
Abstract
Hypoxic-ischaemic brain injury involves increased oxidative stress. In asphyxiated newborns iron deposited in the brain catalyses formation of reactive oxygen species. Glutathione (GSH) and vitamin E are key factors protecting cells against such agents. Our previous investigation has demonstrated that newborn rats, showing physiological low body temperature as well as their hyperthermic counterparts injected with deferoxamine (DF) are protected against iron-mediated, delayed neurotoxicity of perinatal asphyxia. Therefore, we decided to study the effects of body temperature and DF on the antioxidant status of the brain in rats exposed neonatally to critical anoxia. Two-day-old newborn rats were exposed to anoxia in 100% nitrogen atmosphere for 10 min. Rectal temperature was kept at 33 °C (physiological to rat neonates), or elevated to the level typical of healthy adult rats (37 °C), or of febrile adult rats (39 °C). Half of the rats exposed to anoxia under extremely hyperthermic conditions (39 °C) were injected with DF. Cerebral concentrations of malondialdehyde (MDA, lipid peroxidation marker) and the levels of GSH and vitamin E were determined post-mortem, (1) immediately after anoxia, (2) 3 days, (3) 7 days, and (4) 2 weeks after anoxia. There were no post-anoxic changes in MDA, GSH and vitamin E concentrations in newborn rats kept at body temperature of 33 °C. In contrast, perinatal anoxia at elevated body temperatures intensified oxidative stress and depleted the antioxidant pool in a temperature-dependent manner. Both the depletion of antioxidants and lipid peroxidation were prevented by post-anoxic DF injection. The data support the idea that hyperthermia may extend perinatal anoxia-induced brain lesions.
Introduction
Neonatal anoxia is one of the major causes of brain injury in premature infants [Citation1] and may lead to long-lasting disturbances in their development. The oxygen equilibrium restoration after hypoxia-ischaemia results in a burst of free radical production that damages tissues by peroxidation of unsaturated lipids within the cellular membranes [Citation2]. Postponed neurodegenerative processes are closely associated with abnormal iron metabolism that is a cofactor of free-radical reactions.
The average content of free iron in the plasma of prematurely born neonates is much higher than that of timely born ones, which along with low levels of antioxidant enzymes and low-molecular substances, especially reduced glutathione (GSH) and vitamin E, makes the neonates particularly sensitive to free radical-mediated injury [Citation3–5].
GSH has many biological functions, including maintenance of membrane protein sulphydryl groups in the reduced form. The oxidation of them can otherwise cause changes in cellular structure and function [Citation6].
Vitamin E is recognised as another crucial low-molecular antioxidant in the organism, functioning predominantly in maintaining the integrity of biological cell membranes [Citation7]. Thus, maintenance of GSH and vitamin E levels seems to play a pivotal role for cellular defence against anoxia-induced oxidative injury.
Our up-to-date studies unequivocally suggest that an exceptional anoxia tolerance in rat neonates depends upon two defensive reactions: (1) their ability to autoresuscitate due to gasping, and (2) maintaining body temperature at a level of 32–33 °C [Citation8]. Body temperature maintained during brain anoxia/ischaemia and reoxygenation/reperfusion is a very important factor that affects the extent of the organ lesions. Diminished temperature is an outstanding neuroprotective factor – it greatly minimises the intensity of harmful neuron-damaging processes [Citation9]. The inhibition of brain oxidative metabolism seems to be the main cause of extreme efficacy of such a moderate temperature decrease [Citation10]. It may allow the decreased oxygen to match supply under anoxic conditions [Citation11,Citation12]. Moreover, decreased body temperature prevents post-anoxic metabolic acidosis and hyperferraemia [Citation13,Citation14] and it results in significant protection in terms of survival, lesion size, and histological abnormalities [Citation9,Citation15].
Low body temperature recorded during the first several days after parturition may be an important element of defence strategy against perinatal anoxia. Body temperature of 32–33 °C, which is typical of newborn rats, is the same as that considered to be the most effective for preventing brain lesions during therapeutic hypothermia in asphyxiated human neonates [Citation16,Citation17].
On the other hand, maintaining body temperature during anoxia at a level typical of adult rats or higher results in neurological injury of newborn rats [Citation18–21]. We would like to focus on the risk of anoxia-induced changes in oxidative–antioxidative status of the rat’s brain due to overheating (hyperthermia), because in our previous studies [Citation14] we documented cerebral iron accumulation after perinatal anoxia at body temperature elevated to 39 °C. The cerebral iron deposition was not recorded at the neonate’s normal body temperature or at highly hyperthermic body temperature, when the animals were administered deferoxamine (DF, an iron chelator) immediately after the anoxia episode [Citation14]. Used experimentally, DF was efficacious in ameliorating hypoxia-ischaemia-induced reperfusion injury of the brain in newborn lambs [Citation22,Citation23], mice [Citation24] and rats [Citation25].
Assuming that naturally decreased body temperature of newborn rats is a beneficial adaptation, as had been suggested previously [Citation8], we hypothesised that the increase in body temperature to the level typical of adults or higher would constitute an additional metabolic stress to the animal. Therefore, we expected that the perinatal anoxia at body temperature elevated to 37 °C and 39 °C would intensify the post-anoxic oxidative stress in rat pups and deplete the cerebral pool of GSH and vitamin E, whereas physiological body temperature of neonates would prevent cerebral lipid peroxidation and assure an optimal antioxidant protection of their brain. Furthermore, we supposed that blocking the post-anoxic hyperferraemia by means of DF administration would diminish the anoxia-induced oxidative stress and antioxidant defence disturbance.
Accordingly, the aim of the present study was to investigate the effects of neonatal anoxia, hyperthermia and pharmacological treatment with DF on changes in concentrations of MDA and low molecular weight antioxidants: reduced GSH and vitamin E in the brain of newborn rats during the first 2 weeks post-anoxia.
Materials and methods
Animal housing, care and application of experimental procedures have been approved by the Local Committee on the Use and Care of Laboratory Animals in Bydgoszcz, Poland (permission no. 28/2012). Every surgery was performed under ketamine anaesthesia, and all efforts were made to minimise animals suffering.
Animals
A total of 180 newborn Wistar rats of both sexes at 2 days of age, weighing approximately 7–8 g, were used for experiments. The pups (before and after exposure to anoxic or normoxic procedure) were housed, together with their dams, in acrylic cages lined with wood shavings, at a room temperature of 20–22 °C, fed with commercial chow pellets and provided with water ad libitum. The deliveries were spontaneous.
Exposure to anoxia
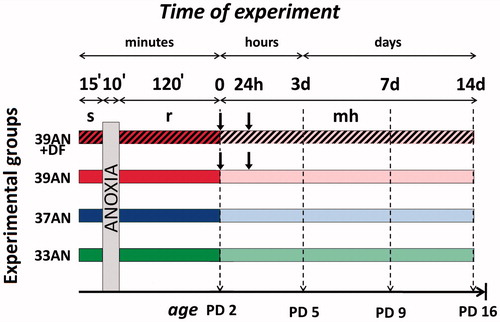
shows details of the experimental design. The litters were assigned to either the anoxic or the corresponding control groups (see below) such that each of them was composed of subjects coming from 2–3 different litters. Animals were randomly divided into eight experimental groups: two normothermic groups, composed of animals maintaining neonatal body temperature of 33 °C (which is the physiological body temperature of newborn rats) exposed to (group 1) normoxic (33C, n = 24) and (group 2) anoxic conditions (33AN, n = 24), and six hyperthermic groups, composed of animals forced to stabilise their body temperature at either 37 °C (which is the body temperature typical of adult individuals) exposed to (group 3) normoxic (37C, n = 24) and (group 4) anoxic (37AN, n = 24) conditions; or animals forced to maintain their body temperature at 39 °C (which is the body temperature typical for febrile adult individuals) exposed to (group 5) normoxic conditions with subcutaneous injection of saline (39C, n = 24), or (group 6) injected with DF (39C + DF, n = 18); and (group 7) exposed to anoxic conditions and injected with saline (39AN, n = 24) or (group 8) DF (39AN + DF, n = 18). The procedures used to elicit anoxia, to manipulate neonatal body temperature, and pharmacological treatment of the rats have been described in detail in our previous work [Citation26]. Concisely, prior to experiments, the miniature (0.1 mm in diameter) copper-constantan thermocouples (Physitemp Instruments, Clifton, NJ, USA), connected to the recording system (3000-E52 Iso-Thermex Interface & Software, Columbus Instruments, Columbus, OH, USA), were inserted to a depth of 5 mm into the colon of the rat pups. The overall accuracy of the measurements was higher than ± 0.05 °C. Then pups were placed individually in 200-mL plethysmographic chambers, which were submerged in a water thermostat under controlled thermal conditions (∼31.5 °C, ∼35.5 °C and ∼37.5 °C) in order to maintain their body temperature at 33 °C, 37 °C and 39 °C, respectively. Rats were exposed to anoxic conditions starting 15 min after body temperatures had reached the scheduled level. To induce anoxia, animals were subjected to 100% nitrogen atmosphere for 10 min. Gasping was continuously recorded using the barometric method [Citation8,Citation13] and none of pups reached the stage of lethal asphyxia (when a critical phase of accelerated and shallow gasping occurred; and continued exposure to anoxia would have led to sudden death within seconds). After anoxia the animals were exposed to atmospheric air at unchanged temperature for 120 min. Control rats were exposed to atmospheric air in the same chambers for the same period of time in the respective thermal conditions. A subcutaneous injection of saline solution or DF (deferoxamine mesylate, Sigma-Aldrich, Steinheim, Germany, 100 mg/kg s.c.) was given into the loose skin of the neck twice: immediately after anoxia and 24 h later (groups 5–8). After the exposure to either anoxia or the control treatment, the pups were returned to their mothers’ cages, where they stayed until decapitation. Pups from each group were observed daily from day 1–14 after the exposure for experimental conditions in order to evaluate their physical development. There was no effect of experimental conditions on the time of eye opening, hair appearance, pinna unfolding, and incisors eruption at any time point post-exposure. Differences in body weight between offspring from different groups were not detectable either. The pups from each experimental group were decapitated under ketamine anaesthesia (100 mg/kg s.c.) at four different time points after exposure to the above mentioned conditions: (1) immediately (except 39C-DF and 39AN-DF groups), (2) 72 h, (3) 7 days and (4) 2 weeks after the experiment. Levels of lipid peroxidation products (MDA), reduced GSH and vitamin E concentrations have been determined in brain homogenates.
Figure 1. Experimental protocol. Control rats were exposed to atmospheric air throughout the same period under respective thermal conditions. Abbreviations: s - stabilisation period; r - reoxygenation period; ![]()
Sample preparation
Homogenates of the collected brains were prepared using a Potter homogeniser (Kleinfeld Labortechnik, Gehrden, Germany) for 2–3 min. The samples were homogenised in 0.9% ice-cold (1:10, w/v) saline and then were centrifuged at 12 000 g for 10 min at 4 °C. Supernatants were collected and kept at −80 °C until they were used for determination of MDA, reduced GSH and vitamin E concentrations.
Protein concentration assay
Total protein concentrations in the homogenates were estimated by Folin phenol methods, described by Lowry et al. [Citation27], using bovine serum albumin (Sigma-Aldrich) as a standard. For each sample 0.5 mL of the homogenate was used. The absorbance of the colour complex was determined at 750 nm. In the blank sample the homogenate was replaced with NaOH.
Malondialdehyde assay
The formation of MDA was used as an indicator of lipid peroxidation. MDA concentrations were measured according to the method described by Mateos et al. [Citation28]. For each sample 500 μL of the brain homogenate was mixed with 100 μL of 0.01% solution of butyl-4-hydroxytoluene in acetone and 500 μL of 5% trichloroacetic acid (TCA).The samples were incubated for 10 min at room temperature and centrifuged for 10 min at 4000 g. Then 500 μL of supernatant was mixed with 4.5 mL of 15% (w/v) TCA solution in 25 mM HCl and 0.375% (w/v) thiobarbituric acid (TBA) in 25 mM HCl (hydrochloric acid) and samples were kept for 20 min in a boiling water bath. After cooling, the samples were centrifuged at 12 000g for 10 min. From each sample 200 µL volume of supernatant was transferred to a vial, then a 50 µL aliquot was injected into the high-performance liquid chromatography (HPLC) system. Excitation and emission wavelengths were of 532 and 553 nm, respectively. The mobile phase was composed of methanol in 50 mM ammonium formate buffer of pH 6.5 (40: 60, v/v), the flow rate was 1 ml/min. MDA concentration was measured by HPLC connected to a fluorescence detector (binary HPLC pump 1525 and 2475 multi λ fluorescence detector, Waters, Milan, Italy), equipped with a Polaris C18 column, 150 × 4.6 mm ID, particle size 5 μm (Varian, USA). The concentration of MDA was expressed in nmol/mg protein.
Reduced glutathione assay
Reduced GSH concentration was determined using the Ellman method [Citation29]. Briefly, 500 µL of the brain homogenate was mixed thoroughly with 1000 µL of a stock solution containing: 500 µL of 10% TCA and 500 µl of 10-mM ethylenediaminetetraacetic acid (EDTA) and then the samples were centrifuged for 5 min at 10 000g. Samples of each supernatant were collected and their 200 µL volumes were used for determination of reduced GSH. The samples were added to 2.3 mL of deionised water, 100 µL of 0.3 M EDTA, 300 µL of 0.32 M tris(hydroxymethyl)aminomethane (TRIS) and 100 µL of 0.086 mM 5,5'-dithiobis-2-nitrobenzoic acid (DTNB), and were maintained at 10 °C for 10 min and then they were analysed spectrophotometrically at 412 nm. The blank sample did not contain the homogenate. The optical density of GSH was measured in the “Cary 100” spectrophotometer (Varian Inc., Palo Alto; CA; USA). GSH concentration was expressed in µmol/g tissue.
Vitamin E assay
Before analysis, 200 µL volumes of brain homogenates were mixed thoroughly with 500 µL of acetonitrile. Then the samples were shaken with 4 mL of hexane and centrifuged at 3000 g for 5 min to remove participated protein. The samples were then frozen at −80 °C for 45 min, decanted to clean tubes, evaporated until dry in a nitrogen atmosphere at 40 °C for 60 min and dissolved by adding 100 µL of mobile phase containing acetonitrile and methanol (95:5) and mixed with the use of ultrasound. As an internal reference α-tocopheryl acetate (Sigma-Aldrich) was used. The concentration of α-tocopheryl was measured with the use of the HPLC system (Varian Inc., Palo Alto; CA; USA) consisting of a ProStar 210 pump, a Kinetex column (2.6 μm C18 75 × 4.6 mm, a flow rate of 2 mL/min) and ProStar 363 fluorescence detector [Citation30]. The level of vitamin E was analysed at 295 nm. The concentration of vitamin E was expressed in µg/mL homogenate.
Data analysis
Results were analysed by two-way ANOVA for multiple group comparison in a 2 × 3 factorial design, where experimental conditions (anoxia vs. control) and temperature (33, 37 and 39 °C) were the factors. Each analysis was followed by multiple comparison using Tukey post hoc test. The effect of treatment with DF in hyperthermic groups was analysed by one-way ANOVA. The significance level was set at p < 0.05 for all statistical tests. Data analyses were performed with the use of IBM SPSS Statistics 22.0.
Results
MDA concentration
MDA levels were significantly higher in both control hyperthermic groups (37C and 39C) 3, 7 and 14 days after the experiment compared with those recorded in control animals maintaining physiological body temperature of 33 °C (33C) (, p < 0.001). In addition, immediately after the exposure to various body temperatures the level of MDA was significantly higher in the 39C group than in animals from the 33C group (, p < 0.001). However, there was no difference in the values of MDA between animals from groups 33C and 37C (). MDA concentration was significantly higher in 39C in comparison to rats from the 37C group immediately and 3 days after the exposure (, p < 0.01, , p < 0.001).
Figure 2. Effects of neonatal anoxia, neonatal body temperature (33 °C, 37 °C and 39 °C) and chelation of iron with deferoxamine (DF) on cerebral concentration of malondialdehyde (MDA) in newborn rats immediately (panel A), 3 (panel B), 7 (panel C) and 14 (panel D) days after exposure to anoxia or to control conditions. Data are presented as means ± SEM. Statistically significant differences between anoxic animals and their control counterparts at the same body temperatures are denoted: #p < 0.05 and ###p < 0.001; and those between the temperature variants of control or anoxic animals are denoted: **p < 0.01 and ***p < 0.001. Statistically significant differences between rats forced to maintain neonatal body temperature of 39 °C injected with saline and their counterparts injected with deferoxamine are denoted: ^^p < 0.01 and ^^^p < 0.001.
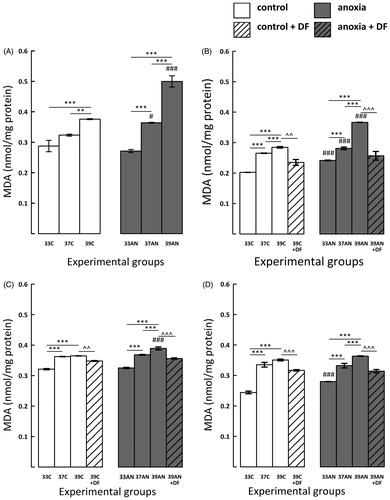
MDA concentration was higher in anoxic hyperthermic rats (39AN) compared with their control counterparts (39C), not only immediately but also 3 and 7 days after the exposure (, p < 0.001). Furthermore, during the first days following anoxia the MDA levels in the 37AN group were significantly higher than those recorded in their normoxic counterparts (37C) (, p < 0.05, , p < 0.001), and later on the difference disappeared ().
Immediately and 7 days after exposure to anoxia the concentration of MDA in rats having physiological body temperature of 33 °C (33AN) did not differ from their normoxic counterparts (33C) (), although 3 and 14 days after the insult MDA levels in 33AN were higher than in the control group (33C) (, p < 0.001).
The peak in the level of MDA in the 39AN group was recorded immediately after exposure to anoxia, then it slowly declined during the following days, but was still significantly higher than in animals from the 37AN and 33AN groups (, p < 0.001). It must be stressed that concentrations of MDA in 37AN animals also remained permanently significantly higher (p < 0.001) than those recorded in animals from the 33AN group ().
DF treatment significantly reduced the increases in MDA concentration in the hyperthermic groups of animals (39C-DF and 39AN-DF) compared to their counterparts injected with saline (39C and 39AN) in all measurement time points ().
GSH concentration
Immediately after the normoxic exposure to different thermal conditions as well as 3 and 7 days later GSH concentration in both control hyperthermic groups (37C and 39C) was significantly lower compared to those observed in the 33C group (, p < 0.001). Lower levels of GSH were also recorded in rats of the 39C group compared to animals from the 37C group during the first three days after the exposure (, p < 0.001). On day 14 after the insult the concentrations of GSH in all control experimental groups were comparable ().
Figure 3. Effects of neonatal anoxia, neonatal body temperature (33 °C, 37 °C and 39 °C) and chelation of iron with deferoxamine (DF) on cerebral concentration of glutathione (GSH) in newborn rats immediately (panel A), 3 (panel B), 7 (panel C) and 14 (panel D) days after exposure to anoxia or to control conditions. Data are presented as means ± SEM. Statistically significant differences between anoxic animals and their control counterparts at the same body temperatures are denoted: #p < 0.05, ##p < 0.01 and ###p < 0.001; and those between the temperature variants of control or anoxic animals are denoted: *p < 0.05 and ***p < 0.001. Statistically significant differences between rats forced to maintain neonatal body temperature of 39 °C injected with saline and their counterparts injected with deferoxamine are denoted: ^p < 0.05 and ^^^p < 0.001.
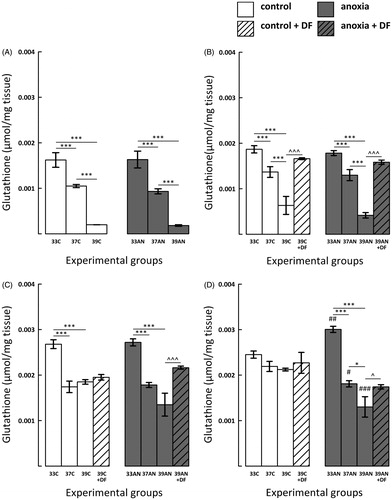
There were no differences in GSH level between control and anoxic groups throughout the first week post-exposure (). On day 14 after the insult in rats having neonatal normal body temperature of 33 °C (33AN) there was a significant increase of GSH concentration (, p < 0.01) compared to their control counterparts (33C). In contrast, the level of GSH was significantly lower in 37AN (p < 0.05) and 39AN (p < 0.001) groups in comparison to their normoxic counterparts ().
Post-anoxic GSH levels were lower in hyperthermic rats (both from 37AN and 39AN groups) in comparison to the 33AN group throughout the entire experimental period (, p < 0.001). Moreover, immediately, as well as 3 and 14 days after exposure to anoxia the concentration of GSH was significantly lower in the 39AN group in comparison to the level of the antioxidant in animals from 37AN group (, p < 0.001; , p < 0.05).
DF administration significantly increased GSH concentration in 39C-DF and 39AN-DF groups 3 days after the experiment (, p < 0.001). In addition, 7 and 14 days post-anoxia the concentration of GSH was also significantly higher in DF-treated anoxic rats (39AN + DF) in comparison to their saline-treated counterparts (39AN) (, p < 0.001, , p < 0.05). On the other hand, there were no significant increases of GSH concentration in DF-treated control hyperthermic rats 7 and 14 days post-exposure ().
Vitamin E concentration
In normoxic hyperthermic rats (39C) concentration of vitamin E was reduced compared with the 33C group in all measurement time points (): immediately (p < 0.05), 3 (p < 0.001), 7 (p < 0.05) and 14 (p < 0.001) days post-exposure. Furthermore, immediately, as well as 3 and 14 days after exposure to different thermal conditions the concentration of vitamin E was significantly lower also in 37C group in comparison to rats from 33C group (, p < 0.05; , p < 0.001).
Figure 4. Effects of neonatal anoxia, neonatal body temperature (33 °C, 37 °C and 39 °C) and chelation of iron with deferoxamine (DF) on cerebral concentration of vitamin E in newborn rats immediately (panel A), 3 (panel B), 7 (panel C) and 14 (panel D) days after exposure to anoxia or to control conditions. Data are presented as means ± SEM. Statistically significant differences between anoxic animals and their control counterparts at the same body temperatures are denoted: #p < 0.05 and ###p < 0.001; and those between the temperature variants of control or anoxic animals are denoted: *p < 0.05,**p < 0.01 and ***p < 0.001. Statistically significant differences between rats forced to maintain neonatal body temperature of 39 °C injected with saline and their counterparts injected with deferoxamine are denoted: ^p < 0.05 and ^^p < 0.01.
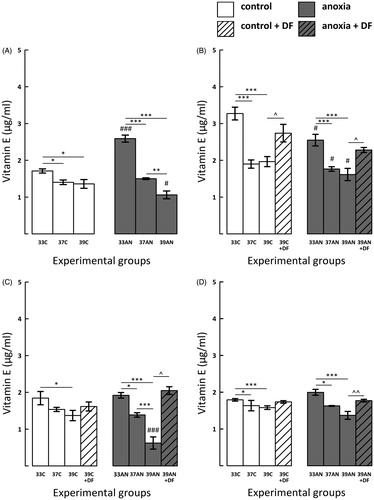
Immediately after anoxia the level of vitamin E in 33AN group was increased in comparison to the 33C group (, p < 0.001), but in the extremely hyperthermic group of rats (39AN) it was significantly lower than their normoxic counterparts (39C) (, p < 0.05). In all anoxic groups concentration of vitamin E was significantly reduced in comparison to their control counterparts 72 h after exposure to anoxia (, p < 0.05). On day 7 post-anoxia the significantly lower level of vitamin E was recorded only in the extremely hyperthermic group of rats (39AN) in comparison to their normoxic counterparts (39C) (, p < 0.001). On day 14 the differences between control animals and their anoxic counterparts were not significant ().
In both hyperthermic groups of rats (37AN and 39AN) post-anoxic concentration of vitamin E was lower than in the 33AN group immediately after the exposure (, p < 0.001); this effect was also maintained 3 days later (, p < 0.001) and persisted for 7 and 14 days post-exposure (; 37AN vs. 33AN, p < 0.05; 39AN vs. 33AN, p < 0.001). Furthermore, vitamin E level in the 39AN group was reduced in comparison to the 37AN group immediately (, p < 0.01) and 7 days (, p < 0.001) post-anoxia.
The significantly increased level of vitamin E was recorded in anoxic DF-treated rats 3, 7 days post-anoxia (, p < 0.05) and 14 days later (, p < 0.01). DF treatment significantly augmented the cerebral pool of vitamin E also in normoxic hyperthermic rats 3 days post-exposure (; p < 0.05), and later on concentration of the vitamin tended to be increased but the differences were non-significant.
Discussion
In the present study we evaluated the effects of body temperature on anoxia-induced changes in MDA, GSH and vitamin E levels in the brain of newborn rats.
Animal studies have indicated that GSH and vitamin E are depleted during hypoxia-ischaemia and that their maintenance prevents the oxidative stress-induced damage [Citation31–36]. For example, GSH level was significantly reduced at 3 and 6 h after hypoxia-ischaemia in the striatum of newborn piglets [Citation32]. Moreover, the concentration of GSH decreased after hypoxia-ischaemia in 7-day-old rats [Citation35,Citation36].
The present data provides new evidence that a normothermic body temperature of newborn rats of 33 °C prevents post-asphyxic disturbances in cerebral oxidant homeostasis. Although increases in MDA level occured also in rats of this temperature group 3 days () and 14 days () post-anoxia, they were clearly less pronounced than those in both hyperthermic groups. A long-term increment of concentration of two important low-molecular antioxidants: GSH and vitamin E ( and ) is more evidence of the beneficial effect of the maintenance of low (33 °C) physiological body temperature during both the anoxia and reoxygenation period. The results of the present study are consistent with the report of Karibe et al. [Citation10] that intra-ischaemic hypothermia (33 °C) applied in adult rats suppressed GSH depletion in their cortex 3 h after the insult.
Maintenance of GSH level is pivotal for cellular defence against oxidative injury and thus for cellular homeostasis. Recovery of total GSH levels has been shown to precede recuperation of neuronal tissue from oxidative stress after cerebral ischaemia [Citation33]. GSH seems to be especially important for newborn rats. It has been shown that oligodendrocyte progenitors have the increased vulnerability to oxidative stress correlated with their greater dependence upon intracellular GSH for survival [Citation4].
A lot of studies have reported that administration of α-tocopherol exerts protective effects against hypoxic-ischaemic events [Citation37,Citation38]. Our results also suggest that both GSH and vitamin E are responsible, at least partially, for decreasing the level of lipid peroxidation in rats allowed to maintain physiological body temperature of 33 °C during the period of anoxic insult and reoxygenation. The action of α-tocopherol is mainly attributed to maintaining the structure and/or metabolic integrity of nerve cells [Citation7,Citation37,Citation38], regulating cell signalling and gene expression [Citation39], and this compound is implicated in the turnover of different neurotransmitters in the rat brain [Citation40].
Our earlier investigations demonstrated that body temperature maintained during and immediately after anoxia could be crucial factor influencing the development of post-anoxic pathological processes in neonatal brain [Citation8,Citation13,Citation14]. The present results suggest that endogenous low-molecular antioxidants play a role in neuroprotection provided by decreased body temperature of newborn rats. The neuroprotective effects of the decreased body temperature should be discussed in terms of shifts of thermoregulatory set-point. Body temperature of newborn rats as low as 32–33 °C during the first days after delivery is their physiological body temperature [Citation8]. It means that body temperatures of rat pups of 33 °C should not be regarded as hypothermia. On the other hand, a forced fixing of body temperature even at the value typical of adult rats should be interpreted as hyperthermia, which would be counterproductive for the delivery of oxygen to the essential organs [Citation11].
The brain is extremely sensitive to slight changes in temperature, and in animal studies even mild hyperthermia worsened all aspects of the neurotoxic cascade [Citation19,Citation21]. Indeed, forcing body temperature of a hypoxic newborn to rise to the hyperthermic value by artificial warming may impose additional metabolic and physiological demands counterproductive to survival [Citation20,Citation41,Citation42]. Among these, a decrease in peripheral vascular resistance (as a thermolytic response used to dissipate heat by diverting blood to the body periphery), an increased cardiac output [Citation42], increased glucose consumption resulting in severe hypoglycaemia [Citation20], overproduction of transition metal ions, which can make electron donations to oxygen, forming superoxide anions [Citation42] and significant increase in plasma adrenocorticotropic hormone and corticosterone levels [Citation18] seem to be worth mentioning. Our own studies revealed that subjecting animals to neonatal anoxia at elevated body temperatures elicits a strong metabolic acidosis and a significant increase of free iron concentration in plasma (hyperferraemia), while during anoxia at normal neonatal body temperature of 32–33 °C these disturbances are not recorded [Citation13].
The present study has demonstrated that cerebral concentrations of antioxidants are sensitive to changes in body temperature both in control and anoxic conditions. Under hyperthermic conditions, in addition to the increased anoxia-induced oxidative stress, we also recorded a serious depletion of cerebral GSH and vitamin E. The most profound changes in the antioxidant levels were observed in extremely hyperthermic (39 °C) animals. However, it should be emphasised that holding the body temperature of newborn rats at a level typical of adults (37 °C) was enough to cause the depletion of GSH and vitamin E. Moreover, 14 days after the exposure of the rats to hyperthermic conditions the differences in GSH and vitamin E levels were more clear-cut between anoxic groups than between control (normoxic) groups, which means that anoxia is an add-on factor influencing the cerebral pool of low-molecular antioxidants ( and ). These data, together with those concerning MDA level (), show that all disturbances recorded in the present study are clearly temperature dependent.
Our results are consistent with those published previously, which demonstrated concomitant decreases in GSH and vitamin E levels, on the one hand, and increases in the level of lipid peroxidation on the other [Citation31–33,Citation43]. In 1-week-old piglets GSH depletion and oxidative stress resulting from hypoxia-ischaemia, promote damage to membrane and cytoskeletal proteins, DNA and RNA, thereby causing neuronal necrosis in the striatum [Citation13]. GSH depletion was found to produce a delay in the postnatal development of the rat [Citation44]. We recorded a long-term depletion in GSH and vitamin E up to day 14 after anoxia ( and ), which suggests that it could be one of the pivotal factors underlying developmental disturbances. The important role of GSH in hypoxia-ischaemia neurotoxicity is supported by the effects of GSH depletion, which causes increased formation of reactive oxygen species and lipid peroxidation [Citation31–33], increased excitotoxic response to NMDA [Citation45], and enlargement and degeneration of mitochondria in neurons of newborn rats [Citation46]. Interestingly, experimental studies also suggest that depletion of GSH can lead to apoptosis or necrosis [Citation32,Citation47]. Since the importance of iron in oxidative stress induction is indisputable, it should be mentioned that α-tocopherol has neuroprotective effects on the rat's hippocampal and nigral neurons against iron-induced neurotoxicity produced by excessive iron injection [Citation7]. Moreover, pretreating hippocampal neurons with α-tocopherol protects them against damages due to Fe2+-mediated oxidation [Citation48,Citation49]. GSH is another factor preventing iron-induced oxidative stress. Endogenous GSH protects cultured newborn rat astrocytes from the iron-mediated toxicity of hydrogen peroxide [Citation50].
We decided also to evaluate the efficacy of DF in the inhibition of depletion of low-molecular antioxidants at body temperature of 39 °C, because in our earlier paper [Citation14] we documented iron accumulation in neurons of the frontal cortex, hippocampus, and striatum after perinatal anoxia at that particular level of body temperature. Present results show that DF is efficacious in preventing lipid peroxidation () and depletion of cerebral low-molecular antioxidants ( and ) in extremely hyperthermic groups, both anoxic and normoxic ones, at all measurement time points. Nevertheless, the treatment was more efficacious in the anoxic animals. The neuroprotective role of DF was also supported by our earlier behavioural experiments. The rats exposed to hyperthermia alone and those exposed to anoxia at elevated body temperatures showed disturbances in behavioural responses to stress and spatial memory impairment during their whole life. The disturbances were not recorded in animals subjected to anoxia at the neonatal rat physiological body temperature of 33 °C or in animals at hyperthermic body temperature that were treated with DF immediately after the insult [Citation26,Citation51].
The inhibition of Fenton chemistry via iron chelation is the most possible mechanism of neuroprotection with DF [Citation23], thus it has significant contribution to prevent free radical-induced damage to the brain after hypoxic-ischaemic insult. However, DF appears to have other less characterised but detectable therapeutic properties: decreasing vascular endothelial free radical generation, scavenging radical species, inhibiting peroxynitrite synthesis, and other iron-dependent reactions leading to oxidative injury [Citation5,Citation52–54]. Treatment with DF, besides inhibiting the hypoxia-ischaemia-induced free iron increase in plasma in newborn lambs aged ∼7.5 days, also had a positive effect on post-hypoxic-ischaemic cerebral perfusion, metabolism and electrocortical brain activity [Citation22,Citation23] as well as inhibiting the plasma membrane depolarisation in rat brain synaptosomes [Citation52].
These findings mentioned above, including those from the present research, suggest a complex action of DF when administered into the immature brain immediately after hypoxia-ischaemia. It is of particular interest that depletion of low-molecular antioxidants can be almost completely prevented by DF ( and ). This implies that DF is a therapeutic agent acting not only via iron chelation. However, further experimental studies are required to evaluate more delayed effects of DF.
In conclusion, the present study highlights the importance of body temperature and treatment with DF for a proper development of an infant’s brain after neonatal anoxia. The up-regulation of GSH and vitamin E in an initial phase after neonatal anoxia might be a part of an adaptive response to counteract the consequences of the insult. However, the down-regulation of the same molecules could set the conditions for increased vulnerability of the brain to damage. Our study suggests that the depletion of low-molecular antioxidants and the increased oxidative stress imposed on anoxic newborns maintained at hyperthermic conditions may have significant short- and long-term consequences. Moreover, our research encourages us to suggest that detailed studies should be undertaken to develop guidelines for the control of body temperature in anoxic human newborns.
Declaration of interest
Research on this paper was supported by grant from the National Science Centre, Poland, number 2011/03/B/NZ7/00682. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Barrett RD, Bennet L, Davidson J, Dean JM, George S, Emerald BS, et al. Destruction and reconstruction: Hypoxia and the developing brain. Birth Defects Res C Embryo Today 2007;81:163–76
- Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med 2006;40:388–97
- Chiswick M, Gladman G, Sinha S, Toner N, Davies J. Prophylaxis of periventricular hemorrhage in preterm babies by vitamin E supplementation. Ann N Y Acad Sci 1989;570:197–204
- Fragoso G, Martínez-Bermúdez AK, Liu H-N, Khorchid A, Chemtob S, Mushynski WE, et al. Developmental differences in HO-induced oligodendrocyte cell death: Role of glutathione, mitogen-activated protein kinases and caspase 3. J Neurochem 2004;90:392–404
- Palmer C. Iron and oxidative stress in neonatal hypoxic-ischemic brain injury. directions for therapeutic intervention. In: Connor JR, ed. Metals and Oxidative Damage in Neurological Disorders. Boston (MA): Springer US, 1997
- Aoyama K, Nakaki T. Impaired glutathione synthesis in neurodegeneration. Int J Mol Sci 2013;14:21021–44
- Bostanci MO, Bas O, Bagirici F. Alpha-tocopherol decreases iron-induced hippocampal and nigral neuron loss. Cell Mol Neurobiol 2010;30:389–94
- Rogalska J, Caputa M. Spontaneously reduced body temperature and gasping ability as a mechanism of extreme tolerance to asphyxia in neonatal rats. J Therm Biol 2005;30:360–9
- Pabello NG, Tracy SJ, Keller RW. Protective effects of brief intra- and delayed postischemic hypothermia in a transient focal ischemia model in the neonatal rat. Brain Res 2004;995:29–38
- Karibe H, Chen SF, Zarow GJ, Gafni J, Graham SH, Chan PH, et al. Mild intraischemic hypothermia suppresses consumption of endogenous antioxidants after temporary focal ischemia in rats. Brain Res 1994;649:12–18
- Mortola JP. How newborn mammals cope with hypoxia. Respir Physiol 1999;116:95–103
- Mortola JP. Implications of hypoxic hypometabolism during mammalian ontogenesis. Respir Physiol Neurobiol 2004;141:345–56
- Caputa M, Rogalska J, Nowakowska A. Effect of temperature on postanoxic, potentially neurotoxic changes of plasma pH and free iron level in newborn rats. Brain Res Bull 2001;55:281–6
- Rogalska J, Danielisova V, Caputa M. Effect of neonatal body temperature on postanoxic, potentially neurotoxic iron accumulation in the rat brain. Neurosci Lett 2006;393:249–54
- Zhu C, Wang X, Cheng X, Qiu L, Xu F, Simbruner G, et al. Post-ischemic hypothermia-induced tissue protection and diminished apoptosis after neonatal cerebral hypoxia-ischemia. Brain Res 2004;996:67–75
- Azzopardi D, Strohm B, Linsell L, Hobson A, Juszczak E, Kurinczuk JJ, et al. Implementation and conduct of therapeutic hypothermia for perinatal asphyxial encephalopathy in the UK – analysis of national data. PLoS One 2012;7:e38504
- Edwards AD, Brocklehurst P, Gunn AJ, Halliday H, Juszczak E, Levene M, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: Synthesis and meta-analysis of trial data. BMJ 2010;340:c363
- Bruder ED, Kamer KJ, Guenther MA, Raff H. Adrenocorticotropic hormone and corticosterone responses to acute hypoxia in the neonatal rat: Effects of body temperature maintenance. Am J Physiol Regul Integr Comp Physiol 2011;300:R708–15
- Ginsberg MD, Globus MY, Dietrich WD, Busto R. Temperature modulation of ischemic brain injury – a synthesis of recent advances. Prog Brain Res 1993;96:13–22
- Guenther MA, Bruder ED, Raff H. Effects of body temperature maintenance on glucose, insulin, and corticosterone responses to acute hypoxia in the neonatal rat. Am J Physiol Regul Integr Comp Physiol 2012;302:R627–33
- Reglodi D, Somogyvari-Vigh A, Maderdrut JL, Vigh S, Arimura A. Postischemic spontaneous hyperthermia and its effects in middle cerebral artery occlusion in the rat. Exp Neurol 2000;163:399–407
- Shadid M, Moison R, Steendijk P, Hiltermann L, Berger HM, van Bel F. The effect of antioxidative combination therapy on post hypoxic-ischemic perfusion, metabolism, and electrical activity of the newborn brain. Pediatr Res 1998;44:119–24
- Shadid M, Buonocore G, Groenendaal F, Moison R, Ferrali M, Berger HM, et al. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neurosci Lett 1998;248:5–8
- Sarco DP, Becker J, Palmer C, Sheldon RA, Ferriero DM. The neuroprotective effect of deferoxamine in the hypoxic-ischemic immature mouse brain. Neurosci Lett 2000;282:113–16
- Papazisis G, Pourzitaki C, Sardeli C, Lallas A, Amaniti E, Kouvelas D. Deferoxamine decreases the excitatory amino acid levels and improves the histological outcome in the hippocampus of neonatal rats after hypoxia-ischemia. Pharmacol Res 2008;57:73–8
- Rogalska J, Caputa M, Piątkowska K, Nowakowska A. Neonatal asphyxia and hyperthermia and cognitive deficits in adult rats: Role of iron. J Therm Biol 2009;34:391–400
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265–75
- Mateos R, Lecumberri E, Ramos S, Goya L, Bravo L. Determination of malondialdehyde (MDA) by high-performance liquid chromatography in serum and liver as a biomarker for oxidative stress. Application to a rat model for hypercholesterolemia and evaluation of the effect of diets rich in phenolic antioxidant. J Chromatogr B Analyt Technol Biomed Life Sci 2005;827:76–82
- Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys 1959;82:70–7
- Wielinski S, Olszanowski A. Simultaneous determination of retinol acetate, retinol palmitate, cholecalciferol, α-tocopherol acetate and alphacalcidol in capsules by non-aqueous reversed-phase HPLC and column backflushing. Chromatographia 1999;50:109–12
- Hota SK, Barhwal K, Singh SB, Ilavazhagan G. Differential temporal response of hippocampus, cortex and cerebellum to hypobaric hypoxia: A biochemical approach. Neurochem Int 2007;51:384–90
- Martin LJ, Brambrink AM, Price AC, Kaiser A, Agnew DM, Ichord RN, et al. Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol Dis 2000;7:169–91
- Shivakumar BR, Kolluri SV, Ravindranath V. Glutathione and protein thiol homeostasis in brain during reperfusion after cerebral ischemia. J Pharmacol Exp Ther 1995;274:1167–73
- Bragin DE, Zhou B, Ramamoorthy P, Müller WS, Connor JA, Shi H. Differential changes of glutathione levels in astrocytes and neurons in ischemic brains by two-photon imaging. J Cereb Blood Flow Metab 2010;30:734–8
- Dafre AL, Arteni NS, Siqueira IR, Netto CA. Perturbations in the thiol homeostasis following neonatal cerebral hypoxia-ischemia in rats. Neurosci Lett 2003;345:65–8
- Wallin C, Puka-Sundvall M, Hagberg H, Weber SG, Sandberg M. Alterations in glutathione and amino acid concentrations after hypoxia–ischemia in the immature rat brain. Dev Brain Res 2000;125:51–60
- Cadir FO, Bicakci U, Tander B, Kilicoglu-Aydin B, Rizalar R, Ariturk E, et al. Protective effects of vitamin E and omeprazole on the hypoxia/reoxygenation induced intestinal injury in newborn rats. Pediatr Surg Int 2008;24:809–13
- Mishima K, Tanaka T, Pu F, Egashira N, Iwasaki K, Hidaka R, et al. Vitamin E isoforms α-tocotrienol and γ-tocopherol prevent cerebral infarction in mice. Neurosci Lett 2003;337:56–60
- Zingg J-M, Azzi A. Non-antioxidant activities of vitamin E. Curr Med Chem 2004;11:1113–33
- Castaño A, Herrera AJ, Cano J, Machado A. Effects of a short period of vitamin E-deficient diet in the turnover of different neurotransmitters in substantia nigra and striatum of the rat. Neuroscience 1993;53:179–85
- Rohlicek CV, Saiki C, Matsuoka T, Mortola JP. Cardiovascular and respiratory consequences of body warming during hypoxia in conscious newborn cats. Pediatr Res 1996;40:1–5
- Belhadj Slimen I, Najar T, Ghram A, Dabbebi H, Ben Mrad M, Abdrabbah M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int J Hyperthermia 2014;30:513–23
- Rabinovic AD, Hastings TG. Role of endogenous glutathione in the oxidation of dopamine. J Neurochem 1998;71:2071–8
- Reyes E, Ott S, Robinson B, Contreras R. The effect of in utero administration of buthionine sulfoximine on rat development. Pharmacol Biochem Behav 1995;50:491–7
- Bridges RJ, Koh J, Hatalski CG, Cotman CW. Increased excitotoxic vulnerability of cortical cultures with reduced levels of glutathione. Eur J Pharmacol 1991;192:199–200
- Jain SK, McVie R, Smith T. Vitamin E supplementation restores glutathione and malondialdehyde to normal concentrations in erythrocytes of type 1 diabetic children. Diabetes Care 2000;23:1389–94
- Li YQ, Guo YP, Jay V, Stewart PA, Wong CS. Time course of radiation-induced apoptosis in the adult rat spinal cord. Radiother Oncol 1996;39:35–42
- Crouzin N, de Jesus Ferreira M-C, Cohen-Solal C, Aimar R-F, Vignes M, Guiramand J. Alpha-tocopherol-mediated long-lasting protection against oxidative damage involves an attenuation of calcium entry through TRP-like channels in cultured hippocampal neurons. Free Radic Biol Med 2007;42:1326–37
- de Jesus Ferreira MC, Crouzin N, Barbanel G, Cohen-Solal C, Récasens M, Vignes M, et al. A transient treatment of hippocampal neurons with alpha-tocopherol induces a long-lasting protection against oxidative damage via a genomic action. Free Radic Biol Med 2005;39:1009–20
- Liddell JR, Robinson SR, Dringen R. Endogenous glutathione and catalase protect cultured rat astrocytes from the iron-mediated toxicity of hydrogen peroxide. Neurosci Lett 2004;364:164–7
- Rogalska J, Caputa M. Neonatal asphyxia under hyperthermic conditions alters HPA axis function in juvenile rats. Neurosci Lett 2010;472:68–72
- Pekun TG, Hrynevich SV, Waseem TV, Fedorovich SV. Role of iron, zinc and reduced glutathione in oxidative stress induction by low pH in rat brain synaptosomes. Springerplus 2014;3:560
- Chiou T. Protection of cells from menadione-induced apoptosis by inhibition of lipid peroxidation. Toxicology 2003;191:77–88
- Liu J, Tang T, Yang H. Protective effect of deferoxamine on experimental spinal cord injury in rat. Injury 2011;42:742–5