Abstract
Background. Lipids are best known for their fundamental role in forming biological membranes and as intracellular signalling molecules. Interactions between proteins and lipids are central to nearly every cellular process yet these crucial relationships often go overlooked. Changes or switches in the lipid profile of a cell drastically affects cellular metabolism and signal transduction. In relationship to cancer, upregulation of lipid metabolism is often observed during the early stages of neoplasia and is a recognised hallmark of many types of cancer. Methods. We performed a comprehensive review of the literature using PubMed regarding lipid metabolism in cancer and the importance of protein-lipid interactions in the function of mitochondria. Results. An increase in the basal rate of de novo lipogenesis generates a substantial rise in the saturated fatty acid content of cellular membranes. The ensuing alteration in the acyl chain profile of phospholipids has severe consequences on the function of organelles and membrane-bound proteins, and result in a host of pathologies including the cardiac disorder Barth Syndrome. Conclusions. Although increased lipogenesis is specifically selected for during cellular transformation it remains unclear if it confers an advantage for survival or is a byproduct of more global changes in cellular metabolism. We discuss the current data regarding the potential of targeting the lipogenic switch as a cancer therapy. In addition, we describe the importance of mitochondrial phospholipid composition during a number mitochondria-driven events observed to have roles in cancer. We specifically highlight the function of cardiolipin in maintaining mitochondrial structure, regulating mitochondrial dynamics and bioenergetics as well as its contributions to mitophagy/autophagy and apoptosis.
Mitochondrial phospholipids are intimately engaged with a diverse array of cellular functions including programmed cell death, respiration, mitochondrial dynamics and autophagy/mitophagy. Membrane phospholipids support the formation of protein complexes by creating nucleation sites and can also provide activation platforms for the initiation of signalling cascades. Lipids are thereby fundamentally involved in directing the movement of many proteins between cellular compartments and organelles as well as in the generation of vesicles per se. Thus, any changes in the production, constitution or localisation of phospholipids would have profound effects on overall cellular behaviour. Recent studies on the alteration of the fatty acid profile of a cell, and more specifically changes in the balance between saturated and unsaturated acyl chains, are providing clues into the role of phospholipids in these dynamic events. Therefore, there is a need to comprehensively understand why many cells are compelled to modify their fatty acid content during the transition to a disease state. For these reasons, fatty acid synthesis and lipid metabolism are quickly becoming attractive targets in the treatment of diabetes, obesity and cancer. Herein, we summarise recent advances in the study of fatty acid metabolism and the lipogenic phenotype during cancer and several different types of mitochondrial-driven cellular processes.
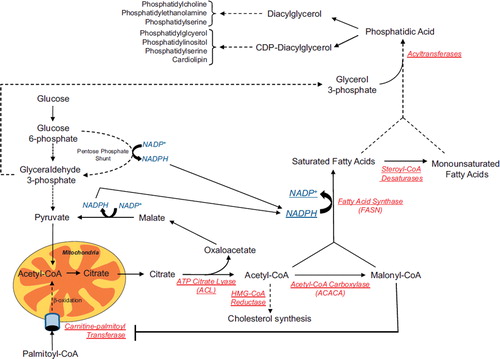
Figure 1. Lipid metabolism. The schematic shows the conversion of metabolic intermediates into free fatty acids and phospholipids. Reducing equivalents, which are required during fatty acid synthesis, can be provided by the pentose phosphate shunt and the conversion of malate to pyruvate. Besides being a carbon source for the production of citrate, glycolysis also provides glycerol 3-phosphate which is used in the formation of triglycerides and phospholipids. Acetyl-CoA can be used in the production of fatty acids through conversion to malonyl-CoA, but can also be used for cholesterol synthesis. The degradation of fatty acids, or β-oxidation, is mediated by the uptake of palmitoyl-CoA across the mitochondrial membrane by carnitine-palmitoyl transferase. This step is regulated by the levels of malonyl-CoA within the cell with high levels being inhibitory. Many of the enzymes in red are often deregulated during tumorigenesis. Dashed lines represent conversions which require multiple steps and enzymes. Solid lines represent direct conversions and/or transport.
Lipogenesis and cancer
Upregulation of de novo fatty acid synthesis in cancer
In 1950s Medes et al. published that neoplastic tissues presented slightly increased de novo lipogenesis, but concluded that these increases were not sufficient to support the high level of proliferation noted in cancer cells [Citation1]. It was not until three decades later when it was determined that the vast majority of total tumour fatty acid content was derived from de novo lipogenesis, at least in some types of cancer [Citation2]. And still another decade before Kuhajda and colleagues identified a gene highly associated with poorer prognosis in breast cancer as a key enzyme in de novo lipogenesis, namely fatty acid synthase (FASN) [Citation3]. Since then there has been a significant rise in the number of publications examining the link between upregulated lipogenesis and cancer. This “lipogenic phenotype” is observed in many types of cancer but is most often associated with hormone and growth factor receptor driven tumours [Citation4]. Steroid hormone receptors and other types of growth factor receptors induce the activation of PI3K-Akt or ERK1/ERK2 pathways, both of which converge on the activation of the transcription factor sterol regulatory element-binding protein 1c (SREBP1c) [Citation5]. SREBP1c upregulates many of the enzymes involved in lipid and cholesterol metabolism including FASN, acetyl-CoA carboxylase (ACACA), HMG-CoA reductase, and steroyl-CoA desaturase. Early studies assumed the increase in de novo lipogenesis would be necessary to satisfy the high demand for membrane biogenesis in rapidly proliferating tumours. However, more recent studies demonstrate that the role of lipogenesis goes beyond providing fatty acids for the formation of membranes. Lipids are potent signalling molecules and their extensive remodelling and modification impacts on a diverse array of cellular processes. This section of the review covers current topics associated with lipid metabolism and cancer.
Inhibition of de novo lipogenesis as a treatment for cancer
The main molecular targets of fatty acid metabolism have been FASN and ACACA with many laboratories using pharmacological inhibition or siRNA mediated knockdown in normal versus tumour tissue to study their individual roles in tumour cell survival. It is widely accepted that knockdown of either FASN or ACACA leads to cell death specifically in cancer cells [Citation6]. However, it was not clear if the mechanism of cell death involved end-product starvation (i.e. loss of palmitate production) or accumulation of biochemical precursors (i.e. malonyl-CoA). Initial studies had suggested it was the specific accumulation of malonyl-CoA that was toxic to the cells [Citation7]. However, these results were challenged when inhibition of ACACA activity was found to be as equally potent at inducing tumour cell death [Citation8,Citation9] as well as ATP citrate lyase [Citation10,Citation11]. Since then there have been numerous reports supporting both possibilities and the topic remains unresolved [Citation12–14]. Further adding to the intrigue is that there is no consensus concerning the mechanism of cell death after de novo lipogenesis inhibition. There are reports supporting malonyl-CoA and carnitine palmitoyltransferase-1 inhibition [Citation7], caspase-mediated apoptosis [Citation8,Citation13,Citation15,Citation16], reduced levels of phospho-Akt [Citation17,Citation18], inhibition of Bcl-2 and Mcl-1 [Citation19], ROS and mitochondrial impairment [Citation14,Citation20], ceramide production [Citation21], endoplasmic reticulum (ER) stress [Citation22], inhibition of mTOR [Citation15], and autophagy [Citation13,Citation23]. Possible reasons for the varied results could be the use of different tissues, cell types and culturing conditions or more likely differences in the metabolic and lipogenic signatures of the individual types of tumour cells. Nevertheless, a comprehensive analysis on the effects of the lipogenic phenotype in tumour cells and the mechanism of cell death associated with its inhibition is warranted.
The lipidomic switch as a therapeutic target in cancer
Outside the role of de novo lipogenesis in providing phospholipid precursors for the increased demand of membrane synthesis, Rysman et al. recently provided an additional reason as to why it might be selected for in certain types of cancer [Citation24]. Given that the typical end product of de novo lipogenesis is palmitate, increases in de novo lipogenesis shift the balance between saturated fatty acids (SFAs) and polyunsatured fatty acids (PUFAs) to one which heavily favours SFAs [Citation25]. This lipogenic switch would not only alter the constitution and biophysical properties of the cellular membranes, but would also decrease susceptibility of phospholipids to peroxidation. Taken together these changes would act as a layer of protection against many of the insults of reactive oxygen species (ROS) and potentially inhibit the uptake of chemotherapeutic drugs [Citation24]. Furthermore, the propensity of phospholipids containing mainly SFAs to segregate into lipid rafts/detergent-resistant membranes would markedly alter signal transduction cascades, vesicular trafficking and cell migration [Citation26].
In tumours the lipogenic switch is suggested to be a consequence of an increase in glycolysis [Citation27]. Increases in de novo lipogenesis coupled with the ability of FASN to consume NADPH may provide the tumour cell, at least during the early stages of transformation, with a mechanism to balance the redox stress produced by the glycolytic switch and when oxygen becomes limited within the growing tumour [Citation28]. Furthermore, SFAs have the ability to compromise the DNA damage response in non-transformed cells, which may lead to defects in cell cycle checkpoints and the accumulation of other genetic mutations [Citation29]. Although DNA-damage induced foci formed normally there were reduced levels of p53 activation in the presence of palmitic acid, which was suggested to be due to an inhibition of ataxia telangiectasia-mutated and ATM-Rad3-related kinases. For these reasons increased lipogenesis could be considered to markedly contribute to tumour progression. In fact, the idea of FASN as an oncogene is not implausible [Citation30]. It will be interesting to see if inhibitors of de novo lipogenesis could be coupled with dietary changes in an attempt to reverse the lipogenic switch and if this might increase susceptibility to chemotherapy. In line with this, inhibition of FASN can act synergistically with 5-FU, trastuzumab, or microtubulin interfering agents in inducing tumour cell death [Citation31–33].
Phospholipid composition and mitochondrial behaviour
The effects of lipids composition on mitochondrial pathophysiology
The first description of the role of lipids in energy-linked ion transport and oxidative phosphorylation in mitochondria was published in 1963 by Green and Fleischer [Citation34]. However, the lipid composition of mitochondrial membranes was not properly analysed until a few years later, revealing the peculiar enrichment of polar phospholipids such as phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and cardiolipin (CL) [Citation35]. The actual function and distribution of phospholipids in the mitochondria is still poorly characterised with the exception of CL, whose role has been extensively studied due to the association between defects in its biosynthesis and the genetic disorder Barth syndrome.
CL and mitochondrial bioenergetics
CL is a negatively charged diphosphatidyl glycerolphospholipid, which is almost exclusively found in biological membranes where the generation of an electrochemical gradient is coupled to ATP synthesis, such as the inner mitochondrial membrane (IMM) and the cytoplasmic membrane of bacteria. The peculiar chemical structure of CL, shared in some aspects with PE, allows its organisation into hexagonal structures with a potent non-bilayer forming activity which has important consequences for membrane morphology and physiology [Citation36,Citation37]. For example, CL is required for the regulation of the permeability of the IMM where it plays a key role in the maintenance of mitochondrial membrane potential and in the regulation of mitochondrial matrix osmolarity [Citation38,Citation39]. In addition, CL can regulate oxidative phophorylation by providing a lipid scaffold for the organisation of respiratory chain complexes into “supercomplexes”, functional macromolecular units which maximise electron flux [Citation40]. In line with this, mitochondria in which CL biosynthesis is impaired have significantly lower levels of oxidative phosphorylation activity [Citation41,Citation42]. Interestingly, the role of CL in the regulation of respiratory chain efficiency is not univocal. Indeed, mitochondria lacking assembly factors for respiratory chain complexes III and IV showed a significant decrease in CL biosynthesis, suggesting a feedback mechanism whereby CL biosynthesis is controlled by mitochondrial activity [Citation43]. CL has been recently found to be required for the assembly of the ATP/ADP carrier [Citation44] and also for the activity of the protein import machinery [Citation45] suggesting an intricate relationship between this phospholipid and protein biogenesis. Thus, protein-lipid interactions are emerging as important parameters for mitochondrial function. These interactions appear to be orchestrated by an important class of proteins of the IMM, the prohibitins. In a recent paper, Osman and colleagues demonstrate that prohibitins, which self organise into ring-like structures on the IMM, act like master regulators of membrane topology promoting the formation of lipid-protein macromolecular structures [Citation46].
CL and mitochondrial ultrastructure
The unique biophysical properties of CL and the presence of large protein clusters have a significant impact on the three-dimensional organisation of the IMM. CL deficient cells show aberrant cristae morphology in several animal and human models [Citation47,Citation48]. Although phospholipid composition contributes significantly to the shape of IMM, cristae morphology is controlled also by the activity of numerous proteins, among which the dynamin-related protein OPA1 has been the most widely investigated so far [Citation49–52]. Intriguingly, the yeast orthologue of OPA1, mgm1p, requires CL for a proper assembly into high order oligomers, suggesting that a close cooperation between lipids and proteins regulates mitochondrial ultrastructure. It is tempting to speculate that the elegant studies of Hackenbrock [Citation53] on the changes in the ultrastructure of mitochondria during different respiratory states could be explained by this tight control between CL abundance, respiratory chain activity, and ultrastructural changes.
CL and mitochondrial dynamics
Phospholipids, and more specifically CL, also contribute to the physicochemical properties of the OMM. Although CL is mostly found in the IMM, recent findings have shown that CL can be relocated to the OMM by phospholipid scramblase 3 [Citation54]. The presence of CL in the OMM has many biological consequences, from the formation of lipid scaffolds for the anchoring of proapoptotic molecules to the regulation of mitochondrial fusion. Interestingly, Choi and colleagues demonstrated that phospholipase D2, an OMM-bound lipid-modifying protein, is required for the fusion of juxtaposed mitochondrial membranes via the formation of the CL metabolite phosphatidic acid [Citation55]. Thus, while the growing family of dynamin related proteins have a clear role in membrane severing, tethering and curvature, lipid composition might directly affect the actual efficiency of lipid exchange between membranes [Citation56].
The lipidomic switch in mitochondria: A gateway for apoptosis
The lipidomic profile of mitochondrial membranes undergoes continuous changes due to fluctuations in biosynthetic processes [Citation57], intra- and inter-organelle lipid exchange [Citation58] and peroxidation. While the mechanism by which lipids are synthesised and imported in the mitochondria is just beginning to be understood, more is known about the lipidomic switch triggered by lipid peroxidation. Mitochondria are the predominant source of reactive oxygen species (ROS) in the cell due to the “leak” of electrons from the activity of the electron transport chain. CL, due to its high prevalence of polyunsaturated acyl chains, is particularly prone to ROS-mediated peroxidation. The detrimental effects of CL peroxidation on mitochondrial structure and function are well characterised hallmarks of numerous pathological conditions, such as neurodegenerative disorders, heart failure, cancer and ageing [Citation59].
Importantly, the peroxidation of CL has an impact that goes beyond mitochondrial bioenergetics. CL is known to bind to cytochrome c and sequester it in the IMM, where cytochrome c typically acts as a carrier of electrons in the repiratory chain [Citation60,Citation61]. However, besides being a vital component of the respiratory chain, cytochrome c is a protein involved in the activation of mitochondrial-dependent cell death. Release of cytochrome c from the IMS to the cytosol leads to the formation of the apoptosome (caspase-9 activation) and the initiation of apoptosis [Citation62]. Cytochrome c is not freely soluble in the IMS as a result of being predominantly bound to CL [Citation60]. Interestingly, Ott and colleagues found that the peroxidation of CL during mitochondrial-dependent apoptosis is a required step in promoting the mobilisation of cytochrome c from the IMM to the intermembrane space (IMS) and its subsequent release after permeabilisation of the OMM [Citation63]. In line with this view, CL-deficient mitochondria present more IMS-soluble cytochrome c [Citation64]. Furthermore, Scorrano and colleagues found that during apoptosis mitochondria undergo profound ultrastructural changes which is characterised by extensive cristae remodelling. This is required to allow the mobilisation of the solubilised cytochrome c from within the cristae, where it is predominantly confined, to the [Citation65]. As described previously, CL affects the ability of OPA1/mgm1p to oligomerise and since OPA1 oligomerisation has been found to be required for cristae remodelling, CL may have a yet uncharacterised role in cristae remodelling [Citation52,Citation66]. However, whether CL can regulate OPA1 complex formation or whether Barth syndrome (see below) patients have aberrant OPA1 processing has not been determined yet.
Barth syndrome is an X-linked recessive disorder characterised at the molecular level by the loss of the expression of an enzyme called tafazzin (TAZ) [Citation67]. Following de novo synthesis of CL, CL undergoes extensive remodelling to form mature CL [Citation68]. During this process CL is deacylated to form monolyso-CL (MLCL), possibly through the activity of calcium-independent phospholipase A2 [Citation69]. MLCL is then reacylated by TAZ to reform CL. However, TAZ preferentially uses polyunsaturated fatty acids during the reacylation process and over a number of cycles ultimately enriches CL with polyunsaturated acyl chains. The high proportion of polyunsaturated acyl chains found in mature CL confers many of the unique properties of CL.
CL has another important role in the activation of the caspase cascade after death receptor ligation [Citation70]. Interestingly, lympoblasts derived from Barth syndrome patients are resistant to death receptor induced apoptosis and after careful analysis it was shown that binding between mature CL and caspase-8 was necessary for a robust activation of caspase-8 [Citation71]. Further studies found that this complex contained not only caspase-8 and CL but the caspase-8 substrate Bid (a proapoptotic BH3-only Bcl-2 family member) [Citation72]. Caspase-8 cleaves Bid into its active cleaved form, tBid, within this complex. tBid in turn induces the permeabilisation of the OMM and activation of the mitochondrial arm of apoptosis. Thus, it seems that CL has a multifaceted role in apoptosis including the activation of caspases, the mobilisation of cytochrome c and control of cristae morphology.
Is the quality control of mitochondra regulated by lipids?
The lipidomic switch observed under many pathological conditions inevitably cause changes in the structure of the mitochondrial reticulum. Mitochondrial fragmentation is a hallmark of apoptosis but it also emerging as an important signal for the clearance of dysfunctional organelles, such as during the specific targeting of mitochondria by the autophagic machinery in a process called mitophagy. Mitophagy, first coined by J. J. Lemasters [Citation73], is a specific form of autophagy in which damaged or dysfunctional mitochondria are degraded. Mitophagy essentially functions as a quality control mechanism by removing potential apoptotic inducing factors, sites of high ROS production and energetic burden. Due to its apparent importance in many disease areas, such as cancer, ageing, metabolic disorders and neurodegenerative disease it has become a subject of increasing interest, much of which has focused on a small set of proteins: PINK1, PARKIN, NIX/BNIP3, NBR1 and p62 [Citation74–83]. A relatively new field of research, much of the specifics of its mechanism remain unclear, and the role of mitophagy in cell survival and death is hotly debated. Other areas for consideration include the role of the biological membranes and more specifically the lipids they are composed of, as well as the seemingly crucial connection between mitophagy and mitochondrial dynamics.
Mitochondrial dynamics during autophagy
The balance between the opposing process of mitochondrial dynamics, fission and fusion appears an essential determinant in mitophagy. Fission appears to be intimately linked to the progression of mitochondrial degradation. Following fission depolarisation occurs which in turn results in mitochondrial engulfment by autophagosomes. Inhibition of FIS-1 and DRP1 (pro-fission proteins) causes failure of mitochondria to fragment and the appearance of mitochondria in the autophagosome is greatly reduced [Citation84]. In addition, under starvation pro-fission proteins are observed to be prevented from localising to the mitochondria, resulting in a biasing of the scales in favour of fusion and mitochondrial extension, which prevents mitochondrial degradation by macroautophagy [Citation85]. It must also be noted that loss of mitochondrial membrane potential is essential for the initiation of mitophagy, with fission events often resulting in one of the resulting daughter mitochondria being depolarised [Citation84]. Using mutant forms of FIS-1, Gomes and Scorrano show that fragmentation of the mitochondria alone is not sufficient to bring about mitochondrial engulfment by the autophagosome, but that loss of functionality, resulting from depolarisation, is also required [Citation86]. Together these groups have demonstrated that both fragmentation of mitochondria by fission and mitochondrial depolarisation are important initator steps in mitophagy. A recent review discusses in greater depth the intricacies of mitochondrial dynamics and the link with mitophagy, where here we move into the role played by lipids during mitophagy [Citation87].
Phosphatidylinositol (4,5) bisphosphate (PtdIns (4,5) P2) has been shown to play a role in mitochondrial fragmentation [Citation88]. Depleting the levels of PtdIns (4,5) P2 on the mitochondria through dephosphorylation by phosphatases, amino acid starvation or by inhibiting its function by masking its headgroup leads to an increase in both mitochondrial fragmentation and mitophagy. The reduction in PtdIns (4,5) P2 levels was found to be associated with a decrease in the activity of a mitochondrially-targeted form of PKC (PKCα) [Citation88]. However, the specific role of PKCα has yet to be established but it is thought that PKCα-targeted proteins on the mitochondria or in the cytosol may be crucial in the control of mitochondrial integrity.
Another lipid which may be involved in mitophagy is CL. Interestingly, CL de novo synthesis was shown to be down-regulated following mitofusin 2 (MFN2) induced mitochondrial fusion [Citation89] and was associated with decreased levels of CL precursors and reduced expression of phosphatidlyglycerol phosphate synthase (PGPS). Twelve hours post-fusion the effects are reversed and there is an increase in de novo synthesis of CL as well as increases in PGPS expression and activity. The mitochondrially localised enzyme TAZ also shows increased expression following fusion. As discussed earlier, TAZ is essential for the remodelling of CL and is responsible for specifically incorporating long polyunsaturated acyl chains into CL and in creating its unique acyl chain profile. It is postulated that the down-regulation of CL levels allows for uninterrupted fusion. However, CL can promote negative membrane curvature, especially in the presence of calcium [Citation90] and would be expected to be pro-fusogenic, given its additional role in the GTPase activity of the yeast orthologue of OPA1, mgm1p [Citation91]. However, formation of phosphatidic acid through mitochondrial phospholipase D-dependent hydrolysis of CL was suggested to promote fusion [Citation55]. Nevertheless, the subsequent up-regulation of CL synthesis post-fusion and the restoration of pre-fusion CL levels most likely are needed to re-establish normal electron transport chain function and/or the exposure of CL on the surface of the mitochondria. Interestingly, CL is intimately involved in the formation of contact sites between the IMM and OMM and its upregulation may be needed to restore these important signalling and transport centres [Citation92]. It may be of interest to look at the effects of preventing the reduction in CL synthesis immediately post-fusion. Will this prevent complete fusion from occurring or not and does calcium play a significant role in this process? Also the fate of mitochondria which fail to increase CL synthesis post-fusion, will this cause mitochondrial dysfunction and mitophagy? While considering the effects of CL, the modifying effects of cholesterol should also be considered, as cholesterol can affect the oxidation state of CL [Citation93].
The lipid source for autophagosomes
In addition to the effects lipids have on the mitochondrial membrane, they also have an important role in forming the autophagosomal membrane required for degradation of mitochondria through mitophagy. The origin of the autophagosomal membrane has proven enigmatic, mostly due to the lack of any organelle specific proteins present on the autophagosomal membrane [Citation94]. It seems plausible that the membrane could originate from any (or a combination) of the organelles within the cells. There is currently support for three potential candidates for membrane donation, namely the mitochondria, the Golgi and the endoplasmic reticulum (ER).
Mitochondria are a major site of phosphatidylethanolamine (PE) synthesis [Citation95]. Given that PE is essential for the binding of LC3 to the autophagosomal membrane [Citation96], it strongly implicates mitochondria as a likely source of PE during autophagosome formation. Furthermore, shortly after the transfer of phosphatidylserine (PS) from the ER to the mitochondria, it is converted to PE [Citation97]. Tracing this transfer using fluorescently-tagged phosphatidlyserine (PS) shows that PS can be specifically transferred from the mitochondria and subsequently to the autophagosomal membrane [Citation98]. Disrupting the interaction between ER and mitochondria by knockout of MFN2 blocks the transfer of PS from the ER to the mitochondria and results in a failure to form autophagosomes. In addition, the presence of OMM markers were observed on the autophagosomes but other organelle markers failed to co-localise. Importantly, this process was considered separate from mitophagy as no IMM or mitochondrial matrix proteins were found to co-localise with the emerging membrane.
In most of the studies looking at the formation of autophagosomes the inducing signal is starvation. While this is a well recognised activator of autophagy, it is not the only means to stimulate autophagy. Hypoxia, oxidative stress and mechanical stress are all potent inducers of autophagy. It should be noted that throughout we have discussed autophagy as a non-specific process, but it is well accepted that selective forms of autophagy exist (e.g. mitophagy, ER-phagy, peroxiphagy, macroautophagy), suggesting that perhaps the origin of the membranes and the type of autophagy being undertaken depend on the stimulus. Bernales et al. hint at this idea in a study in which they observe ER-derived autophagic membrane development which progresses into ER-phagy, but which failed to occur under starvation induced macroautophagy [Citation99]. These results would suggest that the organelle being degraded may provide its own membrane for the process. However, this may not be true in all cases as there are reports that mitochondria can donate membrane components during starvation, but that these donating mitochondria do not then become degraded by autophagy [Citation98]. In another study looking at mitochondrial dynamics during starvation-induced autophagy mitochondria elongate by fusion and avoid degradation by autophagy through a size restraint [Citation85]. It seems in this case that the active fusion may prevent mitophagy, yet it appears they still participate in membrane donation under these circumstances.
In summation, we have alluded to the fact that the membrane may be derived from multiple organelles. The data regarding each organelle is compelling, so perhaps the type of stress dictates the donor membrane or alternatively organelles might work together under certain circumstances, each donating various elements to the emerging autophagosome at various times in its development. Perhaps further investigation of lipid trafficking using labelled lipids may reveal other co-operative relationships between the organelles. It has been suggested [Citation100] that the golgi, in its role as protein and lipid distributor for the cell, may affect how membranes are donated from other organelles depending on what type of lipids they receive from the golgi. Unravelling the origins of the autophagsome will prove invaluable if we are able to exploit it in terms of therapy and even more so if we can target one form of autophagy from another.
Conclusions and perspectives
The title of this review refers to how a musical composition can be compared to the intricate and complex signalling mechanisms of protein-lipid complexes in a cell. For instance, in sheet music, the notes have no meaning without an appropriate staff and time signature. However, when the notes and staff come together they produce harmonies and melodies and support one another in organising complex ideas into a single coherent thought. In this analogy, the notes represent proteins while the staff represents the matrix which holds them all together, in this case the lipid bilayer. Individually the proteins and lipids are non-functional but together they are able to produce an immense array of outcomes. And just as music is able to evoke different emotions, the bilayer/proteins can evoke different cell signalling pathways. For instance, music can move us from a feeling of happiness (normally functioning cell) to one of chaos (dysfunctional signalling and cancer). But if we can re-adjust those notes and put them in the right order, we might be able to ameloriate the situation and bring harmony back to the system.
Acknowledgements
This work was supported by Cancer Research UK. There are no conflicts of interest to be declared.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res 1953;13:27–9.
- Ookhtens M, Kannan R, Lyon I, Baker N. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Physiol Regul Integr Comp Physiol 1984;247:R146–53.
- Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, . Fatty acid synthesis: A potential selective target for antineoplastic therapy. Proc Natl Acad Sci 1994;91:6379–83.
- Swinnen JV, Vanderhoydonc F, Elgamal AA, Eelen M, Vercaeren I, Joniau S, . Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int J Cancer 2000;88:176–9.
- Swinnen JV, Heemers H, Deboel L, Foufelle F, Heyns W, Verhoeven G, . Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway. Oncogene 2000;19:5173–81.
- Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol 2010;6:551–62.
- Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, . Malonyl-Coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res 2000;60:213–8.
- Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-α gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res 2005;65:6719–25.
- Zhan Y, Ginanni N, Tota MR, Wu M, Bays NW, Richon VM, . Control of cell growth and survival by enzymes of the fatty acid synthesis pathway in HCT-116 colon cancer cells. Clin Cancer Res 2008;14:5735–42.
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005;24:6314–22.
- Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, . ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005;8:311–21.
- Thupari JN, Pinn ML, Kuhajda FP. Fatty acid synthase inhibition in human breast cancer cells leads to malonyl-CoA-induced inhibition of fatty acid oxidation and cytotoxicity. Biochem Biophys Res Commun 2001;285:217–23.
- Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, . Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res 2007;67:8180–7.
- Chajes V, Cambot M, Moreau K, Lenoir GM, Joulin V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res 2006;66:5287–94.
- Knowles LM, Yang C, Osterman A, Smith JW. Inhibition of fatty-acid synthase induces caspase-8-mediated tumor cell apoptosis by up-regulating DDIT4. J Biol Chem 2008;283:31378–84.
- Zhou W, Simpson PJ, McFadden JM, Townsend CA, Medghalchi SM, Vadlamudi A, . Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer Res 2003;63:7330–7.
- Wang HQ, Altomare DA, Skele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A, . Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene 2005;24:3574–82.
- Murata S, Yanagisawa K, Fukunaga K, Oda T, Kobayashi A, Sasaki R, . Fatty acid synthase inhibitor cerulenin suppresses liver metastasis of colon cancer in mice. Cancer Sci 2010;101:1861–5.
- Ho TS, Ho YP, Wong WY, Chi-Ming Chiu L, Wong YS, Eng-Choon Ooi V. Fatty acid synthase inhibitors cerulenin and C75 retard growth and induce caspase-dependent apoptosis in human melanoma A-375 cells. Biomed Pharmacother 2007;61:578–87.
- Zecchin KG, Rossato FA, Raposo HF, Melo DR, Alberici LC, Oliveira HC, . Inhibition of fatty acid synthase in melanoma cells activates the intrinsic pathway of apoptosis. Lab Invest 2011;91:232–40.
- Bandyopadhyay S, Zhan R, Wang Y, Pai SK, Hirota S, Hosobe S, . Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Cancer Res 2006;66:5934–40.
- Little JL, Wheeler FB, Fels DR, Koumenis C, Kridel SJ. Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Res 2007;67:1262–9.
- Dowling S, Cox J, Cenedella RJ. Inhibition of fatty acid synthase by Orlistat accelerates gastric tumor cell apoptosis in culture and increases survival rates in gastric tumor bearing mice in vivo. Lipids 2009;44:489–98.
- Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, . De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res 2010;70:8117–26.
- Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: New players, novel targets. Curr Opin Clin Nutr Metab Care 2006;9:358–65.
- Staubach S, Hanisch FG. Lipid rafts: Signaling and sorting platforms of cells and their roles in cancer. Expert Rev Proteomic 2011;8:263–77.
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007;7:763–77.
- Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: A metabolic oncogene in prostate cancer? J Cell Biochem 2004;91:47–53.
- Zeng L, Wu GZ, Goh KJ, Lee YM, Ng CC, You AB, . Saturated fatty acids modulate cell response to DNA damage: Implication for their role in tumorigenesis. PLoS One 2008;3:e2329.
- Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, . Fatty acid synthase: A metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst 2009;101:519–32.
- Vazquez-Martin A, Ropero S, Brunet J, Colomer R, Menendez JA. Inhibition of Fatty Acid Synthase (FASN) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol Rep 2007;18:973–80.
- Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, . Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci U S A 2004;101:10715–20.
- Menendez JA, Vellon L, Lupu R. Targeting fatty acid synthase-driven lipid rafts: A novel strategy to overcome trastuzumab resistance in breast cancer cells. Med Hypotheses 2005;64:997–1001.
- Green DE, Fleischer S. The role of lipids in mitochondrial electron transfer and oxidative phosphorylation. Biochim Biophys Acta 1963;70:554–82.
- Fleischer S, Rouser G, Fleischer B, Casu A, Kritchevsky G. Lipid composition of mitochondria from bovine heart, liver, and kidney. J Lipid Res 1967;8:170–80.
- van den Brink-van der Laan E, Killian JA, de Kruijff B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim Biophys Acta 2004;1666:275–88.
- Cullis PR, de Kruijff B. Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim Biophys Acta 1979;559:399–420.
- Mileykovskaya E, Zhang M, Dowhan W. Cardiolipin in energy transducing membranes. Biochemistry (Mosc) 2005;70:154–8.
- Koshkin V, Greenberg ML. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem J 2002;364:317–22.
- Bianchi C, Genova ML, Parenti Castelli G, Lenaz G. The mitochondrial respiratory chain is partially organized in a supercomplex assembly: Kinetic evidence using flux control analysis. J Biol Chem 2004;279:36562–9.
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, . Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 2003;278:52873–80.
- McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol 2006;361:462–9.
- Gohil VM, Hayes P, Matsuyama S, Schagger H, Schlame M, Greenberg ML. Cardiolipin biosynthesis and mitochondrial respiratory chain function are interdependent. J Biol Chem 2004;279:42612–8.
- Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol 2008;182:937–50.
- Jiang F, Ryan MT, Schlame M, Zhao M, Gu Z, Klingenberg M, . Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem 2000;275:22387–94.
- Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, . The genetic interactome of prohibitins: Coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol 2009;184:583–96.
- Xu Y, Condell M, Plesken H, Edelman-Novemsky I, Ma J, Ren M, . A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A 2006;103:11584–8.
- Ohtsuka T, Nishijima M, Suzuki K, Akamatsu Y. Mitochondrial dysfunction of a cultured Chinese hamster ovary cell mutant deficient in cardiolipin. J Biol Chem 1993;268:22914–9.
- Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, . The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett 2002;523:171–6.
- Darshi M, Mendiola VL, Mackey MR, Murphy AN, Koller A, Perkins GA, . ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J Biol Chem 2011;286:2918–32.
- Rabl R, Soubannier V, Scholz R, Vogel F, Mendl N, Vasiljev-Neumeyer A, . Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. J Cell Biol 2009;185:1047–63.
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, . OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006;126:177–89.
- Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 1966;30:269–97.
- Liu J, Dai Q, Chen J, Durrant D, Freeman A, Liu T, . Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol Cancer Res 2003;1:892–902.
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol 2006;8:1255–62.
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010;11:872–84.
- Gohil VM, Greenberg ML. Mitochondrial membrane biogenesis: Phospholipids and proteins go hand in hand. J Cell Biol 2009;184:469–72.
- Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol 2011;192:7–16.
- Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol 2007;292:C33–44.
- Cortese JD, Voglino AL, Hackenbrock CR. Multiple conformations of physiological membrane-bound cytochrome c. Biochemistry 1998;37:6402–9.
- Nicholls P. Cytochrome c binding to enzymes and membranes. Biochim Biophys Acta 1974;346:261–310.
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996;86:147–57.
- Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A 2002;99:1259–63.
- Choi SY, Gonzalvez F, Jenkins GM, Slomianny C, Chretien D, Arnoult D, . Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ 2007;14:597–606.
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, . A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2002;2:55–67.
- Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, . Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell 2008;31:557–69.
- Barth PG, Scholte HR, Berden JA, Van der Klei-Van Moorsel JM, Luyt-Houwen IE, Van ‘t Veer-Korthof ET, . An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 1983;62:327–55.
- Houtkooper RH, Turkenburg M, Poll-The BT, Karall D, Perez-Cerda C, Morrone A, . The enigmatic role of tafazzin in cardiolipin metabolism. Biochim Biophys Acta 2009;1788:2003–14.
- Malhotra A, Edelman-Novemsky I, Xu Y, Plesken H, Ma J, Schlame M, . Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc Natl Acad Sci U S A 2009;106:2337–41.
- Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta 2009;1788:2022–31.
- Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, . Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol 2008;183:681–96.
- Schug ZT, Gonzalvez F, Houtkooper RH, Vaz FM, Gottlieb E. BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ 2011;18:538–48.
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005;8:3–5.
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008;183:795–803.
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 2009;5:706–8.
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, . PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010;8:e1000298.
- Cherra SJ, 3rd, Dagda RK, Tandon A, Chu CT. Mitochondrial autophagy as a compensatory response to PINK1 deficiency. Autophagy 2009;5:1213–4.
- Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009;284:13843–55.
- Geisler SHKM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/PARKIN – mediated mitophagy is dependant on VDAC1 and p62/SQSTM1. Nature 2010; 12:119–31.
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, . PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2009;107:378–83.
- Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway: A mitochondrial quality control system? J Bioenerg Biomembr 2009;41:499–503.
- Chen H, Chan DC. Mitochondrial dynamics – fusion, fission, movement, and mitophagy – in neurodegenerative diseases. Hum Mol Genet 2009;18:R169–76.
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol 2010;189:671–9.
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, . Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008;27:433–46.
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011;13:589–98.
- Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta 2008;1777:860–6.
- Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 2011;14:1939–51.
- Rosivatz E, Woscholski R. Removal or masking of phosphatidylinositol(4,5)bisphosphate from the outer mitochondrial membrane causes mitochondrial fragmentation. Cell Signal 2011;23:478–86.
- Xu FY, McBride H, Acehan D, Vaz FM, Houtkooper RH, Lee RM, . The dynamics of cardiolipin synthesis post-mitochondrial fusion. Biochim Biophys Acta 2010;1798:1577–85.
- Som A, Yang L, Wong GC, Tew GN. Divalent metal ion triggered activity of a synthetic antimicrobial in cardiolipin membranes. J Am Chem Soc 2009;131:15102–3.
- DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol 2009;186:793–803.
- Epand RF, Tokarska-Schlattner M, Schlattner U, Wallimann T, Epand RM. Cardiolipin clusters and membrane domain formation induced by mitochondrial proteins. J Mol Biol 2007;365:968–80.
- Montero J, Mari M, Colell A, Morales A, Basanez G, Garcia-Ruiz C, . Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim Biophys Acta 2010;1797:1217–24.
- Stromhaug PE, Berg TO, Fengsrud M, Seglen PO. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J 1998;335(Pt 2):217–24.
- Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev 2002;54:101–27.
- Ichimura Y, Imamura Y, Emoto K, Umeda M, Noda T, Ohsumi Y. In vivo and in vitro reconstitution of Atg8 conjugation essential for autophagy. J Biol Chem 2004;279:40584–92.
- Vance JE, Vance DE. Phospholipid biosynthesis in mammalian cells. Biochem Cell Biol 2004;82:113–28.
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, . Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010;141:656–67.
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 2006;4:e423.
- van der Vaart A, Griffith J, Reggiori F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol Biol Cell 2010;21:2270–84.