
Abstract
Purpose: We have evaluated the hyperthermia efficacy of oleic acid-functionalised Fe3O4 magnetic nanoparticles (MN-OA) under in vivo conditions and elucidated the underlying mechanism of tumour growth inhibition. Materials and methods: The efficacy and mechanism of tumour growth inhibition by MN-OA-mediated magnetic hyperthermia therapy (MHT) was evaluated in a murine fibrosarcoma tumour model (WEHI-164) using techniques such as TUNEL assay, Western blotting (WB), immunofluorescence (IF) staining and histopathological examination. In addition, bio-distribution of MN-OA in tumour/other target organs and its effect on normal organ function were studied by Prussian blue staining and serum biochemical analysis, respectively. Results: MN-OA-induced MHT resulted in significant inhibition of tumour growth as determined by measurement of tumour volume, as well as by in vivo imaging of tumour derived from luciferase-transfected WEHI-164 cells. Histopathology analysis showed presence of severe apoptosis and reduced tumour cells proliferation, which was further confirmed by TUNEL assay, reduced expression of Ki-67 and enhanced level of cleaved caspase-3, in tumours treated with MHT. Moreover, expression of heat stress marker, Hsp90 and its client protein, AKT/PKB was reduced by ∼50 and 80%, respectively, in tumours treated with MHT as studied by WB and IF staining. Serum analysis suggested insignificant toxicity of MN-OA (in terms of liver and kidney function), which was further correlated with minimal accumulation of MN-OA in target organs. Conclusions: These results suggest the involvement of apoptosis and Hsp90/AKT modulation in MN-OA-mediated MHT-induced tumour growth inhibition.
Introduction
Despite the advances in therapeutic modalities, cancer remains one of the leading causes of deaths all over the world [Citation1]. Conventional cancer therapy modalities suffer from several limitations, such as intrinsic or acquired resistance to chemotherapeutic agents/radiation, adverse side effects of chemo- or radiation therapy [Citation2,Citation3]. Thus, development of alternative or adjunct therapies, which are minimally invasive and can enhance the therapeutic efficacy of the available conventional modalities, becomes indispensable. One such modality, which is minimally invasive and has fewer side effects, is hyperthermia therapy (HT). HT involves exposing the tumour tissue or whole body to elevated temperatures, which kills the tumour cells directly and/or sensitises them to chemo- or radiation therapy [Citation4]. HT is relatively selective to tumours than normal tissue because of altered anatomical/physiological features of tumour tissue characterised by presence of leaky vasculature, hypoxia, acidosis, poor lymphatic drainage and a high interstitial pressure [Citation5–7]. Conventionally, HT is administered using different modes, i.e. infrared, microwaves, high intensity focused ultrasound, sauna bath or water bath, for example [Citation8]. However, conventional HT suffers from two major drawbacks: 1) non-accessibility of deep-seated tumour sites and 2) non-uniform heating of the tumour tissue and generation of a temperature gradient from surface to core of tumour tissue, which in turn causes insufficient killing of tumour cells and severe side effects such as skin burns, blisters, pain and discomfort to the patient [Citation9]. These drawbacks can be significantly minimised by application of magnetic nanoparticle (MNP)-induced intracellular hyperthermia therapy of tumours [Citation10–12]. These MNPs can be specifically targeted to the tumour site and thus can heat the tumour from within, more uniformly, minimising the development of temperature gradients within the tumour and thus enabling better tumour control [Citation13].
Iron oxide (Fe3O4) MNPs have been extensively used for magnetic hyperthermia therapy (MHT) applications due to their superparamagnetism, high magnetic moment, high specific absorption rates (SAR) and biocompatibility [Citation14,Citation15]. In addition to these unique magnetic properties, Fe3O4 MNPs are also amenable to surface modification with different organic molecules, antibodies, peptides, for example, which further makes them suitable for various diagnostic and targeted cancer therapy applications [Citation16–18]. However, these applications require that the MNPs should have reduced agglomeration, good colloidal stability, and better dispersion in physiological conditions [Citation19]. In our previous work we synthesised oleic acid-functionalised iron oxide MNPs (MN-OA) which showed good colloidal stability and dispersibility in physiological medium. Our formulation, in combination with an alternating magnetic field (AMF), showed significant killing of WEHI-164 tumour cells by apoptosis [Citation20]. In the present work we have evaluated the anti-tumour efficacy of MN-OA-induced MHT using a fibrosarcoma tumour model in BALB/c mice and studied the underlying mechanism of tumour growth inhibition using different molecular techniques. Although, in vivo efficacy of MHT has been evaluated in several studies using different tumour models such as glioma, melanoma [Citation21,Citation22], little is known in the literature about the mechanism of tumour growth inhibition induced by MHT. Moreover, success of MHT in in vivo tumour models depends not only on the superparamagnetic properties of MNPs but also on the dose used for therapy, their tumour retention time after administration/injection as well as their intra-tumoural distribution [Citation23].
Compared to previous studies [Citation20–23], in this work, we have 1) studied the bio-distribution of MN-OA in tumour tissue and other probable target organs such as liver, spleen and kidney, 2) we have used a sensitive technique of in vivo animal imaging for measurement of live tumour cells. Moreover, we have 3) elucidated the probable mechanism of tumour growth inhibition following MHT using different molecular techniques and studied the role of Hsp90/AKT signalling in MHT-induced tumour growth inhibition. In addition, 4) we have used a non-invasive method of thermal imaging using an infrared (IR) camera for measuring tumour temperature during MHT instead of the conventionally used invasive optical sensor temperature probes.
Materials and methods
Reagents and chemicals
D-luciferin sodium salt was procured from Biosynth, Staad, Switzerland. Ketamine hydrochloride was obtained from Neon Laboratories, Mumbai, India, and xylazine from Indian Immunologicals (Hyderabad, India). Dimethyl sulphoxide (DMSO) was procured from S.D. Fine-Chem, Mumbai, India. Antibodies for Hsp90, AKT and GAPDH were obtained from Cell Signaling Technology, Danvers, MA, USA and cleaved caspase-3 and Ki-67 from BD Pharmingen, Piscataway, NJ. G418 was obtained from Sigma (St Louis, MO) and Superfect transfection agent from Qiagen, Valencia, CA, USA.
Cell lines and animals
Mouse skin fibrosarcoma cell line (WEHI-164) was obtained from the National Centre for Cell Sciences (Pune, India). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO, Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS) (Himedia Laboratories, Mumbai, India) and antibiotics (100 U mL−1 penicillin and 100 µg mL−1 streptomycin) in a humidified atmosphere of 5% CO2 at 37 °C.
Female BALB/c mice (6–8 weeks old) were obtained from the animal house facility of Bhabha Atomic Research Centre. The animals were housed in a solid floor cage with adequate bedding. The room temperature was controlled to 20–24 °C and a 14 h of light and 10 h of dark cycle was maintained. Animals received a diet of commercially available pellets and water ad libitum. All experiments were conducted according to our institutional animal ethical committee guidelines.
Establishment of fibrosarcoma tumour model
To establish a fibrosarcoma tumour model, WEHI-164 (mouse fibrosarcoma tumour cells; 1 × 106) were injected intramuscularly into the right hind leg of BALB/c mice. Palpable tumours (∼9 mm in diameter) were obtained on day 8 after injection. On day 8 animals were injected, intra-tumourally with vehicle control (3.5 µL oleic acid dispersed in 0.1 M sodium carbonate) or MN-OA (dispersed in 0.1 M sodium carbonate) as described previously [Citation20], followed by HT, as described in the next section.
In vivo magnetic hyperthermia therapy
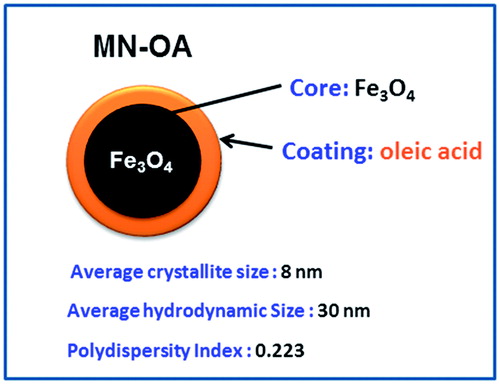
MN-OA having an average crystallite size of 8 nm was synthesised and characterised as described previously [Citation20]. The hydrodynamic size of MN-OA was determined by dynamic light scattering (DLS) and was found to be ∼30 nm with a poly dispersity index (PDI) of 0.22 (Scheme 1 and Supplementary Figure S1). For in vivo MHT, on day 8, 100 µL of MN-OA stock (∼0.44 mg MN-OA) was injected in the tumour in two stages: twice on day 8 and twice on day 10 at an interval of 1 h between the two injections. Thus, total 1.76 mg of MN-OA (equivalent to ∼69.6 µg Fe) was injected intra-tumourally. The whole animal was treated four times (on days 8, 9, 10 and 11) with AMF (400 A, 265 kHz radiofrequency, 10 min; 2 h after the MN-OA injection) using specially designed plastic tubes with sufficient ventilation. This treatment schedule consisted of four groups (eight tumour-bearing mice in each group): Group I: vehicle control (C), Group II: vehicle control and exposed to AMF (C + H), Group III: MN-OA alone (MN-OA), and Group IV: MN-OA followed by exposure to AMF (MN-OA + AMF). The tumour growth of control and treated animals was monitored for another 8 days by measuring the tumour diameter (small and large) using a Vernier caliper. Since the tumour was an oblate spheroid, the volume was calculated using the formula: Tumour volume = 4/3 πabc, since, for an oblate spheroid, a = b > c. Therefore the formula used to calculate the tumour volume is 4/3 πa2c, where a is the larger radius and c the smaller radius. Since we measured the diameter using the Vernier caliper, the formula modifies to: Tumour volume = 4/3 π (A/2)2 (C/2) = 0.523A2C, where A is the larger diameter and C the smaller diameter.
Scheme 1. Scheme for MN-OA formulation and its properties.
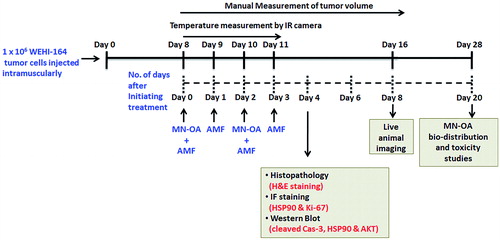
On day 4 after initiation of treatment, mice were sacrificed, tumours were excised, and part of the tumour was fixed in buffered formalin solution (10%) for immunofluorescence and histopathological studies. The remaining part was snap frozen in liquid nitrogen and stored at −80 °C till use. The experimental plan and schedule for MHT is described in Scheme 2.
Scheme 2. Schedule for MHT of fibrosarcoma tumours and experimental plan to study the underlying mechanism of tumour growth inhibition.
Development of luciferase expressing WEHI-164 cells for in vivo imaging
The WEHI-164 cells were transfected with the CMV-FL2-eGFP-pcDNA 3.1(+) plasmid vector by lipofection, as described elsewhere [Citation24]. The transfected clones were selected using G418 selection medium. The luminescence of clones was measured in a luminometer at 550 nm and relative luminescence units (RLU) per µg protein were calculated. Clone expressing highest RLU per µg protein was further cultured in P-100 and maintained in selection medium containing 200 µg per mL of G418 (Supplementary Figure S2). This clone was used for establishment of fibrosarcoma tumours in BALB/c mice, followed by injection of vehicle control or MN-OA and exposure to AMF as described earlier. For in vivo imaging, 3 mg of D-luciferin sodium salt (Biosynth, Staad, Switzerland; stock prepared in saline) was injected intra-peritoneally in animals. This was followed by injection of anaesthesia (ketamine:xylazine::50: 6). Once the animals were anaesthetised (∼5 min after injection) the animals were imaged using the in vivo imaging system (Photon Imager, Biospace, Nesles-la-Vallée, France). Results were expressed as mean luminescence intensity (photons/s/cm2/sr) ± SEM.
Histopathology, TUNEL and immunofluorescence staining
Formalin-fixed tumour tissues were embedded in paraffin wax to obtain tissue blocks, followed by microtomy to obtain 5-µm thick tumour tissue sections. These sections were mounted on pre-treated glass slides and further processed for haematoxylin and eosin (H&E) staining or TUNEL or immunofluorescence (IF) staining. In brief, the sections were de-paraffinised in xylene followed by re-hydration in decreasing grades of alcohol, followed by washing with phosphate buffered saline (PBS). For histopathology studies the slides were stained with H&E. For TUNEL assay, the rehydrated sections were permeabilised with 0.1% Triton X-100 and 0.1% sodium citrate, followed by washing with PBS and incubation in TUNEL reaction mixture (50 µL enzyme + 450 µL label solution) in humidified atmosphere at 37 °C for 1 h. The slides were again washed in PBS followed by mounting with cover slips and observation by fluorescence microscopy at 40 × magnification.
Tumour tissue sections were processed for immunofluorescence staining for Hsp90 and Ki-67 expression. Briefly, the tumour sections were de-paraffinised and re-hydrated in decreasing grades of alcohol as described previously, followed by antigen unmasking by immersing in 10 mM sodium citrate buffer at sub-boiling temperatures (∼80 °C) for 10 min. The slides were allowed to cool at room temperature. Later the slides were immersed in permeabilisation solution (0.1% Triton X-100, 0.1% sodium citrate) for 5 min and further washed with Tris buffered saline with Tween 20 (TBST). The slides were blocked with 5% normal goat serum (NGS) for 2 h at room temperature, followed by incubation with appropriate primary antibody (1:50 dilution in 5% NGS) at 4 °C for overnight. The slides were washed with TBST thrice for 5 min followed by incubation with Alexa Fluor 488/568 labelled anti-mouse/anti-rabbit secondary antibody, respectively for 2 h. The slides were again washed with TBST thrice, followed by mounting with anti-fade containing diamidino-2-phenylindole (DAPI). The slides were observed by fluorescence microscopy at 40 × magnification. To subtract fluorescence due to non-specific binding of secondary antibody, tumour sections were treated with only secondary antibody and their mean fluorescence intensity (MFI) values were subtracted from the MFI of tumour sections treated with primary as well as secondary antibody. Fluorescence intensity was measured by using Image J software and expressed as MFI per field. The values were normalised with fluorescence intensity of DAPI and fold change was calculated as MFI of treated tumours/MFI of control tumours.
Prussian blue staining
Tumours and other organ tissue sections were processed for Prussian blue staining to determine the distribution of MN-OA within the tissue section [Citation20]. Principle of staining is based on reaction of potassium ferrocyanide with Fe3+ under acidic conditions to form ferric ferrocyanide; an insoluble blue-coloured complex, also called as Prussian blue. Thus blue spots correspond to presence of Fe3+. In brief, the paraffin sections were de-waxed in xylene solution, twice for 10 min each, followed by washing with PBS. The sections were then incubated with the staining solution (a 1:1 mixture of 4% potassium ferrocyanide and 4% hydrochloric acid (HCl) at room temperature for 20 min. The slides were again washed with PBS and counter stained with 0.5% neutral red solution and then examined by light microscopy.
Preparation of tumour tissue lysates and Western blotting
For preparation of tumour tissue lysates and protein extraction, the frozen samples were thawed on ice and then transferred to a pre-chilled mortar. The tissue was crushed in presence of liquid nitrogen to a fine powder. The powder was re-suspended in lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM IGEPAL, 1% sodium lauryl sulphate, 1 mM PMSF, pH 7.5) and subjected to 5–7 freeze-thaw cycles with intermittent vortexing (i.e. freezing in liquid nitrogen for 30 s followed by thawing in water for 1 min). Once the lysates appeared clear, it was centrifuged at 4 °C at 32 000 g for 15 min. The protein content in tissue lysates was determined by using the BIO-RAD protein estimation kit following manufacturer’s instructions. Samples of cells/tissue lysates (50 µg of protein) were then subjected to 10% sodium dodecyl sulphate polyacrylamide gel electrophoresis and Western blotting (WB) using the desired primary antibody (1:1000 dilution) and appropriate secondary antibody (1:4000 dilution) conjugated with horse radish peroxidase [Citation25]. The bound secondary antibody was detected using chemiluminiscent detection kit (Roche diagnostics, USA). The intensity of each band was quantified using Image J software. Antibodies against GAPDH (1:4000; CST) were used as loading controls. Intensity of each protein band was divided with respective GAPDH band intensity. The ratio values obtained were used to measure fold change with respect to control.
Measurement of tumour surface temperature using an infra-red camera
The temperature rise at tumour surface was measured by a high resolution infrared (IR) camera (Testo 875-1, South Burlington and Vermont (VT), USA) and the images were analysed by thermography software (Testo IRSoft software, version 3.1). The mean temperature of tumour surface before and after exposure to AMF in both control and treated groups was measured.
Iron estimation
To study the distribution of MN-OA in different organs of mice, the tumour bearing mice were injected with MN-OA or vehicle control as described earlier and then sacrificed on day 20. Different organs (liver, kidney and spleen) and tumour samples were collected and digested in concentrated HCl for 48 h, followed by heating to evaporate the HCl. The samples were diluted in 10 mL of distilled water. The clear solution obtained after digestion was used for estimation of Fe content using phenanthroline method [Citation26]. In brief, 500 µL of sample was mixed with 500 µL of 10% hydroxylamine hydrochloride solution, 250 µL of acetate buffer (0.1 M, pH 4.5), 150 µL ammonia:water (1:1) mixture and 500 µL of 1,10-phenanthroline solution (0.25%) in the same order and incubated at room temperature for 15–20 min. The absorbance was measured at 510 nm. A 70-PPM ferrous ammonium sulphate solution was used as a standard for Fe estimation.
Biochemical analysis
Serum samples were analysed for the levels of creatinine, alanine transaminase (ALT) and alkaline phosphatase (ALP) using automated serum biochemical analyser (Randox, Kearneysville, WV, USA).
Statistical analysis
All values are presented as mean ± SEM and wherever required data was analysed using Student’s t-test. Statistical significance was determined at p < 0.05.
Results
MN-OA induced significant inhibition of tumour growth in combination with AMF
In our previous study [Citation20] we had demonstrated the hyperthermia efficacy of MN-OA in mouse fibrosarcoma (WEHI-164) tumour cells in vitro. To further validate our formulation under in vivo conditions, we established a fibrosarcoma tumour model in BALB/c mice. When the tumour bearing mice were administered with MN-OA, followed by MHT, they showed significant inhibition of tumour growth as determined by tumour volume measurements as compared to control (). Tumours treated with only MN-OA or MN-OA + AMF showed a significant decrease (p < 0.05) in percentage tumour growth () and a tumour growth delay (TGD) of 1.5 and 3 days, respectively. However, there was no significant difference in the tumour volume of control and AMF tumours up to day 8 after treatment.
Figure 1. Tumour growth studies. (A) Tumours were subjected to MHT and tumour volume was measured by Vernier caliper. The graph shows the increase in tumour volume measured on different days after initiating the therapy, and * indicates that values (for MN-OA+AMF) are significant at p < 0.05 with respect to control, for respective days. (B) Images captured by a live in vivo animal imaging system on day 8 after initiating the treatment. (C) WEHI-164 cells expressing the luciferase gene were transplanted in female BALB/c mice to obtain tumours and were further subjected to MHT. The luminescence intensity of tumours was measured on day 8 after initiation of treatment, using an in vivo live animal imaging system. Data is presented as mean ± SEM, N = 5 and * indicates that the values are significant at p < 0.05.
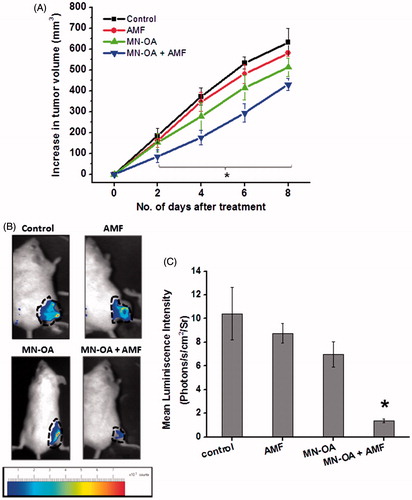
Table 1. Effect of MN-OA-induced MHT on fibrosarcoma tumours expressed in terms of percentage tumour growth.
For a more precise measurement of tumour growth inhibition, WEHI-164 tumour cells, stably expressing the luciferase enzyme, were transplanted in mice to obtain tumours and further subjected to MHT, followed by in vivo imaging on day 8 after initiating the treatment. Tumours treated with MN-OA + AMF showed ∼ seven fold reduction in luminescence intensity as compared to control (). Only MN-OA treatment and only induction heating (AMF) reduced the luminescence intensity by ∼1.5- and 1.2-fold, respectively.
MHT-induced growth inhibition of fibrosarcoma tumours involves apoptotic mechanism
To gain further mechanistic insight about the anti-tumour efficacy of MHT at tissue level, histopathological analyses were performed. Histopathological analysis of control tumour tissue sections exhibited high mitotic index, mild apoptosis, mild hyperchromasia (increased staining intensity of cell nuclei) and anaplasia (reversion of cells to less differentiated state) with infiltration of polymorphonuclear immune cells. On the contrary, tumours treated with either MN-OA or MN-OA + AMF showed presence of moderate and severe apoptotic regions (fragmented nuclei stained with H&E, respectively, along with low mitotic index and moderate hyperchromasia and anaplasia (). To further assess the apoptosis-specific events in tissue sections, TUNEL assay was performed. Results showed presence of severe apoptosis (69.3 ± 15.14 foci per field) in tumours treated with MN-OA + AMF, as against mild apoptosis in control (5.5 ± 0.07 foci per field) and C + H (3.5 ± 0.07 foci per field) tumours. In tumours treated with only MN-OA, moderate apoptosis (42 ± 2.0 foci per field) was observed (). These results suggested the role of apoptotic cell death in MN-OA-mediated MHT induced tumour growth inhibition.
Figure 2. Mechanism of tumour growth inhibition after MHT was studied by (A) histopathology analysis: tumour sections were stained with H&E, where the nucleus appears as dark blue or purple and cytosol appears dark pink in colour. Arrows indicate apoptotic nuclei; (B) TUNEL assay of tumour tissue sections: values in brackets indicate the average number of apoptotic foci per field ± SEM, N = 5 and arrows indicate apoptotic foci; (C) Western blot analysis for expression of apoptosis marker (cleaved caspase-3) in tumour tissue lysates on day 4 after initiating the treatment. Values represent the relative fold change as compared to control after normalising with GAPDH.
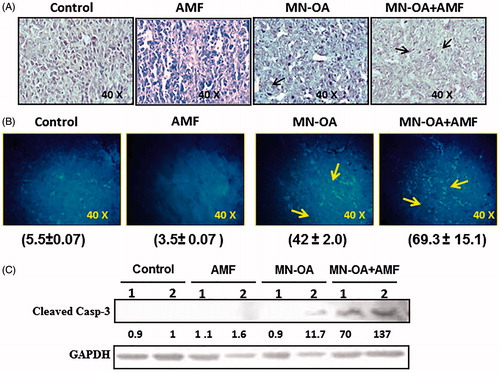
Furthermore, after MHT, the expression of apoptosis marker cleaved caspase-3 was studied by WB in tumour tissue lysates (). Results showed enhanced levels of expression of cleaved caspase-3 (∼ 103 fold, average value of two mice) in tumours treated with MN-OA + AMF, but was substantially lower in tumours treated with only MN-OA, only AMF, or control.
Tumours treated with MHT showed down-regulation of Hsp90 and AKT
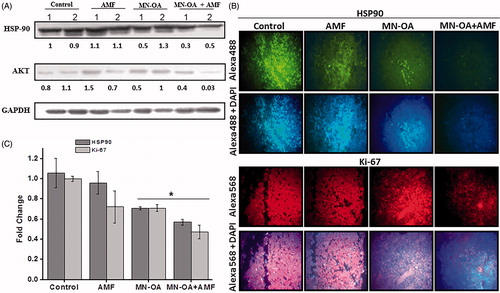
To further identify the signalling molecules that are involved in the induction of caspase-3 dependent apoptosis in tumour cells treated with MHT, expression of two cell survival proteins, heat shock protein 90 (Hsp90) and AKT/PKB was studied by WB and IF staining. Expression of Hsp90 (heat stress marker) did not vary significantly between control and AMF as studied by WB. Although only MN-OA treatment showed down-regulation of Hsp90, it was further significantly enhanced in tumours treated with MN-OA + AMF (∼0.45-fold (average value of two mice) reduction in expression of Hsp90). In addition, ∼0.25-fold (average value of two mice) reduction in the expression of cell survival marker, AKT was observed by WB analysis (). To further substantiate these results, the level of cellular expression of Hsp90 and Ki-67 (cell proliferation marker) was studied by IF staining. Results showed 0.57 ± 0.03-fold and 0.47 ± 0.06-fold reduction in expression of Hsp90 and Ki-67, respectively, for tumours treated with MN-OA + AMF as compared to controls ().
Figure 3. (A) Western blot analysis for the expression of heat shock marker (Hsp90) and cell survival marker (AKT/PKB) in tumour tissue lysates, on day 4 after initiating the treatment. Values represent the relative fold change as compared to control after normalising with GAPDH. (B) Fluorescence microscopy images for tumour sections stained by immunofluorescence staining for expression of cell proliferation marker (Ki-67) and Hsp90 (magnification: 40×). (C) The graph represents fold decrease in the expression of Hsp90 and Ki-67 as compared to control after normalising with MFI of DAPI. * indicates that the values are significant at p < 0.05.
Thermal imaging showed increase in tumour surface temperature after MHT
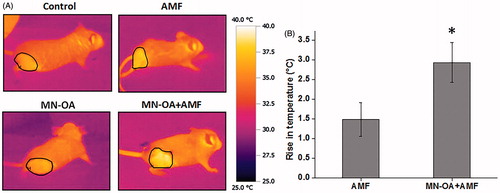
Since MN-OA + AMF showed high anti-tumour efficacy as compared to only MN-OA treatment, we monitored the rise in temperature of the tumour region of the mice after injection of MN-OA and induction heating using an IR camera. Analysis of thermal images of treated tumours (MN-OA + AMF), captured immediately after MHT, showed a significant temperature rise of ∼3 ± 0.5 °C (p < 0.05) at the tumour surface as compared to AMF tumours, which showed a temperature rise of ∼1.5 ± 0.4 °C ().
Figure 4. Measurement of tumour surface temperature using an IR camera. (A) Images for temperature rises at the tumour region in mice treated with only AMF and MN-OA + AMF, before and after the therapy. (B) Bar graph of temperature rises at the tumour region for control and treated mice. Values represent mean ± SEM and * indicates that values are significant at p < 0.05.
MN-OA remains localised mainly at the tumour site with minimal accumulation in other target organs
To study the retention of MN-OA in tumour tissue after intra-tumour injection and its distribution to other organs (liver, spleen and kidney), tumour and organ samples were collected 20 days post-injection and Fe localisation and its concentration were determined by Prussian blue staining and colorimetric estimation of Fe, respectively (). MN-OA-treated tumours showed ∼1.6-fold higher Fe content as compared to control tumours. The excess Fe content (∼50.95 µg/gm of tissue) amounted to ∼73.20% of the injected dose (). Moreover, the Fe content of different organs (liver, kidney and spleen) did not vary significantly between control and MN-OA-treated tumours as confirmed by Prussian blue staining () as well as by colorimetric estimation of iron content (). These results suggest that a major fraction of MN-OA is retained at the tumour site for a prolonged duration.
Figure 5. Study of bio-distribution of MN-OA (A) tissue sections of tumour and different organs (liver, kidney, spleen and lungs) were stained by Prussian blue staining to study localisation of Fe. Blue spots correspond to presence of Fe3+, (B) estimation of Fe content in tumour and different organs of control and treated mice using the phenanthroline method.
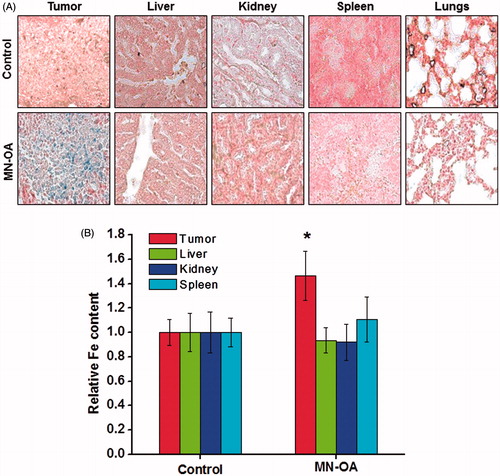
Table 2. Iron content in tumour and different organs in mice treated with MN-OA.
MN-OA showed minimal toxicity to liver and kidney function in mice
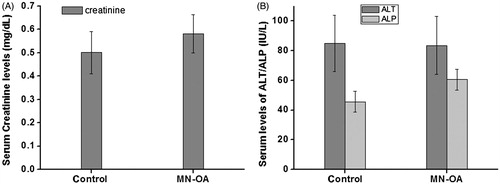
To examine the effect of MN-OA formulation on normal tissue functions, serum samples were assayed for the levels of creatinine and ALT/ALP as a measure of kidney and liver function, respectively (). The levels of these biochemical markers did not vary significantly between control and MN-OA-treated tumours, indicating that MN-OA had an insignificant effect on normal liver or kidney function in the MN-OA-treated mice.
Figure 6. Biochemical analysis of serum for (A) levels of creatinine and (B) levels of ALP and ALT.
Discussion
In the present study we have demonstrated the efficacy of MN-OA for MHT of fibrosarcoma tumours in BALB/c mice, and further investigated the underlying mechanism of tumour growth inhibition at cellular and molecular level. Our previous in vitro data, demonstrated the cancer cell-killing efficacy of MN-OA in combination with AMF in WEHI-164 cells [Citation20]. In the present work we validated these results under in vivo conditions using WEHI-164 cell-derived murine fibrosarcoma tumour tissue. MN-OA-mediated MHT showed significant inhibition of tumour growth ( and ). To evaluate their hyperthermia efficacy in an in vivo tumour model we used a sensitive technique of in vivo imaging, which quantifies the number of metabolically active tumour cells. The tumour burden was determined by measuring the luminescence intensity of transfected WEHI-164 tumour cells using a live animal imaging system. Results showed a significant decrease in tumour luminescence intensity for the animals treated with MHT as compared to only MN-OA treatment (). These results substantiate the MN-OA-induced inhibition of fibrosarcoma tumours in combination with AMF. The tumour growth inhibition measured by in vivo imaging (∼ seven fold) was found to be more prominent as compared to physical determination of tumour size or volume (∼1.5-fold). The difference in the observed efficacy of MN-OA-induced MHT by the above two methods could be explained by the difference in the parameters that are assessed. Live animal imaging technique is based on measurement of the luciferase enzyme activity expressed by viable tumour cells [Citation27]. On the contrary, manual measurements using a Vernier caliper cannot distinguish between viable and necrotic/non-viable tumour cells. In addition, manual measurements cannot account for the contribution of normal tissue in the vicinity of the tumour in tumour volume measurements.
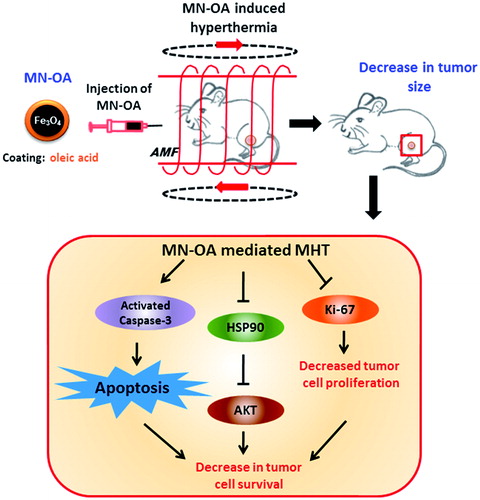
Experiments performed to determine the underlying mechanism of tumour growth inhibition after MHT showed presence of severe apoptosis in tumours-treated with MN-OA + AMF as against lower apoptosis in only MN-OA and control tumours (). This observation was found to be consistent with molecular detection of apoptosis by TUNEL assay, which showed higher apoptotic foci in tumours treated with MHT (). To further validate the mode of cell death, tumour tissue was examined for the expression of apoptotic and cell survival marker proteins (cleaved caspase-3 and AKT, respectively). Enhanced level of cleaved caspase-3 was observed in tumours treated with MN-OA + AMF as compared to only MN-OA treatment or control, confirming that MHT-induced tumour growth inhibition involves caspase-3-dependent apoptosis (). Moreover, decreased expression of AKT (pro-survival marker) and Ki-67 (cell proliferation marker) was observed in tumours treated with MHT (). This result was in accordance with the lower mitotic index observed in MHT-treated tumours (). Expression of Hsp90 was found to be down-regulated in MHT-treated tumours as confirmed by WB analysis as well as by IF staining ().
Over-expression of HSPs has been reported to play a role in development of thermo-tolerance in response to conventional hyperthermia therapy [Citation28,Citation29]. Interestingly, we observed a down-regulation of Hsp90 in response to MHT. These results suggest that MN-OA-mediated MHT may have dual implications in cancer therapy. Firstly, Hsp90-associated thermotolerance may not occur during MHT, and secondly it may also contribute to overcoming the Hsp90-mediated resistance during chemo- and/or radiation therapy. These explanations are consistent with the application of Hsp90 inhibitors such as SNX-2112 [Citation30], geldanamycin, 17-allylaminogeldanamycin (17-AAG) [Citation31], for example, alone or in combination with conventional cancer therapeutic modalities to accomplish better tumour control.
Our results also showed down-regulation of both Hsp90 and AKT in tumours treated with MHT. To further understand the observed Hsp90 regulation after MHT, we conducted in vitro experiments using WEHI-164 cells treated with different concentrations of Hsp90 inhibitor (17-AAG). Treatment with inhibitor showed a dose-dependent increase in Hsp90 and decrease in downstream pro-survival client protein, AKT (Supplementary Figure S3). Interestingly, treatment with MN-OA also showed up-regulation of Hsp90 and down-regulation of AKT. These results suggest that at cellular level MN-OA is inhibiting the Hsp90 function similar to 17-AAG. Hsp90 inhibitors are known to inhibit its function by binding to the ATP/ADP pocket, which in turn causes incomplete refolding of its client proteins, subsequently resulting in growth arrest and apoptosis of the treated cells. Moreover, Hsp90 is reported to be essential for functional maturation of AKT/PKB and thus down-regulation of Hsp90 is known to be associated with degradation of AKT [Citation32,Citation33]. To further test the possibility of MN-OA interaction with Hsp90, we performed co-localisation experiments by confocal microscopy in control and MN-OA-treated tumour tissue. Our results showed co-localisation of MN-OA with Hsp90 in the tumour tissue (Supplementary Figure S4) as compared to control tissue. In view of this observation it is possible that heat generation during MHT may lead to the down-regulation of associated proteins such as Hsp90 (Scheme 3). Under in vitro and in vivo conditions, the levels of Hsp90 were differentially regulated by only MN-OA treatment ( and Supplementary Figure S3). This may be attributed to the difference in MN-OA dose and time period after which the samples were analysed. These observations provide scope for further studies to understand the molecular interactions after MN-OA treatment and their role in tumour toxicity during MHT.
Scheme 3. Proposed mechanism of MHT-induced inhibition of tumour growth.
To measure the tumor surface temperature, a non-invasive method of thermal imaging using IR camera was employed. MHT showed significant increase in tumour surface temperature (∼3 °C) as compared to control (∼1.5 °C) (). Although the tumour surface temperature after MN-OA-mediated MHT did not reach hyperthermic temperature (42 °C), the tumour growth inhibition observed suggests a higher temperature in tumor bulk, which however, needs to be measured using a suitable temperature probe.
Moreover, in the present work we have studied the bio-distribution of MN-OA after intra-tumour injection and determined whether MN-OA has any side effects on the normal organ function in mice. Our iron estimation data showed that ∼73% of injected MN-OA was retained at the tumour site 20 days post-injection, indicating that MN-OA is strongly associated with the tumour microenvironment (). Such a prolonged retention of a significant amount of MN-OA at the target site implies the potential for repeated HT without the need for repeated MN-OA injections. Consequently, the observed insignificant difference in the iron content of liver, kidney and spleen, between control and treated mice, could be a result of such an association of MN-OA with the tumour, which would be interesting to study in further detail. This observation was further corroborated by the results of serum biochemical analysis, which showed that MN-OA had minimal side effects in terms of organ function (no significant difference in levels of creatinine, ALP and ALT between control and treated mice) ().
Conclusion
This study delineates the mechanism of tumour growth inhibition induced by MN-OA-mediated MHT in a preclinical tumour model of fibrosarcoma. MHT induced a significant tumour growth inhibition by apoptosis but showed insignificant toxicity to the normal organ function in mice. The observed decrease in tumour volume seems to involve modulation of Hsp90/AKT signalling and caspase-3-dependent apoptosis. Thus, MN-OA provides a potential formulation for MHT, which however, needs to be further tested for combinatorial therapy along with radiation and chemotherapy, in order to accomplish better tumour control.
Supplementary material avialble online
Supplementary_Figures.pdf
Download PDF (441.4 KB)Acknowledgements
The authors acknowledge the help of Manjoor Ali, Radiation Biology & Health Sciences Division, BARC, Mumbai, India for microscopy, Milind Kamble and P. S. Dange for their assistance during biochemical analysis of serum samples, Hansa Yadav for technical support, and Asmita Sakpal, ACTREC, for technical assistance during transfection experiments.
Declaration of interest
This research work was funded by Bhabha Atomic Research Centre, Department of Atomic Energy, Government of India. The authors alone are responsible for the content and writing of the paper.
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69–90
- Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics 2012;2:3–44
- Chidambaram M, Manavalan R, Kathiresan K. Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J Pharm Pharm Sci 2011;14:67–77
- Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, et al. Hyperthermia in combined treatment of cancer. Lancet Oncol 2002;3:487–97
- Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, et al. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol 2002;43:33–56
- Hegyi G, Szigeti GP, Szasz A. Hyperthermia versus oncothermia: Cellular effects in complementary cancer therapy. Evid Based Complement Alternat Med 2013;2013:672873–84
- Bleehen NM. Hyperthermia in the treatment of cancer. Br J Cancer Suppl 1982;5:96–100
- Cheung AY, Neyzari A. Deep local hyperthermia for cancer therapy: External electromagnetic and ultrasound techniques. Cancer Res 1984;44:S4736–44
- Habash RW, Bansal R, Krewski D, Alhafid HT. Thermal therapy, part 2: Hyperthermia techniques. Crit Rev Biomed Eng 2006;34:491–542
- Huang HS, Hainfeld JF. Intravenous magnetic nanoparticle cancer hyperthermia. Int J Nanomedicine 2013;8:2521–32
- Kobayashi T. Cancer hyperthermia using magnetic nanoparticles. Biotechnol J 2011;6:1342–7
- Kozissnik B, Bohorquez AC, Dobson J, Rinaldi C. Magnetic fluid hyperthermia: Advances, challenges, and opportunity. Int J Hyperthermia 2013;29:706–14
- Sadhukha T, Wiedmann, TS, Panyam J. Inhalable magnetic nanoparticles for targeted hyperthermia in lung cancer therapy. Biomaterials 2013;34:5163–71
- Laurent S, Bridot JL, Elst LV, Muller RN. Magnetic iron oxide nanoparticles for biomedical applications. Future Med Chem 2010;2:427–49
- Liu G, Gao J, Ai H, Chen X. Applications and potential toxicity of magnetic iron oxide nanoparticles. Small 2013;9:1533–45
- Amstad E, Textor M, Reimhult E. Stabilization and functionalization of iron oxide nanoparticles for biomedical applications. Nanoscale 2011;3:2819–43
- Hadjipanayis CG, Machaidze R, Kaluzova M, Wang L, Schuette AJ, Chen H, et al. EGFRvIII antibody-conjugated iron oxide nanoparticles for magnetic resonance imaging-guided convection-enhanced delivery and targeted therapy of glioblastoma. Cancer Res 2010;70:6303–12
- Hansen L, Larsen EK, Nielsen EH, Iversen F, Liu Z, Thomsen K, et al. Targeting of peptide conjugated magnetic nanoparticles to urokinase plasminogen activator receptor (uPAR) expressing cells. Nanoscale 2013;5:8192–201
- Akbarzadeh A, Samiei M, Davaran S. Magnetic nanoparticles: Preparation, physical properties, and applications in biomedicine. Nanoscale Res Lett 2012;7:144–56
- Jadhav NV, Prasad AI, Kumar A, Mishra R, Dhara S, Babu KR, et al. Synthesis of oleic acid functionalized Fe3O4 magnetic nanoparticles and studying their interaction with tumor cells for potential hyperthermia applications. Colloids Surf B Biointerfaces 2013;108:158–68
- Balivada S, Rachakatla RS, Wang H, Samarakoon TN, Dani RK, Pyle M, et al. A/C magnetic hyperthermia of melanoma mediated by iron(0)/iron oxide core/shell magnetic nanoparticles: A mouse study. BMC Cancer 2010;10:119–27
- Silva AC, Oliveira TR, Mamani JB, Malheiros SM, Malavolta L, Pavon LF, et al. Application of hyperthermia induced by superparamagnetic iron oxide nanoparticles in glioma treatment. Int J Nanomedicine 2011;6:591–603
- Kim JS, Yoon TJ, Yu KN, Kim BG, Park SJ, Kim H, et al. Toxicity and tissue distribution of magnetic nanoparticles in mice. Toxicol Sci 2006;89:338–47
- Gaikwad SM, Gunjal L, Junutula AR, Astanehe A, Gambhir SS, Ray P. Non-invasive imaging of phosphoinositide-3-kinase-catalytic-subunit-alpha (PIK3CA) promoter modulation in small animal models. PLoS One 2013;8:e55971
- Desai S, Laskar S, Pandey BN. Autocrine IL-8 and VEGF mediate epithelial-mesenchymal transition and invasiveness via p38/JNK-ATF-2 signalling in A549 lung cancer cells. Cell Signal 2013;25:1780–91
- Rana S, Jadhav NV, Barick KC, Pandey BN, Hassan PA. Polyaniline shell cross-linked Fe3O4 magnetic nanoparticles for heat activated killing of cancer cells. Dalton Trans 2014;43:12263–71
- Jost SC, Collins L, Travers S, Piwnica-Worms D, Garbow JR. Measuring brain tumor growth: A combined BLI/MRI strategy. Mol Imaging 2009;8:245–53
- Kregel KC. Molecular biology of thermoregulation, invited review: Heat shock proteins: Modifying factors in physiologicalstress responses and acquired thermotolerance. J Appl Physiol 2002;92:2177–86
- Carper SW, Duffy JJ and Gerner EW. Heat shock proteins in thermotolerance and other cellular processes. Cancer Res 1987;47:5249–55
- Liu KS, Liu H, Qi JH, Liu QY, Liu Z, Xia M, et al. SNX-2112, an Hsp90 inhibitor, induces apoptosis and autophagy via degradation of Hsp90 client proteins in human melanoma A-375 cells. Cancer Lett 2012;318:180–8
- Okamoto J, Mikami I, Tominaga Y, Kuchenbecker KM, Lin YC, Bravo DT, et al. Inhibition of Hsp90 leads to cell cycle arrest and apoptosis in human malignant pleural mesothelioma. J Thorac Oncol 2008;3:1089–95
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. AKT forms an intracellular complex with heat shock protein 90 (Hsp90) and CDC37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem 2002;277:39858–66
- Sato S, Fujita N, Tsuruo T. Modulation of AKT kinase activity by binding to Hsp90. Proc Natl Acad Sci USA 2000;97:10832–37