Abstract
The diagnostic work-up and treatment of patients with neuroendocrine tumours has undergone a major change during the last decade. New diagnostic possibilities and treatment options have been developed. These Nordic guidelines, written by a group with a major interest in the subject, summarises our current view on how to diagnose and treat these patients. The guidelines are meant to be useful in the daily practice for clinicians handling patients with neuroendocrine tumours.
Epidemiology
Neuroendocrine tumours (NET) are rare but occur with an increasing incidence. Between 1973 and 2004 a total number of 35 618 patients with broncho-pulmonary and gastroenteropancreatic NETs were identified in the North American Surveillance, Epidemiology, and End Results (SEER) Programme. The incidence increased from 1.1/100 000 per year in 1973 to 5.3/100 000 per year in 2004. In particular the incidence of NET in the broncho-pulmonary system, small intestine and rectum increased considerably [Citation1].
The significant increase in incidence over the three decades is partly attributed to an increased awareness among clinicians, in particular pathologists as well as an improved classification system, WHO 2000, and development of improved diagnostic tools such as immunohistochemistry (IHC) for chromogranin A (CgA) and tumour specific hormones, somatostatin receptor scintigraphy (SRS) and multi-detector computed tomography (MDCT). However, a real increase in incidence is also very likely, since some of the NET tumour entities increased markedly, whereas others had an almost stable incidence during the time period.
The prevalence is calculated to 35/100 000, which is much higher than for several other cancer types such as oesophageal, gastric and pancreatic cancer [Citation1]. The higher prevalence of NET is explained by their prolonged survival compared to the other cancers.
For all NETs, 50% were localised, 25% had regional involvement and 25% had distant metastases at time of diagnosis [Citation1].
Classification
Gastroenteropancreatic NETs are classified according to the WHO classification [Citation2,Citation3], see , and the TNM classification stipulated by ENETS [Citation4,Citation5].
Table I. The WHO 2000 classification for endocrine tumours in the gastrointestinal tract.
Diagnostic procedures
Pathology
Formalin-fixed paraffin-embedded 1.2 mm biopsies or surgical tumour specimens are needed for diagnosis. Fine needle aspiration biopsy is not recommended since it is unlikely to give a definite diagnosis but merely a suspicion. Furthermore, it will not generate optimal material for IHC and it is impossible to evaluate proliferation index (PI) by Ki67 on aspiration material.
Certain knowledge of NETs’ many different growth patterns is a prerequisite for suspecting that a tumour may be a NET (). For diagnostic purposes the general NE markers synaptophysin and CgA should be applied. If one of these is strongly positive in most tumour cells the suspicion of a NET is confirmed. Expression of serotonin should also be investigated since a positive staining would suggest a primary tumour in the small intestine. Other hormone markers are optional depending on the organ involved. Assessment of PI is mandatory as it influences the choice of treatment and is an important prognostic parameter [Citation6].
Figure 1. Different growth patterns for NETs. (A) Typical insular pattern of small intestinal NET. (B) ECLoma: discrete small islands of tumour cells in the gastric fundic mucosa. (C) “somatostatinoma” of the duodenum: glandular pattern with psammoma bodies. (D) Goblet cell carcinoid of the appendix. (E) Trabecular pattern in a rectal carcinoids. (F) PDEC (poorly differentiated endocrine carcinoma).
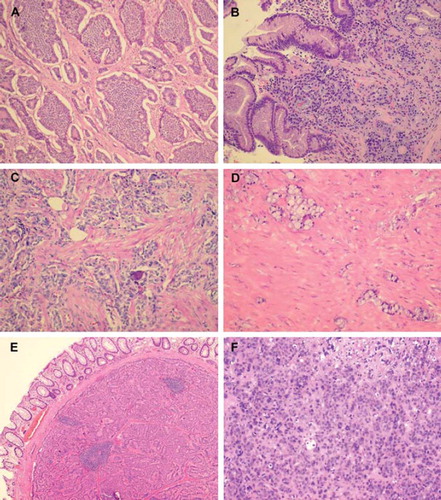
Ki67 should be counted in “hot spot” areas, i.e. areas with highest proliferation. Approximately 20 areas (positive cells per 100 cells) should be counted, as the positive areas often are unevenly distributed. The PI may change during the course of disease and a new tumour biopsy may be required to re-evaluate the PI.
Biochemical markers
CgA is a glycoprotein involved in the regulated secretion from neuroendocrine cells and the majority of NETs display pathologically increased levels [Citation7,Citation8]. The plasma level of CgA correlates with tumour burden in untreated patients. Poorly differentiated endocrine carcinomas (PDECs) tend to secrete relatively less CgA than highly differentiated tumours. Measurements of circulating CgA levels can be used in diagnosis, evaluation of therapy response and to detect progression and recurrence at an early stage [Citation9]. Elevated levels of CgA is, however, not specific for NETs. Endocrine cell hyperplasia in chronic atrophic gastritis, use of proton pump inhibitors, kidney and hepatic failure and some non-neuroendocrine malignancies give rise to elevated levels [Citation10]. Several assays for measurement of CgA exists, these have different sensitivities and specificities. Thus, measurement of CgA from different laboratories cannot be compared directly. Depending on the origin of the primary tumour measurement of specific markers in the blood such as gastrin, insulin, c-peptide, pro-insulin, glucagon, vasoactive intestinal polypeptid (VIP), pancreatic polypeptide (PP), somatostatin, adrenocorticothropine releasing hormone (ACTH) and calcitonin should be done. Urinary 5-hydroxyindoleacetic acid (U-5HIAA) is measured when small intestinal NET is suspected [Citation7].
Radiology
Current imaging techniques include multidetector CT (MDCT) and magnetic resonance imaging (MRI) > 1.5 T to allow for image reconstruction in transversal, coronal and sagittal views (see Facts 1). Proper use of i.v. contrast media for CT (“three phase examination”), MRI [Citation11,Citation12] and ultrasonography (US) (dynamical contrast enhancement) is of fundamental importance to visualise and characterise well and poorly vascularised tumour lesions [Citation13].
Similarly, optimal contrast technique is important in case of fatty infiltration or fibrosis of the liver (that may result from alpha-interferon (IFN), chemotherapy or radionuclide therapy), which may alter imaging conditions. Thus, liver metastases clearly depicted in one phase at baseline examination may be completely disguised at follow-up and instead be visible in another contrast enhancement phase [Citation14].
In order to allow for high quality MRI merely a limited part of the body can be examined within a reasonable time period. This fact, together with the lower sensitivity of MRI for lung metastases, makes MRI less useful for follow-up of thoracic lesions. Also, because of its limited availability compared to CT, MRI is best used as a “problem solving tool” when previous imaging results are equivocal or contradicting. However, in order to decrease the radiation dose to the patient, particularly for those who are young and have a long life expectancy, MRI may still be considered for therapy monitoring.
Facts 1
For MRI, 3D imaging sequences allows for dynamical contrastenhanced MRI performed at a minimum at 30, 70 and 120 seconds and at 3–5 minutes after injection start. Fat suppressed sequences are recommended to increase the MR signal contrast between different tissues. For MRI, not only the conventional extracellular Gd-preparations are available but also the more recently developed hepatocyte- and Kupffer cell specific contrast media to visualise and characterise liver lesions. Mangafodipir trisodium (Mn-DPDP) may also be used for MRI of the pancreas.
By dynamic i.v. contrast-enhanced US it is possible to detect small (≤ 3 to 4 mm) liver metastases and to characterise previously equivocal tumour lesions. Alternate use of US and CT for follow-up may be advantageous in patients where CT and US visualises different tumour lesions, and to decrease radiation dose.
Endoscopic ultrasound (EUS) is highly sensitive for visualisation and staging of pancreatic tumours and tumours located in the oesophageal, gastric and duodenal wall as well as adjacent regional lymph node metastases [Citation15]. In addition, EUS-guided biopsy of the lesion is possible.
Intraoperative US is a useful tool to detect small pancreatico-duodenal tumours, lymph node metastases and liver metastases and to evaluate resectability. Therefore, it is mandatory in hepato-biliary-pancreatic surgery.
Somatostatin receptor scintigraphy (SRS)
SRS is an important tool for detection of NETs, tumour staging, diagnosis of recurrent disease, and to evaluate eligibility for peptide receptor radionuclide therapy (PRRT).
NET visualisation by SRS using 111In-octreotide is based on the presence of somatostatin receptors which are seen in 80–90% of NETs [Citation16]. Tumours that lack these receptors or have a low receptor density may not be detected, e.g. benign insulinomas are often negative. Furthermore, small tumours (∼1–1.5 cm) may not be visualised, especially if located in areas with a normal “high” physiological background uptake as in the liver. Evidence for pausing somatostatin analogue treatment before SRS is lacking.
Somatostatin receptors are also expressed in non-NET tissue (such as tissues with inflammation, wound-healing, infections and non-endocrine malignancies, see Facts 2); therefore differential diagnoses should be considered to avoid “false-positive” findings.
SRS is performed as a whole body investigation and should include SPECT/CT (advantageous as high-dose diagnostic CT) allowing for attenuation correction and more precise localisation and interpretation. The combination increases the diagnostic accuracy of both modalities.
Bone scintigraphy
Bone scintigraphy is complementary to SRS to detect metastases in the skeleton. However, since it is as sensitive as SRS but less specific it is rarely required. SPECT/CT is suggested if planar imaging findings are equivocal.
Positron emission tomography (PET)
PET is a functional technique that provides information on, for example metabolism, proliferation, receptor density and bioamine uptake of NET dependent of which PET tracer and isotope is utilised. With a PET/CT-scanner the functional information is also combined with the morphological information from CT.
Somatostatin receptor imaging with PET using, e.g. 68Ga-DOTATOC may become an alternative to SRS due to the better spatial resolution and sensitivity of PET compared to scintigraphy, and the much faster PET procedure which allows examination already one hour after injection [Citation17]. The 11C-labelled serotonin precursor, 5-hydroxytryptophan (5-HTP), generally shows increased uptake in NETs. The method is more sensitive than SRS and CT in small NETs [Citation18]. 18F-DOPA may also be an alternative PET tracer. A comparative study between SRS and PET showed that 18F-DOPA PET/CT is the optimal imaging modality for staging in small intestinal NETs patients, and 11C-5HTP PET/CT in islet cell tumour patients [Citation19].
18F-deoxyglucose (FDG) uptake reflects glucose metabolism in the tumour. A high metabolism (FDG positive) is found in PDECs, while the large proportion of highly differentiated NETs often are FDG negative. Both FDG-PET and SRS have a prognostic value in that prognosis is worse if a tumour is FDG positive or SRS negative and vice versa [Citation20]. FDG-PET should not be routinely performed in well-differentiated NET, but may be of value in case of tumour progression (increased Ki-67).
In general, the short half-lives of PET isotopes are a drawback and most of the PET tracers are locally produced and not widely available. Furthermore, the methods have not been fully validated. Hence, at present, SRS with 111In-octreotide remains the gold standard for evaluating NET with radionuclide imaging.
Facts 2
Examples of causes for misinterpretation of positive SRS results
Bacterial infections, respiratory infections, granulomas, radiation pneumonitis, surgical scar, common cold, diffuse breast uptake, accessory spleen, gallbladder uptake, nodular goiter, thyroid associated opthalmopathy, meningioma, astrocytoma, lymphoma, sarcoma, melanoma, lung/breast/kidney/thyroid/prostate carcinoma.
Treatment overview
Surgery
A treatment algorithm is presented in . Surgery is the only treatment able to cure the patient. Therefore, all NET patients, regardless of extent of disease, should be considered for surgery. However, curative surgery in malignant NET is possible in probably less than 30%, depending on tumour type, site and spread. Despite radical surgery, recurrence occurs in > 75% of the patients within a time frame up to 15 years.
Figure 2. (A) Algorithm for the treatment of localised or metastasised NET. The treatment depends on localisation of the primary and the proliferation index (Ki-67). Light blue boxes indicate seldom used alternatives. (B) Algorithm for interventional treatment of neuroendocrine liver metastases.
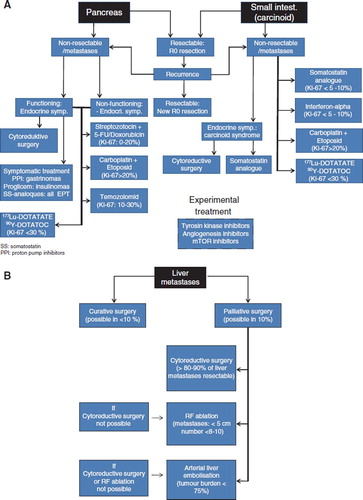
When curative surgery is not possible, usually due to metastatic disease, debulking surgery is often a beneficial treatment for local and endocrine symptoms.
Resection of the primary tumour in patients with disseminated disease may still be an option, e.g. resection of ileal carcinoid tumours, to prevent bowel obstruction [Citation21]. Resection of primary pancreatic, gastric, colonic and other NETs in patients with disseminated disease depends on the local and hormonal symptoms caused by the NET. The benefit of resection should be evaluated in relation to the possible operative complications [Citation22].
Sufficient data for the intervention of surgical treatment of PDEC is lacking.
Surgery may be combined with other treatment modalities, e.g. preoperative down-staging by bio- and chemotherapy, intra-arterial liver embolisation, radiofrequency ablation and peptide receptor radionuclide therapy (PRRT), which may increase the number of patients eligible for surgery.
To prevent excess hormone release during interventional procedures or surgery peri-operative somatostatin analogue treatment should be used – particularly in patients with intestinal carcinoid tumours. Examples are: Sandostatin® sc (300–500 μg) one hour before surgery and repeated as iv injection (300–500 μg) during surgery with short intervals if symptoms of excess hormonal release occurs. Alternatively administrate 500μg Sandostatin in 500 ml NaCl as an infusion, 50–100 μg/hour from before induction of anesthesia until at least four hours postoperatively.
It has long been discussed whether the gall bladder should be removed in patients with NET undergoing treatment with somatostatin analogues (which may cause development of bile stones) or liver embolisation (may accidentally cause embolisation of the cystic artery). As somatostatin-induced bile stones usually are asymptomatic and since liver embolisation techniques have improved considerably, prophylactic cholecystectomy in NET patients is generally not recommended. The risk of complications to cholecystectomy (0.3–3%) should be taken into consideration.
Chemotherapy
The use of chemotherapy in low-proliferating NET is controversial [Citation23]. As the effect of chemotherapy depends on the amount of dividing cells, only a modest effect would be expected when the PI is low [Citation24]. Studies on chemotherapy treated NETs are difficult to interpret as the PI of the tumours are rarely reported and frequently a mixture of different NET tumour types is included. Many studies do not, e.g. differentiate between ileum carcinoids with a Ki67 index of < 2% and pancreatic NETs with a Ki67 index of 5–10%.
Chemotherapy is recommended as first line therapy in metastatic/unresectable pancreatic NET. Usually a combination of streptozotocin (STZ) and 5-fluorouracil (5FU) has been given. The combination starts with an induction course of STZ 500 mg/m2 days 1–5 combined with 5FU 400 mg/m2 bolus days 1–3. After this initial course STZ 1000 mg/m2 and 5FU 400mg/m2 are given every third week [Citation25]. If the patient responds well the interval can be increased after one year of treatment to every four weeks. 5FU can safely be given if neutrophil count is ≥ 1.5 × 109/L and blood platelets ≥ 75 × 109/L, otherwise the treatment should be postponed. STZ is nephrotoxic and regular measurement of glomerular filtration rate (GFR) is mandatory. GFR should be measured by cr-EDTA clearance, plasma cystatin C or other validated methods, but not estimated by the method by Cockroft and Gault using s-creatinine. If baseline GFR is < 50 ml/min, STZ should not be used. If baseline GFR is 50–60 ml/min a three day induction course can be given.
A suggested dose modification schedule for STZ when treatment is ongoing according to renal function is found in .
Table II. Suggested GFR levels for reduction of ongoing Streptozotocin treatment.
Response rates for pancreatic NETs are 15–40% with a progression free survival (PFS) of 12–18 months. Side-effects include nausea, vomiting and renal toxicity. To avoid renal toxicity sufficient intravenous hydration must be given. Chemotherapy has usually been given until progression or toxicity. However, as peptide receptor radionuclide therapy is a possible second line therapy for these patients, renal toxicity must be avoided and a treatment break may be considered after 6–12 months. A treatment alternative is to combine STZ with doxorubicin 40 mg/m2 day 3, a combination which increases response rate but has the potential of myocardial toxicity (maximum cumulative dose 550 mg/m2). Temozolomide has been given to pancreatic NETs as second and third line therapy with response rates of 20–30% with a PFS of 7–9 months [Citation26,Citation27]. Temozolomide was given as 150–200 mg/m2 days 10–14 and capecitabine as 1 500 mg days 1–14 in a four week schedule. The use of temozolomide (200 mg/m2) with capecitabine (1 500 mg/m2) as first-line therapy for pancreatic NET has been reported with a response rate of 70% [Citation28,Citation29]. If these data are consistent, this oral drug combination may evolve to be a future first line therapy option. Toxicity is mild, with some nausea due to temozolomide. A rare but serious complication for which patients must be controlled is grade 4 thrombocytopenia occurring about three weeks after start of temozolomide.
In some patients the tumour shows a de-differentiation with a shift of the clinical course to a more aggressive one. If a new biopsy shows Ki67 > 20% a regimen with cisplatin and etoposide should be tried [Citation30]. Chemotherapy is generally not recommended for ileal NETs. In the rare event where Ki67 is 5–10% in an ileum NET, STZ/5FU is a second line option. In ileal NETs where Ki67 is between 10–20% chemotherapy may be a first line option. More traditional chemotherapies (as gemcitabine) for gastrointestinal tumours have not proven effective in NETs. Patients with metastatic PDECs should be treated with a combination of cisplatin and etoposide [Citation31,Citation32]. There are many different schedules, one frequently used is etoposide 100 mg/m2 days 1–3 and cisplatin 45 mg/m2 days 2–3 every fourth week. Some centres use carboplatin instead of cisplatin due to less toxicity. All schedules have considerable haematological toxicity and peripheral neuropathy may be dose-limiting for patients on cisplatin therapy. The effect of chemotherapy should be assessed approximately every two to three months using the RECIST criteria.
External radiotherapy
Radiotherapy is very effective to decrease the pain from bone metastases. Solitary brain metastases not treated by surgery should be considered for stereotactic radiotherapy or gamma-knife radio surgery. Non-operable brain metastases can be treated with whole-brain radiotherapy. Stereotactic body radiotherapy is an emerging option for unresectable liver metastases.
Alpha-Interferon (IFN) therapy
IFN was introduced in the early 1980s for NET treatment. There are no placebo controlled trials with IFN. However, a large number of phase 1 and 2 trials have been published with more than 670 patients. IFN treatment has proven effective especially in low-proliferating NET with Ki67 < 2% and should generally not be used when Ki67 > 10%. INF affects NET by apoptosis, anti-proliferative and anti-angiogenic effects combined with stimulation of the immune system [Citation33]. The best studied and used formulations are recombinant IFNs (Intron-A, Roferon), but long-acting pegylated interferon formulations are available (Peg-Introna, Pegasys) as well as Multiferon, a human leucocyte IFN. The INF dose should be individually titrated by side-effects. The leucocyte count should be around 3×109/L. The dose of regular IFN should be 3–5 million units sc three to five times per week subcutaneously. Pegylated IFN is administered in doses ranging from 80–150 μg once weekly.
IFN treatment is indicated in low proliferating tumours independent of the magnitude of tumour burden. Symptomatic response is seen in 62% (29–100%), biochemical response 50% (9–100%), tumour reduction 10% (0–25%) and tumour stabilisation 65% (38–94%) of ileal NET with a PFS of more than 36 months [Citation34]. IFN can be used alone or in combination with somatostatin analogues and the combination shows additive effects on tumour response but without proven benefit on survival [Citation33].
Adverse effects include flu-like symptoms and fever during the first one to two weeks of treatment which may be relieved by paracetamol 1 000 mg half an hour before injection. Dose-limiting toxicities are chronic fatigue (30–75%), mental depression (5–10%), neurological disorders (5–10%), and weight loss. Other side-effects are bone marrow depression and increased liver enzymes (10–30%). Severe side-effects include autoimmune reactions (thyroiditis, myositis, etc.) [Citation35]. IFN treatment should be monitored by body weight, blood cell count, liver enzymes, and thyroid function every three to six months.
Somatostatin analogue treatment
Somatostatin is a native hormone that regulates the normal secretion of peptide hormones. It acts through five receptors sst1-5 and inhibits cell proliferation and hormone release from NETs, thereby decreasing the symptoms caused by them. The two somatostatin analogues available for clinical use are octreotide and lanreotide. Octreotide is available as short-acting Sandostatin and as a long-acting release formula Sandostatin LAR. Lanreotide is available as the long-acting Lanreotide PR and Lanreotide Autogel. There are no major differences in symptomatic effects between the two analogues [Citation36].
Clinically, reduction in tumour size is seen in less than 10% of patients but a stabilisation of tumour growth is seen in up to 40%. A biochemical response is seen in 50–70% and a symptomatic response in 70–90%. The clinical effect and improvement of quality-of-life in patients with carcinoid syndrome, VIPomas and glucagonomas is well established. Recently the PROMID study showed that octreotide LAR has an anti-tumour effect with longer time to progression than placebo in ileal NETs with unresectable minor tumour burden [Citation37]. This study included SRS negative patients and a possible effect of somatostatin analogues in SRS negative patients can not be excluded.
Somatostatin analogue treatment should start with 2–3 (100 μg) s.c. injections of the short-acting analogue octreotide before injecting a long-acting release drug since some patients react with severe abdominal cramps and therefore should not be treated. The standard doses of octreotide LAR is 20–30 mg/4 weeks and for Lanreotide Autogel 90–120 mg/4 weeks. If relapse of symptoms occurs during the four weeks between injections, the interval between administrations may be reduced to three weeks. If symptoms persist or reoccur during treatment, or if there is a tendency towards tumour progression, the dose of octreotide LAR or Lanreotide Autogel may be increased to 60 mg/3–4 weeks or 240 mg/3–4 weeks, respectably. In the very rare cases where short-acting release is preferred, doses of 100–1 000 μg tid are used.
Some patients may experience diarrhoea due to reduced secretion of pancreatic enzymes with malabsorption, in these cases replacement therapy is recommended. Common side-effects are abdominal cramps (usually transient), diarrhoea, flatulence, nausea, subcutaneous nodules at the injection site and asymptomatic gall stones. Unusual side-effects include hypocalcemia, bradycardia and hypo/hyperglycaemia. There is no need for blood tests for safety reasons.
Recently a new somatostatin analogue, pasireotide (SOM230), has become available for clinical trials. This new analogue binds with high affinity to somatostatin receptors 1–3 and 5. It seems to be effective in patients with severe carcinoid syndrome despite high doses of the already registered long-acting somatostatin analogues. However, the exact place in the treatment arsenal for this new somatostatin analogue still has to be defined.
Peptide receptor radionuclide therapy (PRRT)
Tumour targeting treatment of NETs with radiolabelled somatostatin analogues has become more frequent during the last five years [Citation38]. Two different preparations, 177Lu -DOTATATE and 90Y -DOTATOC, are most often applied. A high tumour uptake (higher than physiological liver uptake) of 111In-Octreotide in planar SRS is required for treatment.
The treatment protocols differ: For 177Lu-DOTATATE four doses of radioactivity are given with six to ten weeks interval. For 90Y-DOTATOC two doses with eight to ten weeks interval are recommended. Treatment with long-acting somatostatin analogues should be interrupted at least four to six weeks before PRRT in order not to block the somatostatin receptors. Short-acting analogues may be used if severe symptoms occur during this period. Treatment with IFN should be stopped at least one week and chemotherapy at least four weeks before PRRT. The effect on tumours with a Ki67 > 30% is uncertain.
Acute side-effects include nausea, vomiting and pain. Other, usually reversible, side-effects are bone marrow depression and impaired renal function. Patients with huge liver involvement and bone metastases should be evaluated carefully prior to treatment because of the risk of severe hepatic failure and bone marrow toxicity. Some patients experience mild hair loss but alopecia is very rare. Myelodysplastic syndrome is an extremely rare but serious complication [Citation38,Citation39].
The tumour response differs between different tumour groups with pancreatic NETs showing the best results with a median duration of response of 30 months and a PFS of three to four years. Treatment with 177Lu-DOTATATE induced partial response in 30%, stable disease in 50%, and progressive disease in 20% in a series of 310 patients [Citation38,Citation39]. Similar responses have been achieved in smaller series using treatment with 90Y-DOTATOC. Partial tumour response was achieved in 24–33%, stable disease in 52–65%, and progression in 10–19%. Most of the PRRT studies are unfortunately hampered by the fact that the patient status (progressive vs. stable disease) at enrolment to PRRT was partly missing. Furthermore, there are no studies comparing the two isotopes or comparing PRRT to other treatments.
For patients that have responded to PRRT who have a later progression, re-treatment can be considered if kidney and bone marrow function are preserved.
The quality of life improves significantly in the majority of patients after PRRT [Citation38].
Specific part
Inherited syndromes with associated NETs
Pancreatico-duodenal NETs can be part of a familial syndrome. Most common are MEN 1 and von Hippel Lindaus disease (VHL) [Citation40] but also patients with Carneys complex [Citation41] and Neurofibromatosis type 1 (NF1) may develop pancreatic NETs [Citation40].
In MEN1 a mutation in the tumour suppressor gene encoding for the protein menin at 11q13 gives rise to pancreatic-duodenal NETs (multiple), pituitary adenomas and parathyroid hyperplasia. Other rare tumours associated with the syndrome are bronchial and thymic NETs, and adenomas in the adrenal cortex. Twenty-five percent of all gastrinoma patients have MEN 1. In these patients PTH, S-Ca and anterior pituitary hormones should be determined and family history recorded.
VHL has a mutation in 3p25-26 and gives rise to pancreatic NETs and renal cysts, renal cell carcinoma, hemangioblastoma, retinal angioma, pheochromocytoma, cystadenomas in lig. flavum and tumours in the middle ear. The pancreatic NETs are generally non-functioning and may be aggressive. For both MEN 1 and VHL, genetic testing is available and should be performed after obtaining informed consent from the patient.
Oesophageal NET
Clinical presentation. Patients present similarly to patients with other oesophageal cancers with dysphagia and retrosternal pain as the most common symptoms.
Pathology. Oesophageal NETs are rare (< 1% of all NETs) and are generally poorly differentiated small cell carcinomas, often with components of adenocarcinoma, and usually situated in the lower third of the oesophagus.
Treatment. Surgical principles generally follow those for oesophageal adenocarcinoma. Tumours should only be operated if radical resection is possible, and most frequently after preoperative chemoradiation [Citation42]. Medical treatment should follow the same principles as for other PDECs.
Gastric NETs
ECL cell hyperplasia. ECL-cell hyperplasia is a common finding in gastric biopsies. Biopsies should be taken from all parts of the stomach and duodenum for diagnosis. ECL cell hyperplasia should not be considered for endoscopic or surgical resection unless tumours develop. Follow-up gastroscopy with biopsies may be considered but evidence for this is lacking. Gastric NETS are divided into type I, II, III and PDECs.
Type I and II gastric NETs
Clinical presentation. Most type I and II gastric NETs are incidental findings at gastroscopy performed for other causes and rarely give rise to any symptoms themselves.
Pathology. The tumours arise from ECL-cells and comprise 85% of NETs in the gastric mucosa. They have the same pathophysiological background with an elevated gastrin as the cause of ECL-cell hyperplasia, which in time leads to tumour development. In type I gastric NETs the cause of hypergastrinemia is destruction of parietal cells due to autoimmune chronic atrophic gastritis (with concomitant hypo- or achlorhydria). Type II gastric NETs arise in patients with gastrin-producing tumours (and concomitant hyperchlorhydria). These patients may have an associated MEN 1 syndrome. Gastric NETs are usually multiple, < 2 cm with Ki67 < 2% in type I and 2–5% in type II. Multiple biopsies from different parts of the gastric mucosa should be examined to separate the two types.
Biochemistry. P-CgA and s-gastrin as well as gastric pH or gastric acid secretion (basal and pentagastrin-stimulated) should be measured. In type I gastric NET antibodies against intrinsic factor may result in decreased cobalamine which therefore should be measured.
Imaging. Gastroscopy with biopsies and EUS with evaluation of tumour invasion is mandatory. Other imaging procedures have a minor role in the diagnostic work-up. However, in case of type II gastric NET a search for the underlying gastrinoma(s) must be performed and signs of MEN-1 must be investigated.
Treatment. Tumours, single or multiple, < 1 cm should be biopsied and surveilled, as the lesions are usually benign. Tumours > 1 cm should be locally resected by endoscopy or surgery depending on the number of lesions, and whether there appears to be invasion into the muscularis propria. Treatment with somatostatin analogues can reduce tumour volume and numbers [Citation43]. However, such treatment is generally not recommended unless the patient has endocrine symptoms (histamine induced flush – the atypical carcinoid syndrome). For some patients histamine receptor type 2 blockers (H2-receptor blockers) may give symptomatic relief.
Follow-up. Patients should be followed by repeated endoscopy, initially every sixth month and after three years annually. The follow-up should include measurements of p-CgA, s-gastrin and cobalamine. EUS, CT/MRI and SRS should only be performed when tumour growth or metastases are suspected.
Prognosis. In type I gastric NETs local lymph node metastases are seen in < 2%, and liver metastases are almost never seen. In type II gastric NETs local metastases are found in 5–10% and liver metastases in < 2%. The prognosis is good with a five year survival of 100% in type I and 95% in type II gastric NETs.
Type III gastric NETs and PDEC
Clinical presentation. Symptoms of anaemia, GI-bleeding, dyspepsia and eventually gastric obstruction are seen.
Pathology. Type III gastric NETs are sporadic and constitute about 10–20% of all gastric NETs. The tumour is usually solitary and larger than 2 cm in diameter with Ki67 2–20% and are not associated with hypergatrinemia.
PDECs constitute about 5% of gastric NETs.
Biochemistry. There are no specific biochemical markers but p-CgA may be measured. S-gastrin levels and gastric acid secretion are normal.
Imaging. CT thorax and abdomen is performed for staging of the tumour. In type III gastric NETs and PDECs, SRS and FDG-PET are recommended.
Treatment. Surgical resection with lymph node dissection should be performed when possible. Preoperative down-staging by chemotherapy may be considered if radical resection is not possible. For histamine producing tumours, somatostatin analogues and H2-receptor blockers may be used to reduce symptoms. In low-proliferating tumours IFN can be used as treatment.
Follow-up. Measurement of p-CgA, if elevated initially, and imaging (CT/MRI) should be done every three to six months.
Prognosis. In type III gastric NETs local or liver metastases are seen in more than half of the patients at diagnosis while almost all patients with PDEC have advanced disease. Five year survival is 50% and close to 0%, respectively.
Duodenal NETs
Clinical presentation. Gastrinomas may induce the Zollinger-Ellison syndrome, whereas other duodenal NETs rarely give rise to endocrine symptoms. Jaundice due to obstruction of the common bile duct, GI-bleeding, anaemia and dyspepsia are seen in 20–35%.
Pathology. Endoscopic biopsies are crucial in distinguishing between the five different types of duodenal NETs.
Type 1: Gastrin-producing tumours (40%) occur mainly in the proximal duodenum and are associated with the Zollinger-Ellison syndrome and often with MEN 1.
Type 2: Somatostatin-producing tumours (30%) are associated with neurofibromatosis type 1 in 50% of cases. They are usually located around the papilla of Vateri and have an adenomatous growth pattern with psammoma bodies.
Type 3: Gangliocytic paragangliomas have an endocrine component which often stains with antibodies against somatostatin and pancreatic polypeptide (PP). They are almost always benign (Ki67 < 2%).
Type 4: Serotonin-, calcitonin and PP-producing tumours are rare, usually small and benign (Ki67 < 2%) and localised away from the ampullar region.
Type 5: PDECs are rare and highly malignant.
Biochemistry. S-gastrin, p-calcitonin, p-CgA, U-5HIAA should be measured. Gastric pH or gastric acid secretion (basal and stimulated) should be measured if a gastrinoma is suspected. A secretin test may support the gastrinoma diagnosis [Citation44].
Imaging. Gastroscopy with biopsies, EUS with biopsy, dedicated pancreatico-duodenal CT or MRI, and SRS are recommended for staging of the disease.
Treatment. In selected small tumours endoscopic resection may be possible. Otherwise, local resection(s), pancreaticoduodenectomy or pancreatic sparing duodenectomy with reimplantation of the ampulla of Vateri may be performed. Intraoperative US of the liver, pancreas and duodenum is mandatory to identify additional tumours and metastases. Gastroscopy and EUS may be performed intraoperatively if required to localise small tumours. The routine procedure for small tumours is longitudinal duodenotomy, digital exploration by the surgeon and submucosal resection. Surgical treatment of gastrinomas in patients with MEN1 is controversial, since these tumours are generally multiple and rarely metastasise to the liver. These patients should be thoroughly evaluated for concomitant pancreatic NETs. If operated, duodenotomy with a thorough search for multiple tumours in the entire duodenum and pancreas is preferred.
Very little evidence is present to guide therapy of other duodenal NETs. However, 40–60% have lymph node metastases and 10% have liver metastases at diagnosis. If there are no signs of lymph node metastases at EUS, CT or MRI, and the tumour is < 1 cm, endoscopic resection may be performed. Otherwise, or when endoscopic resection is not feasible, surgical resection is recommended and follow the procedures mentioned above.
Follow-up. Measurement of p-CgA, and tumour specific hormones, endoscopy, EUS and imaging (CT/MRI) every three, six or 12 months should be done; the frequency depends on the malignant status of the tumour.
Prognosis. The prognosis is good with a median survival of more than 100 months for local and regionally metastasised tumours. For tumours with distant metastasises the median 5-year survival is around 60% [Citation1].
Pancreatic NETs
Clinical presentation. Non-functioning tumours constitute about 65% of pancreatic NETs and may present with abdominal pain, anorexia, weight loss and jaundice, similar to pancreatic adenocarcinomas. However, in many cases the patient may be remarkably devoid of symptoms. The remaining 35% are functioning tumours, which release a hormone causing a specific syndrome. Among these 15% produce insulin, 15% produce gastrin, and the rest produce a wide variety of hormones, such as glucagon, VIP and somatostatin, which give rise to specific symptoms/syndromes. Even small tumours of a few mm may give rise to fulminate symptoms.
Pathology. NETs in the pancreas account for approximately 1–2% of all pancreatic neoplasms. They usually present as solitary brownish tumours 1–5 cm in diameter. Apart from insulinomas where less than 10% are malignant, NETs in the pancreas all have a significant malignant potential. A variety of hormones may be tested on tissue sections by IHC. If the patient is young it is important to exclude a solid pseudopapillary tumour (positive vimentin at IHC). Ki67 is generally 5–15%. The tumours can be aggressive and are likely to be diagnosed at an advanced stage (14% localised, 22% regional and 64% distant) [Citation45].
Biochemistry. Measurement of the following markers is recommended: p-CgA, s-insulin, s-c-peptide, s-proinsulin, s-gastrin, p-VIP, p-glucagon, s-calcitonin., s-pancreatic polypeptide and p-somatostatin. For the diagnosis of insulinoma a 24–72-hour fasting test is recommended [Citation46]. For the diagnosis of gastrinoma, measurement of pH or basal and maximal gastric acid output is recommended to distinguish from secondary hypergastrinaemia. A secretin test may support the gastrinoma diagnosis [Citation44].
Imaging. A dedicated pancreatico-duodenal CT or MRI, EUS and SRS should be performed before surgery is considered [Citation47]. Functioning pancreatic NETs may not be localised by these procedures due to their small size. Benign insulinomas (in contrast to malignant ones) are often negative at SRS due to their small size but also because of lack of somatostatin receptor expression. In these cases angiography with arterial stimulation by calcium (for stimulation of insulin secretion) or secretin (stimulation of gastrin secretion) and venous sampling (ASVS) as well as portal venous sampling (PVS) may be performed to localise the tumour. Alternatively, PET using 68Ga-DOTATOC or 11C-5HTP PET/CT and 18F-DOPA-PET can help with the localisation. Intra-operative US of the pancreas and liver is mandatory and is especially important to localise multiple tumours in MEN1.
Surgery. Since 90% of the insulinomas are benign, enucleation or resection is feasible and can often be performed laparoscopically. However, enucleations of insulinomas close to the pancreatic duct should be avoided as pancreatic fistulas may occur. Insulinomas in the pancreatic tail are preferably treated by distal pancreatic resection, and those in the head may need a pancreaticoduodenectomy. Blind resection is not recommended instead repeated imaging, as well as evaluation by PVS and ASVS should be performed. The differential diagnosis of pancreatic hypoglycemia syndrome (NIPHS or nesidioblastosis) may be considered. The malignant insulinomas should be treated by aggressive pancreatic surgery, including vascular resection. Repeated surgery for metastatic disease should always be considered. If radical surgery is not possible debulking procedures should be performed in order to reduce severe endocrine symptoms. 18F-DOPA-PET might help to localise the most active area in nesidioblastosis [Citation48].
Sporadic pancreatic gastrinomas have more often spread to the liver than duodenal gastrinomas at diagnosis. The majority originate in the pancreatic head. Pancreatic surgery, combined with hepatic surgery, radiofrequency ablation or embolisation of liver metastases should be considered depending on tumour burden. Other functioning tumours have often metastasised at the time of diagnosis. The same principles as for gastrinomas may be used.
Identical surgical principles are indicated for non-functioning and functioning pancreatic NETs. Due to late development of symptoms, non-functioning NETs are often large and have metastasised at diagnosis. These tumours may be attached to surrounding vessels and organs, but this should not be a contraindication for surgery. In some cases preoperative chemotherapy or PRRT may downstage the tumour and make resection feasible.
Medical treatment. There is no evidence for the effect of adjuvant treatment of radically operated patients. If the tumour and/or its metastases can not be completely resected medical treatment is required. When the tumour has a Ki67 < 20% chemotherapy with STZ and 5FU is first line treatment. 5FU can be replaced by doxorubicin if Ki67 is in the higher range. Second line treatment is usually PRRT or Temozolomide alone or in combination with Capecitabine. In the future PRRT may be applied as first line treatment due to promising results in well differentiated pancreatic NET. IFN may be used in pancreatic NET with a Ki67 < 2%, although data is limited, and can be of value in VIP-omas with symptoms that are not controlled by somatostatin analogues. However, somatostatin analogues have no proven effect on tumour growth.
Recently, everolimus (RAD001), an oral inhibitor of mammalian target of rapamycin (mTOR), showed activity in patients with advanced pancreatic NETs after failure of prior systemic chemotherapy [Citation49]. In this study, patients progressing on or after prior chemotherapy achieved a response or stabilisation of the disease in 84–100% of the cases with a PFS of 9.7 (everolimus alone) and 16.7 months (everolimus + octreotide LAR). In a phase III study of sunitinib (an oral multitargeted tyrosine kinase inhibitor) vs. placebo for treatment of advanced pancreatic NETs the active drug showed an increase of PFS from 5.5 to 11.4 months [Citation50]. All included patients had progressive disease and 70% had received prior systemic chemotherapy. Both everolimus and sunitinib are therefore second and third line treatment options in pancreatic NET.
Symptomatic treatment. For patients with gastrinomas proton pump inhibitors, often administered in high doses, control the symptoms of increased acid secretion. For patients with insulinomas frequent small meals may temporarily reduce the hypoglycaemic attacks. Diazoxide, which inhibits insulin release, may stabilise B-glucose but has several severe side-effects. Somatostatin analogues may control hypoglycaemia but the treatment should be closely followed since it may aggravate the hypoglycemia due to concomitant inhibition of glucagon, growth hormone and ACTH secretion. In some cases with severe and frequent attacks of hypoglycemia constant infusion with glucose or glucagon may be necessary. Glucocorticoids are not recommended unless other treatments have failed, since the secretion of endogenous corticoids from the adrenals is suppressed and the protective counter-regulatory mechanism is blocked in case of hypoglycaemic attacks. Calcium channel blockers may also be used. Somatostatin analogues are efficient at stopping or reducing VIP induced diarrhoea and electrolyte derangements in up to 80% of patients.
Necrolytic migratory erythema and other glucagon induced symptoms disappear in 40–90% of cases within days to weeks following treatment with somatostatin analogues.
Follow-up in radically operated patients. A benign insulinoma should have a postoperative clinical and biochemical control but does not need further follow-up. Other patients should be followed with biochemical markers and CT/MRI every 6–12 months for more than ten years.
Follow-up in metastatic disease. Patients should be monitored closely during treatment, initially every three months, with biochemical markers, CT/MRI and when indicated by SRS. Annually, or if the patient reports new endocrine symptoms, all pancreatic hormones and tumour markers should be measured, since the tumours may switch profile and consequently the patient may need a change of treatment.
Prognosis. Median overall survival in a large cohort of 324 patients was 99 months [Citation51]. The 5- and 10-year survival was 64% and 44%. Good prognostic factors were radical surgery and Ki67 < 2%. Prognosis correlated well with both the TNM and WHO classifications. Median survival in the SEER data from 2000–2004 for advanced disease was 27 months [Citation1].
The prognosis for patients with resected benign insulinomas is excellent and mimics that of the normal population. For patients with malignant insulinomas the median survival is two years. For patients with gastrinomas the 5-year survival is close to 100% in the absence of liver metastases. In the presence of liver metastases the 5-year survival is reduced to 50%.
Small intestinal NETs (classical carcinoid tumours)
Clinical presentation. The primary tumour may give rise to abdominal discomfort for years, diarrhoea (due to partial bowel obstruction or ischemia), acute small bowel obstruction and rarely to gastrointestinal bleeding, intestinal intussusceptions and malnutrition. Tumours are often found incidentally during operation for small bowel obstruction or for an abdominal malignancy, in particular colorectal cancer. The carcinoid syndrome with flushing and diarrhoea requires the presence of liver metastases or large retroperitoneal masses draining into the caval vein and is seen in less than 30% of the patients at diagnosis. Between 10% and 30% of these patients also suffer from right-sided heart failure due to tricuspid valve insufficiency and pulmonary valve stenosis provoked by hormone-induced (probably serotonin) fibrosis.
Pathology. Approximately 25–30% of GI NETs arise in the small bowel, mainly in the ileum, the incidence being 1.5/100 000 per year. The primary tumour is usually located in the distal part of the ileum. There may be multiple tumours in the intestinal mucosa but these are most probably mucosal metastases. Most tumours exceed 2 cm in diameter and about 60% have metastasised at the time of diagnosis. Even small tumours may give rise to extensive metastases. The growth pattern is typically insular and the tumours will stain positive for serotonin, CgA and synaptofysin. Most tumours have a Ki67 < 2%. Peritumoural fibrosis is often found, producing kinking of the bowel wall and strangulation of vessels which may give rise to bowel obstruction and ischemia.
Biochemistry. P-CgA and U-5HIAA are generally increased and should be monitored.
Imaging. In patients who are not operated, US with tru-cut biopsy of metastases may give the final diagnosis. Staging of regional and distant metastases is performed by thoraco-abdominal CT, which also provides information on possible metastatic encasement of the superior mesenteric vessels for preoperative planning. SRS should always be performed. Imaging of the small bowel by conventional plain film barium examination or capsule endoscopy is rarely needed. Echocardiography should initially be performed in all patients with metastatic disease to evaluate right-sided heart disease, and repeated at least annually in those with proven heart failure.
Surgery. Small intestinal carcinoid tumours should ideally be resected with sufficient margins and the central mesenteric lymph node(s) included in the specimen if possible. Intra-operative palpation of the entire small intestine should be performed to identify multiple tumours. In case of extensive central mesenteric affection and retroperitoneal spread, which compromise the major mesenteric vessels, no attempts of emergency surgery in this area should be performed. However, the primary tumour should be resected and merely intestinal by-pass avoided. If residual tumour or metastases are identified after emergency surgery, definitive surgery of mesenteric root metastases may be attempted at a second operation for curative or palliative intent.
Surgery of liver metastases should always be considered. If not possible, RF ablation in cases with few liver metastases < 5 cm may be used. In cases of multiple liver metastases arterial embolisation may be useful especially in patients with the carcinoid syndrome and limited extra hepatic tumour burden. PRRT should be considered in patients with progression despite optimal surgery and somatostatin analogue and IFN therapy.
In severe cases of carcinoid heart disease thoracic surgery with valvular replacement using biological valves is indicated, and should be performed before hepatic surgery to reduce the blood pressure in the hepatic veins.
Medical treatment. There is no evidence for the effect of adjuvant treatment of radically operated patients. In the presence of non-resectable residual tumour/metastases medical treatment is indicated and a somatostatin analogue is the primary choice for most patients with or without the carcinoid syndrome. Treatment with Sandostatin-LAR has recently been shown to prolong progression free survival in patients with unresectable but limited tumour burden [Citation37].
IFN treatment is indicated in low proliferating tumours independent of the magnitude of tumour burden. Combined therapy with IFN and somatostatin analogues may have an additive effect on impending tumour growth [Citation52].
Chemotherapy with STZ and 5-FU is generally not recommended in low proliferating small intestinal NET. However, in the rare cases of Ki67 > 10% chemotherapy may be first line treatment, and it may be a second line option in tumours which progress rapidly when on biotherapy.
Follow-up. In asymptomatic patients who have undergone radical surgery, follow-up is recommended every 6–12 month for at least ten years with measurements of p-CgA and imaging to locate recurrence if indicated by rise in biomarkers. A rise in p-CgA is often the first evidence of recurrence [Citation9]. Patients with disseminated disease should be examined every three to six months with measurements of p-CgA, U-5HIAA and CT/MRI. SRS should be performed when whole body imaging is required or radionuclide treatment considered.
Prognosis. Five-year survival for localised or regional disease is 75% but recurrence is frequent, up to 75% after 15 years. In the presence of distant metastases the 5-year survival is reduced to 50%. Median survival is > 100 months for patients with local and regional disease and around 60 months for distant disease. However, a much longer survival may be seen, even in patients with disseminated disease.
Appendix NETs
NETs in the appendix can be divided into three different categories; carcinoids with a growth pattern and IHC similar to the small bowel NETs, Goblet cell carcinoids and tubular carcinoids.
Appendix carcinoid
Clinical presentation. These tumours very rarely give rise to symptoms and are almost always found by the pathologist in the tissue specimen after appendectomy for acute appendicitis.
Pathology. Most (85%) appendix NETs are morphologically similar to small bowel tumours and serotonin positive. More than 60% are < 1 cm and belong to WHO group 1 with Ki67 ≤ 2%.
Biochemistry. Biochemical markers (P-CgA and U-5HIAA) are usually normal postoperatively.
Imaging. Imaging follow-up is not necessary when the tumour is ≤ 2 cm and has clear resection margins. CT and SRS are recommended after surgery to diagnose recurrent or metastatic disease in patients with tumours > 2 cm.
Treatment. Appendectomy is sufficient treatment in > 90% of cases. Right-sided hemicolectomy with lymph node dissection should be performed if the tumour is > 2 cm, or if the pathologist reports tumour involved resection margins, location at the base of the appendix, infiltration into mesentery or vessels, spread to regional lymph nodes or if Ki67 > 10%.
In metastatic disease, medical treatment is similar to that for small intestinal carcinoid tumours.
Follow-up. In patients where appendectomy is considered sufficient p-CgA and U-5HIAA may be performed after three to six months. If normal the patients do not need further follow-up. However, follow-up is required in patients who have undergone hemicolectomy, and is then identical to that of small intestinal carcinoid tumours, including p-CgA, U-5HIAA and imaging.
Prognosis. For tumours < 2 cm without spread the 5-year survival is 100%. For tumours > 2 cm with spread and malignant behaviour five year survival is less than 70% [Citation53].
Goblet cell carcinoids
Clinical presentation. In tumours localised to the appendix the presentation is similar to that of other appendicial carcinoids. In disseminated cases they may present as malignant ovarian neoplasms with diffuse carcinomatosis but rarely with liver metastases.
Pathology. Approximately 10% of appendiceal NETs are goblet cell carcinoids. The histogenesis of this tumour is unclear and the appearance varies from uniform goblet cells arranged in small groups and single files to tumours with few goblet cell areas mixed with areas that are indistinguishable from an adenocarcinoma. Prognosis worsens with increasing adenocarcinoma areas [Citation54].
Biochemistry. P-CgA and U-5HIAA are usually normal and should not be used in follow-up as a routine. S-CEA, s-CA19-9 and s-CA125 may be elevated and can serve as tumour markers.
Imaging. Irrespective of size, all patients should undergo CT or MRI of the abdomen for staging of regional and distant metastases. SRS is usually negative and of minor value. FDG-PET might be positive and add diagnostic information.
Treatment. Localised Goblet cell appendix carcinoids should be treated with right-sided hemicolectomy with lymph node dissection. Routine hystero-salpingo-oophorectomy should not be performed unless ovarian metastases are present. Metastatic disease may be treated aggressively, including peritonectomy and intraoperative chemotherapy. Medical treatment (both adjuvant and for metastatic disease) follows that of colon adenocarcinomas.
Follow-up. Analysis of tumour markers and CT/MRI should be performed every three to six months.
Prognosis. For localised and regional disease the 5-year survival is 80%, which drops to below 20% in disseminated tumours [Citation53].
Tubular carcinoids
Clinical presentation. As for other appendix carcinoids. Pathology. Less than 5% of appendix NETs are tubular carcinoids. This tumour is a small (3–10 mm) benign tumour, which is serotonin negative and glucagon positive. It is usually situated at the tip of the appendix. Its main significance is that some pathologists classify this tumour as an adenocarcinoid together with goblet cell carcinoids thereby confusing a totally benign tumour with a potentially malignant tumour.
Biochemistry, imaging, treatment, follow-up and prognosis. As for appendix carcinoids.
Colon NETs
Clinical presentation. Carcinoid tumours of the coecum present with symptoms resembling small bowel carcinoids. Tumours in the remaining part of the colon are usually PDECs and rarely induce endocrine symptoms, but give rise to symptoms similar to colon adenocarcinomas (see PDEC chapter). Neuroendocrine tumours in colon and rectum comprise less than 1% of colorectal cancers.
Pathology. Well-differentiated colon NETs, are mainly located in the coecum, and occur with an incidence of 0.15/100 000. They show an insular growth pattern with CgA, synaptophysin and serotonin positive tumour cells.
Biochemistry. P-CgA and U-5HIAA should be measured in well-differentiated tumours.
Imaging. Colonoscopy with biopsy is mandatory. CT of the thorax and abdomen and SRS are performed for staging of regional and distant metastases.
Treatment. The surgical treatment of well-differentiated NET includes bowel resection with lymph node dissection. The medical treatment depends on the Ki67 index of the tumour, and includes IFN and/or somatostatin analogues or chemotherapy.
Follow-up and prognosis. In well-differentiated tumours as for small intestinal carcinoids.
Rectal NETs
Clinical presentation. The rectal NETs are usually small polyps that never cause any symptoms and are found incidentally at routine sigmoideoscopy. Larger NET polyps may cause rectal bleeding and the few rectal PDEC may present with symptoms as rectal adenocarcinomas and rarely with liver metastases. Hormone related endocrine symptoms are never seen.
Pathology. Rectum NETs comprise approximately 20% of GI NETs. More than 95% are benign and usually have a trabecular growth pattern with glucagon positive and serotonin negative tumour cells. Most are < 1 cm in diameter and Ki67 is usually < 2%.
Biochemistry. P-CgA and U-5HIAA are usually normal.
Imaging. Endoscopy with biopsies/resection is recommended. In tumours > 2 cm the extent and diagnosis of regional lymph node metastases is performed by MRI or rectal US. For complete staging and for detection of distant metastases, CT of the thorax and abdomen and SRS are used.
Treatment. Rectal NET polyps < 1 cm can be radically resected by endoscopy. Tumours between 1–2 cm are treated as small polyps if radical excision can be assured. Tumours > 2 cm may be resected by endoscopy or transendoscopic mucosectomy (TEM) or with a low anterior resection depending on size and Ki67 index.
Metastases are more frequent in tumours > 2 cm. Metastatic patients with a positive SRS may respond well to PRRT. IFN may stabilise tumours with low proliferation and chemotherapy in high proliferating tumours.
Follow-up. Radically resected polyps < 2 cm do not need further surveillance except for one endoscopic follow-up. Larger lesions that are resected should be followed with endoscopy, transrectal US and CT/MRI preferably at least annually.
Prognosis. Five-year survival of localised disease is > 90%, regional disease 50% and distant disease 30–40%.
Poorly differentiated endocrine carcinoma (PDEC)
Clinical presentation. Specific symptoms are related to the organ from which the carcinoma develops. Hormone-induced endocrine symptoms almost never occur.
Pathology. Approximately 10% of GI NETs are PDECs. They are mainly located in the esophagus, stomach, pancreas and colon but can be found in all locations [Citation55,Citation56]. Morphologically there are two types: a small cell carcinoma, resembling small cell carcinoma of the lung, and a large cell pleomorphic carcinoma. Awareness of the latter is essential as these tumours often are indistinguishable from poorly differentiated non-NET carcinomas [Citation57]. Usually only synaptophysin will be positive in IHC while staining for CgA is frequently negative. Ki67 is per definition > 20% but is more likely to be between 50% and 100%.
Biochemistry. Screening for CgA should be done but is usually negative. The tumour markers CEA, CA125 and CA19-9 may be elevated.
Imaging. CT of the neck, thorax and abdomen is performed for staging. With a Ki67 > 15% FDG-PET/CT is positive in 90%, whereas SRS is positive in 70% [Citation20]. Therefore, both imaging methods are recommended. Brain CT/MRI should be performed if signs of neurological involvement are present.
Treatment. For patients with disease localised within an anatomic region (limited disease), initial therapy with chemoradiotherapy or chemotherapy is recommend, followed by surgery if no distant metastases are identified and the locoregional disease is resectable [Citation56]. No convincing data exist concerning adjuvant postoperative chemotherapy. Most patients have metastatic disease at the time of diagnosis or rapid recurrence after surgery.
Debulking surgery and surgery for liver metastasis are generally not recommended, leaving palliative chemotherapy as the only option. Patients with metastatic disease have rapid tumour growth and clinical deterioration. Referral to an oncologist should be rapid before performance status is to poor for chemotherapy. Cisplatin (at some centres carboplatin) and etoposide are standard first-line treatment for patients with good performance status. Response rates are between 41–67%, response duration 8–11 months and median survival 15–19 months [Citation31,Citation32]. Progression after first-line chemotherapy is usually very aggressive with a short survival. There are no published studies on second line chemotherapy. PRRT may be an option if Ki-67 < 30– (Citation50)% and SRS shows a high uptake.
Follow-up. Radically operated patients should be monitored every three months initially with CT as most patients will have a rapid recurrence of the disease. Patients on chemotherapy should be monitored by CT every second month.
Prognosis. The aggressiveness of PDEC is similar to small-cell lung cancer, with a median survival of less than six months without treatment [Citation1]. The prognosis is poor; 5-year survival is less than 5%.
Management of neuroendocrine liver metastases
Single or few hepatic metastases may be resected with curative intent, which is possible in < 10% as most metastases from NETs are multiple at diagnosis. Palliative debulking surgery is indicated to minimise severe hormone or local symptoms, and to reduce the target area for other therapies. The surgical procedures may include atypical resection, segmentectomy, regular or extended hemihepatectomy. In some cases, preoperative embolisation of the portal vein of the most severely metastatic liver lobe may be performed to induce growth of the other liver lobe. This may be performed one to two months before resection of the most severely metastatic liver lobe. Surgery of liver metastases from all types of NETs causes symptom relief in > 90% with a median duration of 45 months, time to recurrence range from 21 to 50 months and survival range is 50–95 months [Citation58–60].
Surgical resection remains the golden standard. However, radiofrequency ablation can be combined with surgical resection, or used alone, when surgery is not possible, with significant symptom relief in midgut carcinoid patients [Citation61]. When surgery or RF-ablation is impossible and the majority of the tumour burden is localised to the liver, liver metastases may be treated by embolisation of the hepatic artery or its branches, occasionally combined with local chemotherapy. Beneficial effects on hormone induced symptoms are seen in 50–90%, with duration of 14–17 months. Median tumour response is 55% with duration of 10–24 months, and median survival of 50 months. Stereotactic body radiotherapy has evolved as an option for unresectable tumours.
Orthotopic liver transplantation may only be used in carefully selected young patients without extrahepatic disease, with severe uncontrollable endocrine symptoms and a NET with a Ki67 < 10%. Approximately 20% are recurrence free after five years and the 5-year survival rate may be as high as 90%, if proper selection is performed.
Neuroendocrine metastases from unknown primary
In about 10–15% of patients, liver or lymph node metastases from a NET are found without known localisation of the primary tumour. In these patients it is important to identify and diagnose the type of primary NET in order to initiate the most suitable treatment.
Clinical presentation. Metastases are often found incidentally by CT or US in patients examined for abdominal discomfort. Endocrine symptoms are less frequently found, but a careful patient history and clinical examination must be undertaken asking for tumour specific endocrine symptoms.
Pathology. After assessment of morphology a suspicion of a NET may be confirmed by applying general NET markers such as synaptophysin and chromogranin A. Once the NET diagnosis is established, a further characterisation of the tumour is performed by extensive IHC for specific hormones and markers. Markers such as serotonin and Ki67 are essential. A positive serotonin will suggest a primary localisation in the midgut area, most likely in the ileum. TTF-1 positivity will suggest a primary tumour in the lung. CDx2 positivity might suggest a primary in the GI tract. Furthermore specific hormones such as gastrin, somatostatin, glucagon, proinsulin, insulin, PP, VIP, calcitonin and CCK may be examined.
Biochemistry. P-CgA and a wide variety of tumour specific hormones as well as U-5HIAA are mandatory since elevation of these may further indicate the localisation of the primary tumour.
Imaging. Thoraco-abdominal CT and depending on histology, SRS or FDG-PET can be helpful. If SRS is negative, 68Ga-DOTATOC-PET/CT is recommended, as the primary tumour may be small. 18F-DOPA or 11C-5HTP PET may also be applied. Endoscopy, capsule endoscopy and EUS are performed accordingly, when localised gastric, intestinal or pancreatico-duodenal NET is suspected.
Treatment. The indications for surgery follows those mentioned for NET liver metastases. Surgery is only possible in less than 20% of the patients. In some cases the primary is found at surgical exploration and can be resected together with resection of liver- and lymph node metastases.
In cases where the diagnostic procedures give no indication of the localisation of the primary tumour the patient should be treated medically according to the Ki67 index, i.e. patients with low proliferation index should be treated with somatostatin analogues and IFN and patients with higher proliferation index with chemotherapy as previously described. PRRT treatment may be indicated as second or third line treatment in patients with positive SRS. Likewise, interventional treatment with radiofrequency ablation or liver embolisation may be indicated as previously described (section liver metastases).
Follow-up. Should be carried out as mentioned for intestinal carcinoid tumours, as most of these tumours may have their origin in the small bowel.
Prognosis. Since most disseminated NETs with unknown primary probably originate in the ileum their prognosis will generally follow the prognosis for these tumours, but in general prognosis will depend on the biological nature of the tumour, including Ki67 and tumour specific hormones evaluated by IHC and by measurement of plasma hormones and U-5HIAA.
Acknowledgements
This work was supported by an unrestricted grant from Novartis Oncology and Ipsen.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, . One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008;26:3063–72.
- Capella C, Heitz PU, Hofler H, Solcia E, Kloppel G. Revised classification of neuroendocrine tumours of the lung, pancreas and gut. Virchows Arch 1995;425:547–60.
- Kloppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: The WHO classification. Ann N Y Acad Sci 2004;1014:13–27.
- Rindi G, Kloppel G, Alhman H, Caplin M, Couvelard A, de Herder WW, . TNM staging of foregut (neuro)endocrine tumors: A consensus proposal including a grading system. Virchows Arch 2006;449:395–401.
- Rindi G, Kloppel G, Couvelard A, Komminoth P, Korner M, Lopes JM, . TNM staging of midgut and hindgut (neuro) endocrine tumors: A consensus proposal including a grading system. Virchows Arch 2007;451:757–62.
- Cunningham JL, Grimelius L, Sundin A, Agarwal S, Janson ET. Malignant ileocaecal serotonin-producing carcinoid tumours: The presence of a solid growth pattern and/or Ki67 index above 1% identifies patients with a poorer prognosis. Acta Oncol 2007;46:747–56.
- de Herder WW. Biochemistry of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab 2007;21:33–41.
- Helle KB, Corti A, Metz-Boutigue MH, Tota B. The endocrine role for chromogranin A: A prohormone for peptides with regulatory properties. Cell Mol Life Sci 2007; 64:2863–86.
- Welin S, Stridsberg M, Cunningham J, Granberg D, Skogseid B, Oberg K, . Elevated plasma chromogranin A is the first indication of recurrence in radically operated midgut carcinoid tumors. Neuroendocrinology 2009;89:302–7.
- Tropea F, Baldari S, Restifo G, Fiorillo MT, Surace P, Herberg A. Evaluation of chromogranin A expression in patients with non-neuroendocrine tumours. Clin Drug Investig 2006;26:715–22.
- Thoeni RF, Mueller-Lisse UG, Chan R, Do NK, Shyn PB. Detection of small, functional islet cell tumors in the pancreas: Selection of MR imaging sequences for optimal sensitivity. Radiology 2000;214:483–90.
- Dromain C, de Baere T, Lumbroso J, Caillet H, Laplanche A, Boige V, . Detection of liver metastases from endocrine tumors: A prospective comparison of somatostatin receptor scintigraphy, computed tomography, and magnetic resonance imaging. J Clin Oncol 2005;23:70–8.
- Mork H, Ignee A, Schuessler G, Ott M, Dietrich CF. Analysis of neuroendocrine tumour metastases in the liver using contrast enhanced ultrasonography. Scand J Gastroenterol 2007;42:652–62.
- Sundin A, Vullierme MP, Kaltsas G, Plockinger U. ENETS Consensus Guidelines for the standards of care in neuroendocrine tumors: Radiological examinations. Neuroendocrinology 2009;90:167–83.
- Rappeport ED, Hansen CP, Kjaer A, Knigge U. Multidetector computed tomography and neuroendocrine pancreaticoduodenal tumors. Acta Radiol 2006;47:248–56.
- Kwekkeboom DJ, Krenning EP, Scheidhauer K, Lewington V, Lebtahi R, Grossman A, . ENETS Consensus Guidelines for the standards of care in neuroendocrine tumors: Somatostatin receptor imaging with (111)In-pentetreotide. Neuroendocrinology 2009;90:184–9.
- Gabriel M, Decristoforo C, Kendler D, Dobrozemsky G, Heute D, Uprimny C, . 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: Comparison with somatostatin receptor scintigraphy and CT. J Nucl Med 2007;48:508–18.
- Orlefors H, Sundin A, Garske U, Juhlin C, Oberg K, Skogseid B, . Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: Comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab 2005;90:3392–400.
- Koopmans KP, Neels OC, Kema IP, Elsinga PH, Sluiter WJ, Vanghillewe K, . Improved staging of patients with carcinoid and islet cell tumors with 18F-dihydroxy-phenyl-alanine and 11C-5-hydroxy-tryptophan positron emission tomography. J Clin Oncol 2008;26:1489–95.
- Binderup T, Knigge U, Loft A, Federspiel B, Kjaer A. 18F-fluorodeoxyglucose positron emission tomography predicts survival of patients with neuroendocrine tumors. Clin Cancer Res 2010;16:978–85.
- Hellman P, Lundstrom T, Ohrvall U, Eriksson B, Skogseid B, Oberg K, . Effect of surgery on the outcome of midgut carcinoid disease with lymph node and liver metastases. World J Surg 2002;26:991–7.
- Hodul PJ, Strosberg JR, Kvols LK. Aggressive surgical resection in the management of pancreatic neuroendocrine tumors: When is it indicated? Cancer Control 2008;15: 314–21.
- Delaunoit T, Neczyporenko F, Rubin J, Erlichman C, Hobday TJ. Medical management of pancreatic neuroendocrine tumors. Am J Gastroenterol 2008;103:475–83; quiz 84.
- Vilar E, Salazar R, Perez-Garcia J, Cortes J, Oberg K, Tabernero J. Chemotherapy and role of the proliferation marker Ki-67 in digestive neuroendocrine tumors. Endocr Relat Cancer 2007;14:221–32.
- Eriksson B, Annibale B, Bajetta E, Mitry E, Pavel M, Platania M, . ENETS Consensus Guidelines for the standards of care in neuroendocrine tumors: Chemotherapy in patients with neuroendocrine tumors. Neuroendocrinology 2009;90:214–9.
- Ekeblad S, Sundin A, Janson ET, Welin S, Granberg D, Kindmark H, . Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res 2007;13:2986–91.
- Kulke MH, Stuart K, Enzinger PC, Ryan DP, Clark JW, Muzikansky A, . Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol 2006;24:401–6.
- Strosberg JR, Choi J, Gardner N, Kvols L. First-line treatment of metastatic pancreatic endocrine carcinomas with capecitabine and temozolomide. J Clin Oncol 2008;26(ASCO Annual Meeting Proceedings Part 1, 15S): Abstract 4612.
- Isacoff WH, Moss, RA, Pecora, AL, Fine, L. Temozolomide/capcitabine therapy for metastatic neuroendocrine tumors of the pancreas. J Clin Oncol 2006;24(ASCO Annual Meeting Proceedings Part 1, 18S): Abstract 14023.
- Fjallskog ML, Granberg DP, Welin SL, Eriksson C, Oberg KE, Janson ET, . Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer 2001;92:1101–7.
- Moertel CG, Kvols LK, O'Connell MJ, Rubin J. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer 1991;68:227–32.
- Mitry E, Baudin E, Ducreux M, Sabourin JC, Rufie P, Aparicio T, . Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br J Cancer 1999;81:1351–5.
- Fazio N, de Braud F, Delle Fave G, Oberg K. Interferon-alpha and somatostatin analog in patients with gastroenteropancreatic neuroendocrine carcinoma: Single agent or combination? Ann Oncol 2007;18:13–9.
- Plockinger U, Wiedenmann B. Neuroendocrine tumors. Biotherapy. Best Pract Res Clin Endocrinol Metab 2007; 21:145–62.
- Pavel ME, Baum U, Hahn EG, Schuppan D, Lohmann T. Efficacy and tolerability of pegylated IFN-alpha in patients with neuroendocrine gastroenteropancreatic carcinomas. J Interferon Cytokine Res 2006;26:8–13.
- Modlin IM, Pavel M, Kidd M, Gustafsson BI. Review article: Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther 2010;31:169–88.
- Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, . Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J Clin Oncol 2009;27:4656–63.
- van Essen M, Krenning EP, Kam BL, de Jong M, Valkema R, Kwekkeboom DJ. Peptide-receptor radionuclide therapy for endocrine tumors. Nat Rev Endocrinol 2009;5:382–93.
- Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP, . Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: Toxicity, efficacy, and survival. J Clin Oncol 2008;26:2124–30.
- Toumpanakis CG, Caplin ME. Molecular genetics of gastroenteropancreatic neuroendocrine tumors. Am J Gastroenterol 2008;103:729–32.
- Boikos SA, Stratakis CA. Carney complex: Pathology and molecular genetics. Neuroendocrinology 2006;83:189–99.
- Ku GY, Minsky BD, Rusch VW, Bains M, Kelsen DP, Ilson DH. Small-cell carcinoma of the esophagus and gastroesophageal junction: Review of the Memorial Sloan-Kettering experience. Ann Oncol 2008;19:533–7.
- Fykse V, Sandvik AK, Qvigstad G, Falkmer SE, Syversen U, Waldum HL. Treatment of ECL cell carcinoids with octreotide LAR. Scand J Gastroenterol 2004;39:621–8.
- Berna MJ, Hoffmann KM, Long SH, Serrano J, Gibril F, Jensen RT. Serum gastrin in Zollinger-Ellison syndrome: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. Evaluation of diagnostic criteria, proposal of new criteria, and correlations with clinical and tumoral features. Medicine (Baltimore) 2006;85:331–64.
- Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): Incidence, prognosis and recent trend toward improved survival. Ann Oncol 2008;19:1727–33.
- de Herder WW, Niederle B, Scoazec JY, Pauwels S, Kloppel G, Falconi M, . Well-differentiated pancreatic tumor/carcinoma: Insulinoma. Neuroendocrinology 2006;84:183–8.
- Anderson MA, Carpenter S, Thompson NW, Nostrant TT, Elta GH, Scheiman JM. Endoscopic ultrasound is highly accurate and directs management in patients with neuroendocrine tumors of the pancreas. Am J Gastroenterol 2000;95:2271–7.
- Kauhanen S, Seppanen M, Minn H, Gullichsen R, Salonen A, Alanen K, . Fluorine-18-L-dihydroxyphenylalanine (18F-DOPA) positron emission tomography as a tool to localize an insulinoma or beta-cell hyperplasia in adult patients. J Clin Endocrinol Metab 2007;92:1237–44.
- Yao JC, Lombard-Bohas C, Baudin E, Kvols LK, Rougier P, Ruszniewski P, . Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J Clin Oncol 2010;28:69–76.
- Raymond E, Niccoli-Sire P. Bang Y-J, Borbath I, Lombard-Bohas C, Valle J, . Updated results of the phase III trial of sunitinib versus placebo for treatment of advanced pancreatic neuroendocrine tumors. Gastrointestinal Cancer Symposium 2010:Abstract 127.
- Ekeblad S, Skogseid B, Dunder K, Oberg K, Eriksson B. Prognostic factors and survival in 324 patients with pancreatic endocrine tumor treated at a single institution. Clin Cancer Res 2008;14:7798–803.
- Kolby L, Persson G, Franzen S, Ahren B. Randomized clinical trial of the effect of interferon alpha on survival in patients with disseminated midgut carcinoid tumours. Br J Surg 2003;90:687–93.
- McGory ML, Maggard MA, Kang H, O'Connell JB, Ko CY. Malignancies of the appendix: Beyond case series reports. Dis Colon Rectum 2005;48:2264–71.
- Tang LH, Shia J, Soslow RA, Dhall D, Wong WD, O'Reilly E, . Pathologic classification and clinical behavior of the spectrum of goblet cell carcinoid tumors of the appendix. Am J Surg Pathol 2008;32:1429–43.
- Formica V, Wotherspoon A, Cunningham D, Norman AR, Sirohi B, Oates J, . The prognostic role of WHO classification, urinary 5-hydroxyindoleacetic acid and liver function tests in metastatic neuroendocrine carcinomas of the gastroenteropancreatic tract. Br J Cancer 2007;96:1178–82.
- Brenner B, Tang LH, Klimstra DS, Kelsen DP. Small-cell carcinomas of the gastrointestinal tract: A review. J Clin Oncol 2004;22:2730–9.
- Shia J, Tang LH, Weiser MR, Brenner B, Adsay NV, Stelow EB, . Is nonsmall cell type high-grade neuroendocrine carcinoma of the tubular gastrointestinal tract a distinct disease entity? Am J Surg Pathol 2008;32:719–31.
- Azimuddin K, Chamberlain RS. The surgical management of pancreatic neuroendocrine tumors. Surg Clin North Am 2001;81:511–25.
- Sarmiento JM, Heywood G, Rubin J, Ilstrup DM, Nagorney DM, Que FG. Surgical treatment of neuroendocrine metastases to the liver: A plea for resection to increase survival. J Am Coll Surg 2003;197:29–37.
- Frilling A, Li J, Malamutmann E, Schmid KW, Bockisch A, Broelsch CE. Treatment of liver metastases from neuroendocrine tumours in relation to the extent of hepatic disease. Br J Surg 2009;96:175–84.
- Eriksson J, Stalberg P, Nilsson A, Krause J, Lundberg C, Skogseid B, . Surgery and radiofrequency ablation for treatment of liver metastases from midgut and foregut carcinoids and endocrine pancreatic tumors. World J Surg 2008;32:930–8.