogen activation by staphlokinase. J Biol Chem 1991;266:11826–32
- Alexandrov AV. Current and future recanalization strategies for acute ischemic stroke. J Intern Med 2010;267:209–19
- Yeo LLL, Sharma VK. The quest for arterial recanalization in acute ischemic stroke – The past, present and the future. J Clin Med Res 2013;5:251–265
- Tapson VF. Thrombolytic therapy for acute pulmonary embolism. Semin Thromb Hemost 2013;39:7
- Block HS, Biller J. Commonly asked questions: Thrombolytic therapy in the management of acute stroke. Expert Rev Neurother 2013;13:157–65
- Stefanadis C, Diamantopoulos L, Vlachopoulos C, Tsiamis E, Dernellis J, Toutouzas K, et al. Thermal heterogeneity within human atherosclerotic coronary arteries detected in vivo – A new method of detection by application of a special thermography catheter. Circulation 1999;99:1965–71
- Toutouzas K, Synetos A, Stefanadi E, Vaina S, Markou V, Vavuranakis M, et al. Correlation between morphologic characteristics and local temperature differences in culprit lesions of patients with symptomatic coronary artery disease. J Am Coll Cardiol 2007;49:2264–71
- Armstrong JK, Wenby RB, Meiselman HJ, Fisher TC. The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys J 2004;87:4259–70
- Scherphof G, Roerdink F, Waite M, Parks J. Disintegration of phosphatidylcholine liposomes in plasma as a result of interaction with high-density lipoproteins. Biochim Biophys Acta 1978;542:296–307
- Shabbits JA, Chiu GNC, Mayer LD. Development of an in vitro drug release assay that accurately predicts in vivo drug retention for liposome-based delivery systems. J Control Release 2002;84:161–70
- Hossann M, Syunyaeva Z, Schmidt R, Zengerle A, Eibl H, Issels RD, et al. Proteins and cholesterol lipid vesicles are mediators of drug release from thermosensitive liposomes. J Control Release 2012;162:400–6
- Shaw GJ, Meunier JM, Huang SL, Lindsell CJ, McPherson DD, Holland CK. Ultrasound-enhanced thrombolysis with tPA-loaded echogenic liposomes. Thromb Res 2009;124:306–10
- Vanderschueren S, Dens J, Kerdsinchai P, Desmet W, Vrolix M, DeMan F, et al. Randomized coronary patency trial of double-bolus recombinant staphylokinase versus front-loaded alteplase in acute myocardial infarction. Am Heart J 1997;134:213–19
- Mandi N, Soorapaneni S, Rewanwar S, Kotwal P, Prasad B, Mandal G, et al. High yielding recombinant staphylokinase in bacterial expression system-cloning, expression, purification and activity studies. Protein Expr Purif 2009;64:69–75
- Prasad B, Salunkhe SS, Padmanabhan S. Novel self-cleavage activity of staphylokinase fusion proteins: An interesting finding and its possible applications. Protein Expression Purification 2010;69:191–7
- Kumar S, Maheshwari N, Sahni G, inventors. Mutants of streptokinase and their covalently modified forms. US Patent 20100034804 A1, 2010
- Collen D, Bernaerts R, Declerck P, DeCock F, Demarsin E, Jenne S, et al. Recombinant staphylokinase variants with altered immunoreactivity. 1. Construction and characterization. Circulation 1996;94:197–206
- Collen D, Moreau H, Stockx L, Vanderschueren S. Recombinant staphylokinase variants with altered immunoreactivity. 1. Thrombolytic properties and antibody induction. Circulation 1996;94:207–16
- Collen D, DeCock F, Demarsin E, Jenne S, Lasters I, Laroche Y, et al. Recombinant staphylokinase variants with altered immunoreactivity. 3. Species variability of antibody binding patterns. Circulation 1997;95:455–62
- Liu J, Wang Z, He J, Wang G, Zhang R, Zhao B. Effect of site-specific PEGylation on the fibrinolytic activity, immunogenicity, and pharmacokinetics of staphylokinase. Acta Biochim Biophys Sin 2014;46:782–91
- Edgell T, McEvoy F, Webbon P, Gaffney PJ. Monoclonal antibodies to human fibrin: Interaction with other animal fibrins. Thromb Haemostasis 1996;75:595–9
- Klegerman ME, Zou Y, McPherson DD. Fibrin targeting of echogenic liposomes with inactivated tissue plasminogen activator. J Liposome Res 2008;18:95–112
- Sen Gupta A, Huang G, Lestini BJ, Sagnella S, Kottke-Marchant K, Marchant RE. RGD-modified liposomes targeted to activated platelets as a potential vascular drug delivery system. Thromb Haemostasis 2005;93:106–14
- Peters D, Kastantin M, Kotamraju VR, Karmali PP, Gujraty K, Tirrell M, et al. Targeting atherosclerosis by using modular, multifunctional micelles. Proc Natl Acad Sci USA 2009;106:9815–19
- Frenkel V, Kimmel E, Iger Y. Ultrasound-induced cavitation damage to external epithelia of fish skin. Ultrasound Med Biol 1999;25:1295–303
- Nightingale K, Soo MS, Nightingale R, Trahey G. Acoustic radiation force impulse imaging: In vivo demonstration of clinical feasibility. Ultrasound Med Biol 2002;28:227–35
- Stone MJ, Frenkel V, Dromi S, Thomas P, Lewis RP, Li KC, et al. Pulsed high intensity focused ultrasound enhanced tPA mediated thrombolysis in a novel in vivo clot model: A pilot study. Thromb Res 2007;121:193–202
- Frenkel V, Oberoi J, Stone MJ, Park M, Deng C, Wood BJ, et al. Pulsed high-intensity focused ultrasound enhances thrombolysis in an in vitro model. Radiology 2006;239:86–93
- Lin PH, Annambhotla S, Bechara CF, Athamneh H, Weakley SM, Kobayashi K, et al. Comparison of percutaneous ultrasound-accelerated thrombolysis versus catheter-directed thrombolysis in patients with acute massive pulmonary embolism. Vascular 2009;17:S137–47