Abstract
The SBG 2000-1 trial is a randomised study that investigates if dose-tailored adjuvant FEC therapy based on the individual's leukocyte nadir value can improve outcome. The study has included 1535 women with medium and high-risk breast cancer. Patients and methods. After a first standard dosed FEC course (5-fluorouracil 600 mg/m2, epirubicin 60 mg/mg2 and cyclophosphamide 600 mg/m2), patients who did not reach leukopenia grade III or IV were randomised to standard doses (group standard) or doses tailored to achieve grade III leukopenia (group tailored) at courses 2–7. Patients who achieved leukopenia grade III or more after the first course were not randomised but continued on standard doses (group registered). Results. Both planned and actually delivered number of courses (seven) were the same in all three arms. The relative dose intensity was increased by a factor of 1.31 (E 1.22, C 1.43) for patients in the tailored arm compared to the expected on standard dose. Ninety percent of the patients in the tailored arm achieved leukopenia grade III–IV compared with 29% among patients randomised to standard dosed therapy. Dose tailoring was associated with acceptable acute non-haematological toxicity with more total alopecia, nausea, vomiting and fatigue. Conclusion. Dose tailoring according to leukopenia was feasible. It led to an increased dose intensity and was associated with acceptable excess of acute non-haematological toxicity.
Numerous studies and overviews have demonstrated benefits of adjuvant chemotherapy in early breast cancer and that anthracycline (doxorubicin or epirubicin) based regimens are among the most efficacious [Citation1]. The optimal dosing of these regimens is still to be determined although lower than normal doses yield inferior results [Citation2,Citation3]. Whereas a large randomised study on dose-escalation of doxorubicin failed to show any benefits in the adjuvant setting [Citation4], there is evidence that increased doses of epirubicin as an integral part of the FEC-regimen is more efficacious. This has been demonstrated in advanced breast cancer in terms of increased response rate [Citation2,Citation5] and time to progression [Citation6]. In adjuvant treatment of high-risk node-positive breast cancer six cycles of epirubicin 100 mg q 3W in combination with cyclophosphamide 500 mg q 3W and 5-FU 500 mg q 3W (FEC 100) was superior to epirubicin 50 mg q 3W with the same combination (FEC50) regarding both DFS and OS [Citation7]. Despite improved results of cancer chemotherapy, the methods of dose optimisation and calculation have been criticised repeatedly [Citation8,Citation9]. The current dosing method is with very few exceptions based on body surface area (BSA), a method that results in a large inter-individual variation of systemic exposure [Citation10–13]. Although acute toxicity of the FEC-regimen generally is manageable there is a non-negligible long-term risk of cardiac morbidity [Citation3,Citation14] and development of secondary leukaemia [Citation15]. The risk of cardiac toxicity after FEC (1.36% at 7 years) was not evidently dose dependent within the commonly used dosing range (300–600 mg/m2) in the French study [Citation3]. For secondary leukaemia there is a dose risk relationship: the higher the total dose of epirubicin and cyclophosphamide the higher is the risk of leukaemia [Citation15]. Keeping these severe side-effects in mind it is obvious that the pursuit for increasingly effective treatment must be combined with efforts to avoid the use of unnecessary high doses.
The knowledge that systemic exposure of chemotherapy is correlated with bone marrow toxicity [Citation11,Citation13] gives an opportunity to individually tailor the dose according to the grade of myelosuppression. This view is supported by several retrospective studies [Citation16–20] indicating that bone marrow toxicity not only mirrors systemic exposure but also predicts anti-tumoural effect in early breast cancer.
The idea of using bone marrow toxicity during chemotherapy as a marker for systemic exposure in order to perform individual dose-tailoring has been tested in a previous Scandinavian study (SBG 9401) on high risk early breast cancer (eight or more positive lymph nodes or at least five positive nodes and nuclear grade two to three and hormone receptor negativity) [Citation21]. The study showed superiority of nine cycles of tailored FEC supported with granulocyte colony stimulation factor (G-CSF) and antibiotics compared with three cycles of standard FEC followed by high dose chemotherapy with autologous bone marrow support. A serious drawback of the tailored arm of the SBG 9401 study was the 4% incidence of secondary myeloic malignancies at long-term follow-up [Citation22], most likely due to too high total doses of epirubicin and cyclophosphamide. This is supported by the fact that a modified G-CSF supported FEC regimen (G-CSF only given day 5–12, the maximal cyclophosphamide dose was reduced to 1.2 mg/m2 and the number of courses were limited to six) no secondary AML/MDS were reported in the EORTC p53 study, with this regimen as one of the FEC options [Citation22].
The rationale for leukopenia based dose tailoring still seems strong since this concept could potentially improve outcome for patients who otherwise would have suboptimal treatment if dosing is based on BSA only. At the same time the concept might decrease the risk of unnecessary dose escalation in patients showing a marked leukopenia, indicating adequate systemic exposure, already at standard doses. Prospective randomised studies comparing the concept of dose-tailored adjuvant chemotherapy based on grade of leukopenia with standard dosed chemotherapy are therefore highly needed. The present study (SBG 2000-1) is a randomised trial comparing standard FEC with tailored FEC in primary breast cancer. The aim of the present analysis is to evaluate dose-intensity, and how haematological toxicity responds to dose tailoring. Non-haematological toxicity among patients in the different treatment arms will also be analysed.
Patients and methods
Study design
The SBG 2000-1 trial is a prospective randomised study involving 33 centres nationwide in Sweden and Denmark, that investigates if dose tailoring of chemotherapy for primary breast cancer based on patients’ grade of leukopenia can improve outcome. A total of 1500 patients were to be included according to the statistical considerations that were based on distant disease-free survival (DDFS), which is the primary endpoint of this study. DDFS was defined as freedom from distant (non-local) recurrence and death of any cause. Women with lymph node positive or high risk node negative primary breast cancer were eligible. Patients should have undergone radical surgery at maximum two months prior to inclusion. High risk node negative was defined by local criteria, however, most Swedish centres required two of the following risk factors: Tumour size > 20 mm, ER and/or PR negativity, Elston grade III (8-9p) and/or high proliferation. Inclusion criteria in Denmark partly differed from those in Sweden and included premenopausal status, axillary nodal metastases or node negative status with one of three of the following factors: tumour size > 2 cm, grade two or three, or negative Er and PgR receptor status. Other major inclusion criteria were: age 18–60 years, performance status 0–1 according to WHO, no other major morbidity, adequate liver-and kidney function. All patients had to give their written informed consent. The study was approved by the ethical committees with jurisdiction for all participating centres.
A total of seven FEC courses every three week were given to the patients. The first course was standard dosed for all patients (5-fluorouracil 600 mg/m2, epirubicin 60 mg/m2, and cyclophosphamide 600 mg/m2). Blood counts including WBC were measured on days 10, 12, 13 and 15 after the first course. Patients who did not reach leukopenia grade III or more (i.e. those who had WBC ≥ 2) after the first course were randomised between continuing standard dosed therapy (the “standard” group) or tailored dosed therapy according to a dose escalation scheme (the “tailored” group), in which epirubicin (E) doses was escalated to 75 or 90 mg/m2 and cyclophosphamide (C) to 900 or 1200 mg/m2 (). Patients in the tailored group escalated to level 2 () not achieving grade III leukopenia were allowed to have doses escalated to level 3 for subsequent courses. Doses were reduced by one step at leukopenia grade IV in all arms. The protocol did not include prophylaxis with antibiotics or G-CSF. Patients who reached leukopenia grade III or IV (WBC < 2.0) after the first course were not randomised but registered in the study and allocated to further standard dosed FEC or dose reduction if experiencing leukopenia grade IV (the “registered” group) (). This third group of patients was to be monitored for toxicity and efficacy in the same way as the randomised groups. After courses 2–7, blood counts were only taken once, and this was at the same day (10, 12, 13 or 15) as the nadir WBC after course 1. The non-haematological toxicity (nausea, vomiting, diarrhoea, alopecia, fatigue and mucositis) was reported by the patients themselves with a questionnaire according to the NCI/CTC criteria for every course. Serious adverse events (SAE) were reported by the responsible local investigator in the Swedish centres alone to the data managing office. An adverse event was registered as serious if it was leading to hospitalisation, permanent disability or was life threatening. Danish centres reported only unexpected serious adverse events to the authorities and data centre. For this reason the comparison of SAEs between the three groups was done in Swedish patients alone.
Tabel I. Dose escalation and reduction scheme (mg/m2).
Table II. Consort diagram.
Statistical methods
The relative dose intensity was calculated as: The given dose delivered in the originally expected time/the expected dose in the expected time. The expected time for seven courses is 21 weeks in this setting. This method has been widely used in other chemotherapy studies [Citation24–25]. The median cumulative doses of cyclophosphamide and epirubicin given in the different treatment arms were calculated in relation to the expected dose for each group. This was performed both for courses 1–7 and for courses 2–7 as the patients randomised to tailored FEC received course 1 with standard doses.
Haematological toxicity was quantified in terms of the maximal grade of leukopenia attained at any point during courses 2–7. The non-haematological toxicity was quantified as the maximal grade according to NCI/CTC criteria for each side-effect during courses 2–7 and the proportion of patients in each treatment group who experienced toxicity at least once. The incidence of haematological and non-haematological toxicity for the three treatment groups was compared with the two-sided Mann-Whitney test comparing the tailored group to either standard or registered patients. Differences in leukocyte nadirs at each of the courses 2–7 was tested by the two-sided t-test on logarithm-transformed nadir values. Nadir values were none-normally distributed, but the values were made to be normally distributed after logarithmic transformations. The differences in serious adverse effects (SAE) were tested with Fisher's exact test.
Results
Patients
A total of 1535 women in Sweden and Denmark entered the study between February 2001 and August 2003. Patient inclusion in the two randomised study arms and the registered arms, as well as patient exclusion from the present analysis is showed in . The number of patients included in this analysis is 1448. Of these patients 526 were randomised to standard dosed FEC courses 2–7, 521 were randomised to tailored FEC and 401 were only registered and allocated to continued standard dosed therapy courses 2–7 due to leukopenia grade III or more after the first course. Four patients randomised despite grade III leukopenia after course one are excluded from the analysis. One patient in the registered group had unknown BSA and could not be included in the analysis for dose intensity. Thirteen of the 521 patients in the tailored group (2.5%) were not treated with escalated doses despite leukocyte nadirs ≥ 2 × 109/l, six patients in the standard group were at some occasion given escalated doses (1.3%) and one patient in the registered group (0.3%) received dose escalated FEC. All these patients are included in the analyses.
The three treatment groups were well balanced for known prognostic factors such as tumour size, number of positive axillary lymph nodes and hormone receptor status. Furthermore, there were no significant differences in age and menopausal status ().
Table III. Patient characteristics.
Dose intensity
The median cumulative dose of C per m2 for courses 1–7 was 143% (6023 for mg/m2) in the tailored group compared to the planned doses for standard FEC giving a dose intensity of 287 mg/m2/w. The median cumulative doses for E per m2 in the tailored group was correspondingly 122% (520 mg/m2) compared to standard FEC giving a dose intensity of 24.4 mg/m2/w. The median relative dose intensity for C and E, defined as the given dose delivered in the originally expected time/the expected dose in the expected time was close to 100% in all three groups (). The relative dose intensity was increased by a factor of 1.32 (C 1.43, E 1.22) for patients in the tailored arm compared to the expected standard dose. The median cumulative dose of 5-FU was identical in all arms (4200 mg/m2). The treatment was well tolerated; more than 95% of patients received all seven courses, and treatment delays for more than one day were rare ().
Table IV. Relative dose intensity.
Table V. Treatment feasibility: number of treatments, number of treatment delays and omitted treatments.
Haematological toxicity course 2–7
The proportion of patients with leukopenia grade III–IV was significantly higher in the tailored group compared with those randomised to standard dosed FEC, 90% and 29% respectively (). In the registered group 88% reached leukopenia grade III–IV, thus the proportion of patients with leukopenia grade III–IV were similar for the registered and tailored groups but grade IV was more common in the tailored group. Cumulative distribution curves for leukocyte nadir at courses 1–7 are shown in . The distribution of leukopenia was similar in the tailored and registered arms at course 2–7, however, especially at later courses leukopenia tended to be somewhat more severe in the tailored arm. Although the tailored group showed significantly more pronounced leukopenia than the registered groups, particularly at later courses, the differences in geometrical mean values of leukocyte nadirs were small between the tailored and the registered groups (1.50 and 1.75, respectively at course 7) compared to the standard group (2.65 at course 7). The frequency of grade three to four thrombocytopenia was low in all arms, 0, 1.2% and 0.5% in the standard, tailored and registered arms, respectively. The differences in thrombocytopenia between the tailored and standard, and tailored and registered arms where nevertheless statistically significant, p < 0.005 for both comparisons (Mann-Whitney). Anaemia was significantly more common in the tailored (p < 0.001) and registered arms (p = 0.0001), but the incidence of grade three to four anaemia was low, 0.8% in the tailored arm and 0 in the two other study arms.
Figure 1. Distribution of nadir leukocytes after courses 1–7. Cumulative percentage of patients in the three study arms on the x-axis with nadir values below value as indicated on the y-axis.
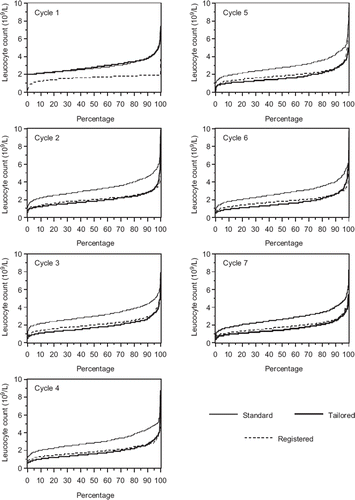
Table VI. Max grade of leukopenia during courses 2–7.
Non-haematological toxicity
The total number of patients where the grade of toxicity was known at least once during courses 2–7 for the treatment groups is shown in the table () (The patients who had not reported grade of a certain side-effect excluded). There were no large differences in non-haematological toxicity, however the patients in the tailored group experienced significantly more mucositis, nausea, vomiting, alopecia and fatigue than the standard group, and similar but smaller differences were seen compared to the registered group.
Table VII. Non-haematological side-effects during courses 2–7.
Serious adverse events (SAE)
Of the 932 included Swedish patients, 96 experienced a serious adverse event at least once, 14% and 6% among patients who received dose tailored treatment and standard dosed therapy respectively and 11% of the registered patients. The difference in SAE between the tailored and standard groups was statistically significant (p < 0.0007), while there were no significant difference between the tailored and the registered groups (p = 0.32). The most common event was hospitalisation due to leukopenia and fever/infection. Febrile leukopenia was more common in the tailored group (7.9%) compared with the standard group (0.9%, p < 0.0007), but similar to the frequency in the registered group (5.9%, p = 0.42). One patient in the tailored group died of cardiac arrest 10 days after the sixth course of chemotherapy.
Discussion
A number of previous retrospective studies has shown, that breast cancer patients given adjuvant chemotherapy but not attaining al least moderate haematological toxicity have a worse prognosis than those with more toxicity [Citation16–20]. The most plausible explanation may be the considerable pharmacokinetic and pharmacodynamic interindividual variations between patients demonstrated in several studies of patients treated with chemotherapeutic regimens commonly used as adjuvant treatment [Citation11–13]. This exposes a proportion of patients to risk of severe, potentially fatal toxicity, while some patients may be undertreated. Three previous European adjuvant breast cancer studies have shown that doses of epirubicin, and potentially cyclophosphamide are important for achieving maximal benefit of adjuvant chemotherapy [Citation3,Citation7,Citation23]. The French study FASG-01 compared three courses of the FEC regime with doses of 500–50–500 mg/m2, respectively with doses of 500–75–500 mg/m2, respectively [Citation7]. A later trial (FASG 05) compared six courses of FEC 500–50–500 with six courses of FEC 500–100–500 [Citation7]. Finally, a Belgian study compared eight courses of EC 60–500 with the same number of EC 75–900 [Citation23]. Both the FASG 05 and the Belgian study showed significant benefit of dose escalation, while the FASG 01 study showed only a marginal non-significant trend towards better outcome with the higher dose of E, possibly due to the suboptimally short adjuvant treatment (three courses). Although dose escalation of epirubicin from the previous standard of 50–60 mg/m2 seems to be beneficial it may also expose some patients to unnecessary serious toxicity. One of the main findings of the SBG 2000-1 trial was that 28% of patients reached maximum tolerated haematological toxicity already at FEC doses of 600–60–600, and might not be dose-escalated without serious risk of leukopenic infections. Thus a more rational way of achieving optimal doses might be to start at a more moderate dose level, and escalate only those patients, whose bone marrow can tolerate such escalation.
The SBG-trial is the first prospective randomised trial that evaluates the concept of individually dosed tailored FEC based on haematological toxicity, compared with standard dosed chemotherapy based on patients’ BSA. This report shows that an increased total dose and intensity of epirubicin and cyclophosphamide can be achieved by dose tailoring. The dose intensity for E and C was increased with a factor of 1.31 compared with the expected standard dose. The present is not the first randomised study utilising the concept of doses adjusted to haematological tolerance. The Scandinavia trial SBG 9401 recruiting breast cancer patients with very high expected risk of relapse randomised patients to either high-dose chemotherapy or dose escalated and tailored FEC [Citation22]. The median total doses of E and C in the present study are considerably lower than the doses in the previous SBG 9401 study [Citation21] 512 mg/m2 compared with 780 mg/m2 for E and 6030 mg/m2 versus 10 238 mg/m2 for C. This difference is of course no surprise since the tailored FEC of the SBG 9401 was supported with G-CSF and prophylactic antibiotics [Citation7]. Recently, a Scandinavian randomised phase 2 trial, SBG 2004-1, has demonstrated, that dose tailoring and escalation in a two-weekly schedule with the aid of G-CSF is manageable also with the combination of anthracyclines and docetaxel [Citation24], presently studied further in the ongoing randomised European (PANTHER) study with more than 1650 included patients. One could argue that the standard dose of E in our study is in the low range compared to the 100 mg dose of the FACS 05 study. However the choice of doses in this study reflects our concern of the severe long-term toxicity that has been associated with the high doses of FEC in the SBG 9401 study. Possibly is the lower dose of E compared with the FEC100 regimen counter balanced by the higher doses of C and 5-FU in our study. Moreover, for a substantial proportion of patients these doses are the maximum tolerated doses without CSF support.
The dose escalating procedure in the present SBG 2000-1 study was designed to achieve leukopenia grade III (WBC < 2.0), which also succeeded for the majority. Further support of the dose tailoring concept was the finding that the incidence of grade III leukopenia after courses 2–7 was almost identical in the group randomised to tailored FEC and the group of non-randomised patients achieving grade III leukopenia after the first course.
Febrile leukopenia was more common in the tailored FEC arm compared with the patients randomised to standard FEC, but not higher that in patients achieving leukopenia grade III–IV with standard doses. This is of course expected since achieving leukopenia grade III was an object to the tailored arm. Moreover, no deaths were attributable to infections with or without leukopenia in our study. The risk of febrile neutropenia in the FASG 05 study was surprisingly low, 2.7% [Citation25], considering that neither G-CSF nor antibiotics were used routinely. A possible explanation for the lower figure in the French study could be the use of lower doses of C and 5-FU. In the SBG 9401 febrile neutropenia was not reported separately but the incidence of grade III–IV infections was 19.5% in the tailored FEC arm [Citation21]. This was despite the use of G-CSF and prophylactic ciprofloxacin. The incidence of severe infections with leukopenia was 7% in the tailored arm of our study. Comparisons between the studies must obviously be done with extreme caution since there are differences in how toxicity has been reported.
The acute non-haematological toxicity in our study was comparable to what has been reported in earlier adjuvant trials involving FEC therapy. Dose escalation of courses 2–7 resulted in acceptable excess toxicity compared with continued standard dosed therapy with more total alopecia, mucositis, nausea, vomiting and fatigue.
The large proportion of patients showing a planned leukopenia response is encouraging and shows that individual dose tailoring according to leukopenia is feasible, reproducible and associated with only moderately increased non-haematological toxicity. Whether this will be reflected in an improved outcome, will be subject to a later analysis of efficacy data. If so, this study may change the principles of chemotherapy dosing.
References
- Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005;365:1687–717.
- Budman DR, Berry DA, Cirrincione CT, . Dose and dose intensity as determinants of outcome in the adjuvant treatment of breast cancer. The Cancer and Leukemia Group B. J Natl Cancer Inst 1998;90:1205–11.
- Fumoleau P, Roche H, Kerbrat P, . Long-term cardiac toxicity after adjuvant epirubicin-based chemotherapy in early breast cancer: French Adjuvant Study Group results. Ann Oncol 2006;17:85–92.
- Henderson IC, Berry DA, Demetri GD, . Improved outcomes from adding sequential Paclitaxel but not from escalating Doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. J Clin Oncol 2003;21:976–83.
- Focan C, Andrien JM, Closon MT, . Dose-response relationship of epirubicin-based first-line chemotherapy for advanced breast cancer: A prospective randomized trial. J Clin Oncol 1993;11:1253–63.
- Brufman G, Colajori E, Ghilezan N, . Doubling epirubicin dose intensity (100 mg/m2 versus 50 mg/m2) in the FEC regimen significantly increases response rates. An international randomised phase III study in metastatic breast cancer. The Epirubicin High Dose (HEPI 010) Study Group. Ann Oncol 1997;8:155–62.
- Bonneterre J, Roche H, Kerbrat P, . Epirubicin increases long-term survival in adjuvant chemotherapy of patients with poor-prognosis, node-positive, early breast cancer: 10-year follow-up results of the French Adjuvant Study Group 05 randomized trial. J Clin Oncol 2005;23:2686–93.
- Baker SD, Verweij J, Rowinsky EK, . Role of body surface area in dosing of investigational anticancer agents in adults, 1991–2001. J Natl Cancer Inst 2002;94:1883–8.
- Gurney H. Developing a new framework for dose calculation. J Clin Oncol 2006;24:1489–90.
- Gurney HP, Ackland S, Gebski V, Farrell G. Factors affecting epirubicin pharmacokinetics and toxicity: Evidence against using body-surface area for dose calculation. J Clin Oncol 1998;16:2299–304.
- Jakobsen P, Bastholt L, Dalmark M, . A randomized study of epirubicin at four different dose levels in advanced breast cancer. Feasibility of myelotoxicity prediction through single blood-sample measurement. Cancer Chemother Pharmacol 1991;28:465–9.
- Sandstrom M, Freijs A, Larsson R, . Lack of relationship between systemic exposure for the component drug of the fluorouracil, epirubicin, and 4-hydroxycyclophosphamide regimen in breast cancer patients. J Clin Oncol 1996;14:1581–8.
- Sandstrom M, Lindman H, Nygren P, Johansson M, Bergh J, Karlsson MO. Population analysis of the pharmacokinetics and the haematological toxicity of the fluorouracil-epirubicin-cyclophosphamide regimen in breast cancer patients. Cancer Chemother Pharmacol 2006;58:143–56.
- Meinardi MT, Van Der Graaf WT, Gietema JA, . Evaluation of long term cardiotoxicity after epirubicin containing adjuvant chemotherapy and locoregional radiotherapy for breast cancer using various detection techniques. Heart 2002;88:81–2.
- Praga C, Bergh J, Bliss J, . Risk of acute myeloid leukemia and myelodysplastic syndrome in trials of adjuvant epirubicin for early breast cancer: Correlation with doses of epirubicin and cyclophosphamide. J Clin Oncol 2005; 23:4179–91.
- Cameron DA, Massie C, Kerr G, Leonard RC. Moderate neutropenia with adjuvant CMF confers improved survival in early breast cancer. Br J Cancer 2003;89:1837–42.
- Colleoni M, Price K, Castiglione-Gertsch M, . Dose-response effect of adjuvant cyclophosphamide, methotrexate, 5-fluorouracil (CMF) in node-positive breast cancer. International Breast Cancer Study Group. Eur J Cancer 1998;34:1693–700.
- Mayers C, Panzarella T, Tannock IF. Analysis of the prognostic effects of inclusion in a clinical trial and of myelosuppression on survival after adjuvant chemotherapy for breast carcinoma. Cancer 2001;91:2246–57.
- Poikonen P, Saarto T, Lundin J, Joensuu H, Blomqvist C. Leucocyte nadir as a marker for chemotherapy efficacy in node-positive breast cancer treated with adjuvant CMF. Br J Cancer 1999;80:1763–6.
- Saarto T, Blomqvist C, Rissanen P, Auvinen A, Elomaa I. Haematological toxicity: A marker of adjuvant chemotherapy efficacy in stage II and III breast cancer. Br J Cancer 1997;75:301–5.
- Bergh J, Wiklund T, Erikstein B, . Tailored fluorouracil, epirubicin, and cyclophosphamide compared with marrow-supported high-dose chemotherapy as adjuvant treatment for high-risk breast cancer: A randomised trial. Scandinavian Breast Group 9401 study. Lancet 2000;356:1384–91.
- Wilking N, Lidbrink E, Wiklund T, . Long-term follow-up of the SBG 9401 study comparing tailored FEC-based therapy versus marrow-supported high-dose therapy. Ann Oncol 2007;18:694–700.
- de Azambuja E, Paesmans M, Beauduin M, . Long-term benefit of high-dose epirubicin in adjuvant chemotherapy for node-positive breast cancer: 15-year efficacy results of the Belgian multicenter study. J Clin Oncol 2009;27:720–5.
- Margolin S, Bengtsson NO, Carlsson L, . A randomised feasibility/phase II study (SBG 2004-1) with dose-dense/ tailored epirubicin, cyclophoshamide (EC) followed by docetaxel (T) or fixed dosed dose-dense EC/T versus T, doxorubicin and C (TAC) in node-positive breast cancer. Acta Oncol 2011;50:35–41.
- Benefit of a high-dose epirubicin regimen in adjuvant chemotherapy for node-positive breast cancer patients with poor prognostic factors: 5-year follow-up results of French Adjuvant Study Group 05 randomized trial. J Clin Oncol 2001; 19:602–11.