Abstract
Introduction. Gemcitabine based regimens have been widely used in patients with advanced cholangiocarcinoma (CC), but no standard therapy exists. In this study we aimed to find the maximally tolerated dose (MTD) of a two-week schedule of fixed dose rate (FDR) gemcitabine (G), oxaliplatin (O) and capecitabine (C), and evaluate the safety and efficacy of this regimen in patients with advanced cholangiocarcinoma (CC). Methods. In the Phase I part of the study a dose-escalation schedule of FDR G, O and C, administered every two weeks, was performed in patients with solid tumours and no other treatments or advanced CC. In the Phase II part response rate, toxicity, progression-free survival (PFS) and overall survival was evaluated in patients with newly diagnosed advanced CC. Results. Thirty-six patients entered the Phase I part and G 1 000 mg/m2 day 1 and 15, O 60 mg/m2 day 1 and 15, and C 1 000 mg/m2 BID day 1–7 and day 15–21 were established as MTD. In the Phase II part, 41 patients with advanced CC were included. Overall response rate was 34% and 51% had stable disease, resulting in a clinical benefit rate of 85%. Grade III and IV adverse events were rare. Median survival was 12.5 months (95% CI 9.2–15.9) and median progression-free survival (PFS) was 6.9 months (95% CI 5.1–8.6). Conclusions. This outpatient regimen was very feasible with significant activity and a favourable safety profile. Further studies will explore this combination with addition of newer targeted agents.
Cholangiocarcinomas (CC) are relatively rare tumours with a poor prognosis. At the time of diagnosis they are most often inoperable and palliative therapy is the only option. The prognosis of untreated advanced CC is poor with a median survival of only 3.9 months [Citation1]. Many chemotherapy regimens have been tested in Phase II trials but no standard regimen exists.
Most recent studies use doublet combination regimens containing a platinum, gemcitabine and/or fluoropyrimidines. Response rates in Phase II studies vary from a few percent to over 50%. Since studies have included small cohorts of patient with varying population characteristics it is difficult to compare results. In a recent analysis of pooled data from 104 trials of chemotherapy for CC, mainly Phase II trials, Eckel and Schmid found that doublet therapies seemed to be superior to single agent regimens. Gemcitabine and platinum had superior survival compared to combination therapies containing fluoropyrimidines and platinum [Citation2].
Capecitabine has been investigated in combination with various other chemotherapeutic agents such as oxaliplatin, gemcitabine, and cisplatin with response rates of 17–40 [Citation3–11] and improved QOL in patients with advanced CC [Citation10].
Gemcitabine has shown promising activity against advanced CCs, with response rates (RRs) in the range of 12 to 41% when used in combination with agents such as fluoropyrimidines, cisplatin, oxaliplatin, and mitomycin C [Citation3,Citation5–8,Citation12].
Gemcitabine is administered as an inactive prodrug, which intracellularly is converted into its main active triphosphate. Clinical studies demonstrated that a lower infusion rate of gemcitabine was associated with higher intracellular levels of the active metabolite in blood mononuclear and leukemic cells. The prolonged infusion of gemcitabine at a fixed dose rate (FDR) of 10 mg/m2 per minute was associated with a higher intracellular accumulation of the active triphosphate, greater toxicity, and a higher response rate than with the standard 30-minute infusion of gemcitabine in patients with pancreatic cancer in a randomised Phase II study by Tempero et al. [Citation13]. Using this administration seemed feasible in a two-week schedule. However, superiority of FDR has not been confirmed in Phase III studies.
Oxaliplatin has been tested in advanced CC with encouraging results. Response rates of 7 to 41% have been observed in clinical trials of oxaliplatin alone or in combination with gemcitabine, fluoropyrimidines and/or cetuximab to patients with advanced CC. Several triplet combinations with gemcitabine, platinum and fluoropyrimidines have been proposed [Citation14–17].
In the present Phase I–II study we planned to find an optimal dosing schedule for the triplet of gemcitabine FDR, capecitabine and oxaliplatin in a two-week schedule, similar to a proposal by Correale et al. [Citation18]. In order to test a two-week schedule we used capecitabine one week on alternating with one week off, as proposed by Scheithauer et al. [Citation19]. With this regimen we wanted to explore response rates, progression free survival and safety.
Patients and methods
The study was conducted at two sites in Denmark; the Phase I part at the department of Oncology at Rigshospitalet, Copenhagen, and the Phase II part at the Department of Oncology at Rigshospitalet, Copenhagen and at the Department of Oncology at Vejle Hospital, Vejle. The trial was approved by the local ethical committee and the Danish Medicines Agency and performed according to ICH-GCP and the Helsinki declaration (Registered ClinicalTrial.gov NCT00350961).
Study objectives
This was an open label Phase I/II trial. The primary endpoint of the Phase I part of the trial was to find the maximal tolerated dose (MTD) and recommended phase two dose (RPTD) for the combination therapy with gemcitabine, oxaliplatin, and capecitabine in patients with solid tumours. The primary endpoint of the Phase II part of the trial was response rate in patients with advanced CC. Secondary endpoints were toxicity, duration of response, progression-free survival (PFS) and overall survival.
Eligibility
Patients with solid malignancies with no other treatment option were eligible for the Phase I part of the trial. Only patients with histologically confirmed CC (intra- and extra-hepatic bile duct carcinoma and carcinoma of the gall bladder) were eligible for the Phase II part of the trial. Other eligibility criteria included age 18–75 years; measurable or evaluable disease according to the RECIST criteria; WHO performance status ≤2; no severe, uncontrolled, concomitant illness; and no known deficiency of dihydropyrimidine dehydrogenase. Laboratory requirements for eligibility were neutrophils count ≥1.5 × 109 cells/L, platelets ≥100 × 109 cells/L, Cr-EDTA clearance >50 mL/min, transaminases ≤3x the upper limit of normal range, billirubin ≤1.5x the normal range for the Phase I part and ≤3x the normal range for the Phase II part of the trial. Prior chemotherapy, radiotherapy or immunotherapy had to be completed at least four weeks prior to inclusion in the Phase I part, and prior chemotherapy or radiotherapy was not allowed in the Phase II part of the study. All patients gave signed informed consent.
Patients had a baseline medical history, physical examination, complete blood counts (CBC) with differential and chemistry panel (electrolytes, creatinine, transaminases, calcium), and CA-19.9. Patients had baseline computed tomography scan of the abdomen and chest x-ray with reassessment every eight weeks.
Treatment
One treatment cycle was defined as 28 days. Oxaliplatin and gemcitabine was administered intravenously on days 1 and 15 of each cycle, gemcitabine as fixed dose rate infusion of 10 mg/m2/min doses ranging from 600–1 200 mg/m2 and oxaliplatin as a two hour infusion at doses ranging from 60–85 mg/m2. Capecitabine was administered orally twice daily at doses of 1 000 and 1 250 mg/m2 on days 1–8 and 15–22. The planned treatment duration was six cycles, but treatment could continue at the discretion of the investigator.
Study assessments
Dose escalation. Patients who received at least one dose of gemcitabine, oxaliplatin, and capecitabine were assessable for toxicity analysis. DLT was defined as any grade 3 or 4 treatment-related non hematological toxicity (National Cancer Institute Common Terminology Criteria of Adverse Events, version 3.0), excluding nausea, vomiting, and alopecia; any grade 4 treatment-related hematological toxicity; or any grade 3 treatment-related hematological toxicity requiring a treatment delay of more than two weeks. Three patients were enrolled at each dose level starting at dose level 1. If no DLT was observed in cycle 1, three patients were enrolled at the next dose level. If DLT was observed in one patient, this dose level was expanded to six patients. If DLT was observed in two patients or more, the MTD was exceeded and the previous dose level was expanded to six patients. The recommended Phase II dose was the highest dose level, at which one of six patients experienced a DLT. Five dose levels were planned (). No intrapatient dose escalations were allowed. The recommended Phase II dose was further evaluated in the Phase II part of the study, where the planned sample size was 39 patients.
Table I. Dose levels in Phase I Part.
Efficacy assessment. Objective tumour evaluation for response was performed according to RECIST. A baseline radiographic tumour evaluation was performed within two weeks before treatment started and repeated every eight weeks until progression. Additional evaluations were performed, if progression was suspected. All included patients with CC cancer are included in the efficacy analyses, but complete adverse event listing is only presented for patients in the Phase II part of the study, receiving the recommended dose for Phase II.
Statistics
The Phase II part was based on Simon's two-stage minimax design. With an intended response rate of >30%, 19 evaluable patients were to be included in the first stage. If more than seven patients achieved partial remission further 20 patients should be included to a total of 39 evaluable patients in the Phase II part. Responses had to be confirmed at least four weeks after initial documentation (confirmed responses), and all data are presented as intention-to-treat. Survival time was defined as time from first study drug infusion to time of death. PFS was defined as the time from first study drug infusion to first objective documentation of tumour progression or death. Distributions of time to event were estimated using the Kaplan-Meier method.
Results
Results of Phase I
Thirty six patients were included in the Phase I part of the study from June 2004 to November 2005. shows the planned dose levels of the three drugs. All patients had solid tumours with no other treatments options, and most patients suffered from CC (24 patients, of which 21 were previously untreated). Other tumour types included hepatocellular carcinoma (five patients), soft tissue sarcoma (two patients), pancreatic adenocarcinoma (two patients), esophageal squamous cell carcinoma (two patients), and one patient with adenoid cystic carcinoma.
No DLT was observed in level 1, DLT as grade III non-hematological toxicity (pulmonary embolism) was observed in 1/6 in level 2, no DLT was observed in level 3 or 4. Grade 3 diarrhoea was observed in 1/3 in level 5, thus another three patients were entered in this cohort, of which 1/3 developed grade 3 diabetes mellitus. Therefore, another three patients were entered in level 4. However, one of these developed grade III diarrhoea. This would result in reduction to level 3. As diarrhoea had been the main DLT in level 4 and 5 it seemed most appropriate to reduce capecitabine rather than oxaliplatin, and an alternative level 3 (designated level 3A) was therefore introduced as a protocol amendment (). Hematological toxicity (grade 3 thrombocytopenia and grade 4 neutropenia) was seen in 1/3 patient, and therefore additional three patients were included at this level. However, another patient also had DLT with febrile neutropenia so dosing was reduced to level 2 with inclusion of additional three patients. None of these experienced DLT, and level 2 was therefore recommended for the Phase II part of the study.
Five previously untreated patients with CC obtained PR, and two of these were down-staged and had radical surgery performed. However, one patient later relapsed with bone metastases, whereas the other patient later relapsed with liver metastases.
Results of Phase II
Patient characteristics. From March 2006 to February 2008 a total of 41 patients with advanced CC (16 men and 25 women) with a median age of 61 (range 35–75), was enrolled into the Phase II part of the trial. Patient characteristics and demographics are detailed in . A total of 217 cycles was administered in the Phase II part, and the patients received a median of five cycles (range 1–10 cycles). Two patients did not complete the first treatment cycle due to early progression and deterioration within 12 days (one patient) and development of myocardial infarction after five days (one patient).
Table II. Demographics of the patients in the Phase II part.
Safety. Grade III and IV non-hematologial toxicities were generally rare (≤2%) and manageable, with only a few incidences of nausea, vomiting, diarrhoea, fatigue and palmo-plantar erythrodysaestesia (PPE). Grade 3 sensory neurotoxicity was observed in four patients (10%). Most frequent grade I–II adverse events were fatigue (86%), nausea (56%), vomiting (19%), diarrhoea (47%), PPE (12% and sensory neuropathy (69%). shows the distribution of the most frequent adverse events related to the number of patients. A total of 23 patients experienced grade III–IV neutropenia (56%). However, only six patients (15%) had infection associated with neutropenia, and only five patients (12%) had grade III–IV thrombocytopenia.
Table III. Adverse events, Phase II part, by Common Toxicity Criteria version 3.0.
Efficacy. Response rates of the patients in the Phase I and II parts of the study are presented separately. In the Phase I part 21 patients had previously untreated CC. However, only 15 of these were evaluable for response. None achieved complete remission (CR), five had partial remission (PR), eight had stable disease (SD) and two had progressive disease (PD) as best response. In the Phase II part, three patients had CR (7%), 11 had PR (27%), resulting in an overall response rate of 34% in the Phase II part (). A total of 21 patients had SD (51%) and only three patients had PD (7%), whereas three patients were not evaluable for response (). Two of the three patients with CR are still without evidence of disease +12 months after discontinuation of therapy, and one of these had surgical resection of the primary tumour in the gall bladder. The pathology report showed no viable tumour cells indicating that a pathological CR was achieved.
Table IV. Response rates, Phase II part (intention-to-treat).
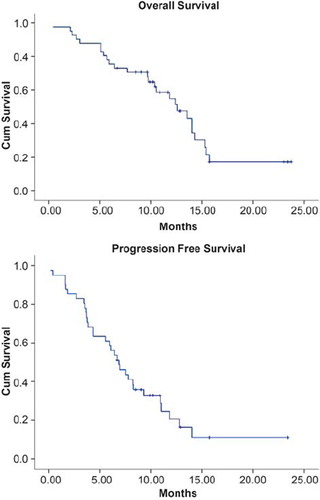
show the survival data. Median survival was 12.5 months (95% CI 9.2–15.9), and median progression-free survival was 6.9 months (95% CI 5.1–8.6).
Figure 1. Kaplan-Meier plot of overall survival and progression-free survival (PFS) of 41 patients treated in the Phase II part of the study. Median survival was 12.5 months (95% CI 9.2–15.9) and median PFS was 6.9 months (95% CI 5.1–8.6).
Discussion
Advanced CC is a relatively rare disease with an incidence of 1–2 per 100 000 [Citation20]. Among untreated patients with advanced CC the median survival is only 3.9 months [Citation1]. Data from presented Guidelines of British Association of Liver Diseases in 2002 indicated that single agent fluoropyrimidine derivatives induced response rates of 10–20%, single agent gemcitabine 20–30%, and combinations with gemcitabine and platinum derivatives induced response rates of 30–50%. In this consensus report it was stated that achieving stable disease should be considered beneficial, as this might be translated into both length and quality of life. This is especially important because lesions in the perihilar region are often difficult to demark, and consequently difficult to evaluate. In the present study, two patients with initially measurable disease turned out to be not evaluable for response [Citation21].
Prior to our study, several two-drug combinations with gemcitabine, oxaliplatin or capecitabine had been presented [Citation5,Citation12,Citation22]. Most of these were initiated in the same period as our trial, which was initiated in order to provide a feasible regimen with a favourable safety profile fit for ambulatory patients.
Simultaneously with our study, a Phase I trial of this biweekly schedule of oxaliplatin, gemcitabine and capecitabine was performed by Tan and colleagues. They found that this combination was well tolerated and deserved further investigation in CC and pancreatic malignancies [Citation23]. MTD for this regimen was declared to be oxaliplatin at 100 mg/m2 i.v., on days 1 and 15, gemcitabine at 800 mg/m2 administered at a constant infusion rate of 10 mg/m2/min on days 1 and 15 plus capecitabine at 800 mg/m2 orally twice a day on days 1–7 and 15–21 [Citation23].
In our regimen, we also incorporated fixed dose rate gemcitabine at a rate of 10 mg/m2/min. It has been hypothesised that prolonged infusion time with lower infusion rates of gemcitabine, would increase intracellular levels of the active metabolite. This has been shown by Tempero et al. [Citation13], who in a randomised Phase II trial in patients with advanced pancreatic cancer showed that fixed dose rate resulted in improved survival as compared to 30-minutes infusion of gemcitabine. However, this was at the expense of increased hematological toxicity, and it was shown that intracellular triphosphate gemcitabine was two-fold increased in the fixed dose rate arm. These data have not been confirmed subsequently.
The administration of capecitabine on days 1–7 and 15–21 in increased doses as compared to the regularly used regimen on day 1–14 every three weeks was explored by Sheithauer et al. in patients with advanced colorectal cancer [Citation18]. In a randomised Phase II trial with 89 patients it was shown that the experimental arm induced increased response rates and progression-free survival, without increased toxicity. In our hands, the schedule of capecitabine on days 1–7 and 15–21 did not result in increased dose intensity, as diarrhoea was DLT in the Phase I part, and the recommended dose was much lower than the dose in the study by Sheithauer et al.. However, at the recommended dose combined with FDR gemcitabine and oxaliplatin, we feel that our combination has a significant activity and a favourable safety profile. In the Phase II part the median number of cycles administered was five, and 12 patients received more than six cycles (seven to ten cycles).
In this Phase II trial a response rate of 34% was achieved. In addition, 51% of the patients had stable disease at four months as best response. This results in a clinical benefit rate of 85%. The median survival was 12.5 months (95% CI 9.2–15.9) and median PFS was 6.9 months (95% CI 5.1–8.6). In a recent published Phase II study by Andre et al. with gemcitabine and oxaliplatin administrated every two weeks, the overall response rate was only 15% and the frequency of stable disease was 36% [Citation24]. In addition, median progression-free survival was only 3.4 months and median survival was 8.8 months. Our data seem somewhat better, but this may be a result of selection bias. This could possibly be explored in a randomised Phase III multinational study.
Most recently, a British randomised Phase III trial of gemcitabine vs. gemcitabine and cisplatin was published by Valle et al. [Citation25]. These data have not yet been published, but showed that addition of cisplatin to gemcitabine resulted in an increased survival with a hazard ratio of 0.68 (95% confidence intervals (0.53–0.86). The median survival was 11.7 months for the combined arm vs. 8.2 months, p =0.002. Also PFS was improved with a hazard ratio of 0.70 (0.56–0.88), and median PFS was 8.5 months and 6.5 months, respectively (p=0.003). This was achieved without increased toxicity, except for slightly increased neutropenia [Citation25]. According to these data, gemcitabine and cisplatin could be considered as standard treatment for advanced CC.
However, it is not determined whether cisplatin or other platinum compounds are to be chosen. Oxaliplatin plays a significant role in gastrointestinal cancer, but different toxicity profiles and cost should be taken into consideration, and it may be difficult to conduct a trial large enough to show any survival difference between cisplatin and other platinum compounds. The role of fluoropyrimidines also needs to be established along with new biological agents that may contribute to improved survival. K-ras status has been evaluated in CC and was mutated in 45% of the patients in a study by Tannapfel et al. [Citation26]. In addition, EGFR overexpression seems to be a prognostic factor [Citation27], and smaller studies with both erlotinib and cetuximab have been reported [Citation28,Citation29]. VEGF inhibition may also play a role [Citation30], and a combination of bevacizumab and FDR gemcitabine and oxaliplatin was recently published, with results similar to what we present here [Citation31].
The question of choosing this best planinum analogue, the addition of fluoropyrimidines and the addition of targeted therapies in CC are awaited in randomised Phase III studies, with gemcitabine and cisplatin as the control arm.
Acknowledgements
The study was supported by research grants from Roche and Sanofi Aventis. Preliminary data of the Phase I part has been presented in abstract form at the World Congress in Gastrointestinal Cancers in 2005, Barcelona, Spain. The Phase II data was presented in abstract form in the Proceedings of the ASCO GI Symposium in 2009, San Francisco, USA.
Declaration of interest: The corresponding author UL (sponsor and investigator) has received research grants from Roche and Sanofi-Aventis. No other author has any conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Park J, Kim M, Kim J, Lee J. Survival time and its prognostic factors in advanced unresected cholangiocarcinoma without chemoradiation. J Clin Oncol 2009;27(Suppl; Abstract e15681).
- Eckel F, Schmid RM. Chemotherapy in advanced biliary tract carcinoma: A pooled analysis of clinical trials. Br J Cancer 2007;96:896–902.
- Kornek GV, Schuell B, Laengle F, Gruenberger T, Penz M, Karall K, . Mitomycin C in combination with capecitabine or biweekly high-dose gemcitabine in patients with advanced biliary tract cancer: A randomised phase II trial. Ann Oncol 2004;15:478–83.
- Cho JY, Nam JS, Park MS, Yu JS, Paik YH, Lee SJ, . A Phase II study of capecitabine combined with gemcitabine in patients with advanced gallbladder carcinoma. Yonsei Med J 2005;46:526–31.
- Cho JY, Paik YH, Chang YS, Lee SJ, Lee DK, Song SY, . Capecitabine combined with gemcitabine (CapGem) as first-line treatment in patients with advanced/metastatic biliary tract carcinoma. Cancer 2005;104:2753–8.
- Riechelmann RP, Townsley CA, Chin SN, Pond GR, Knox JJ. Expanded phase II trial of gemcitabine and capecitabine for advanced biliary cancer. Cancer 2007;110:1307–12.
- Park SH, Park YH, Lee JN, Bang SM, Cho EK, Shin DB, . Phase II study of epirubicin, cisplatin, and capecitabine for advanced biliary tract adenocarcinoma. Cancer 2006;106:361–5.
- Hong YS, Lee J, Lee SC, Hwang IG, Choi SH, Heo JS, . Phase II study of capecitabine and cisplatin in previously untreated advanced biliary tract cancer. Cancer Chemother Pharmacol 2007;60:321–8.
- Iyer RV, Gibbs J, Kuvshinoff B, Fakih M, Kepner J, Soehnlein N, . A phase II study of gemcitabine and capecitabine in advanced cholangiocarcinoma and carcinoma of the gallbladder: A single-institution prospective study. Ann Surg Oncol 2007;14:3202–9.
- Koeberle D, Saletti P, Borner M, Gerber D, Dietrich D, Caspar CB, . Patient-reported outcomes of patients with advanced biliary tract cancers receiving gemcitabine plus capecitabine: A multicenter, phase II trial of the Swiss Group for Clinical Cancer Research. J Clin Oncol 2008;26:3702–8.
- Nehls O, Oettle H, Hartmann JT, Hofheinz RD, Hass HG, Horger MS, . Capecitabine plus oxaliplatin as first-line treatment in patients with advanced biliary system adenocarcinoma: A prospective multicentre phase II trial. Br J Cancer 2008;98:309–15.
- Andre T, Tournigand C, Rosmorduc O, Provent S, Maindrault-Goebel F, Avenin D, . Gemcitabine combined with oxaliplatin (GEMOX) in advanced biliary tract adenocarcinoma: A GERCOR study. Ann Oncol 2004;15:1339–43.
- Tempero M, Plunkett W, Ruiz VH, Hainsworth J, Hochster H, Lenzi R, . Randomized phase II comparison of dose-intense gemcitabine: Thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol 2003;21:3402–8.
- Goetz MP, Erlichman C, Windebank AJ, Reid JM, Sloan JA, Atherton P, . Phase I and pharmacokinetic study of two different schedules of oxaliplatin, irinotecan, Fluorouracil, and leucovorin in patients with solid tumors. J Clin Oncol 2003;21:3761–9.
- Goel S, Bulgaru A, Hochster H, Wadler S, Zamboni W, Egorin M, . Phase I clinical study of infusional 5-fluorouracil with oxaliplatin and gemcitabine (FOG regimen) in patients with solid tumors. Ann Oncol 2003;14:1682–7.
- Ychou M, Conroy T, Seitz JF, Gourgou S, Hua A, Mery-Mignard D, . An open phase I study assessing the feasibility of the triple combination: Oxaliplatin plus irinotecan plus leucovorin/5-fluorouracil every 2 weeks in patients with advanced solid tumors. Ann Oncol 2003;14:481–9.
- Souglakos J, Mavroudis D, Kakolyris S, Kourousis C, Vardakis N, Androulakis N, . Triplet combination with irinotecan plus oxaliplatin plus continuous-infusion fluorouracil and leucovorin as first-line treatment in metastatic colorectal cancer: A multicenter phase II trial. J Clin Oncol 2002;20:2651–7.
- Correale P, Messinese S, Marsili S, Ceciarini F, Pozzessere D, Petrioli R, . A novel biweekly pancreatic cancer treatment schedule with gemcitabine, 5-fluorouracil and folinic acid. Br J Cancer 2003;89:239–42.
- Scheithauer W, Kornek GV, Raderer M, Schull B, Schmid K, Kovats E, . Randomized multicenter phase II trial of two different schedules of capecitabine plus oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 2003;21:1307–12.
- Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD. Cholangiocarcinoma. Lancet 2005;366:1303–14.
- Khan SA, Davidson BR, Goldin R, Pereira SP, Rosenberg WM, Taylor-Robinson SD, . Guidelines for the diagnosis and treatment of cholangiocarcinoma: Consensus document. Gut 2002;51(Suppl 6):VI1–VI9.
- Dingle BH, Rumble RB, Brouwers MC. The role of gemcitabine in the treatment of cholangiocarcinoma and gallbladder cancer: A systematic review. Can J Gastroenterol 2005;19:711–6.
- Tan BR, Brenner WS, Picus J, Marsh S, Gao F, Fournier C, . Phase I study of biweekly oxaliplatin, gemcitabine and capecitabine in patients with advanced upper gastrointestinal malignancies. Ann Oncol 2008;19:1742–8.
- Andre T, Reyes-Vidal JM, Fartoux L, Ross P, Leslie M, Rosmorduc O, . Gemcitabine and oxaliplatin in advanced biliary tract carcinoma: A phase II study. Br J Cancer 2008;99:862–7.
- Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, . Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81.
- Tannapfel A, Sommerer F, Benicke M, Katalinic A, Uhlmann D, Witzigmann H, . Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 2003;52:706–12.
- Yoshikawa D, Ojima H, Iwasaki M, Hiraoka N, Kosuge T, Kasai S, . Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer 2008;98:418–25.
- Paule B, Herelle MO, Rage E, Ducreux M, Adam R, Guettier C, . Cetuximab plus gemcitabine-oxaliplatin (GEMOX) in patients with refractory advanced intrahepatic cholangiocarcinomas. Oncology 2007;72:105–10.
- Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G. Phase II study of erlotinib in patients with advanced biliary cancer. J Clin Oncol 2006;24:3069–74.
- Yoshikawa D, Ojima H, Kokubu A, Ochiya T, Kasai S, Hirohashi S, . Vandetanib (ZD6474), an inhibitor of VEGFR and EGFR signalling, as a novel moleculartargeted therapy against cholangiocarcinoma. Br J Cancer 2009;100:1257–66.
- Zhu AX, Meyerhardt JA, Blaszkowsky LS, Kambadakone AR, Muzikansky A, Zheng H, . Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: A phase 2 study. Lancet Oncol 2009.