Abstract
Background. Patients with chemotherapy refractory metastatic colorectal cancer and KRAS mutations have no effective treatment option. The present study evaluated the efficacy of temsirolimus in chemotherapy refractory mCRC with KRAS mutations. Furthermore, we wanted to investigate if resistance to temsirolimus could be reversed by the addition of irinotecan. Finally, we analyzed pre-treatment blood samples for KRAS mutations to investigate the association between quantitative measures of KRAS mutated alleles and clinical outcome. Material and methods. Patients received weekly temsirolimus 25 mg until progression. Thereafter patients were treated with combination therapy comprising biweekly irinotecan 180 mg/m2 and weekly temsirolimus. A polymerase chain reaction method was used to quantify the KRAS mutated alleles in plasma (pKRAS). Results. Sixty-four patients were included. Treatment was well tolerated. Thirty-eight percent achieved stable disease on monotherapy and 63% on combination therapy. Four and eight patients had a minimal response, respectively. Median overall survival was 160 days. Median time to progression was 45 and 84 days, respectively. The concordance between KRAS status in tumor and plasma was 82%. All patients with tumor reduction had low levels of pKRAS. Patients with high pKRAS had a 77% risk of early progression on monotherapy compared to 43% in patients with lower levels. Multivariate survival analysis confirmed that pKRAS was a strong prognostic factor. Conclusion. Temsirolimus has limited efficacy in chemotherapy resistant KRAS mutant disease, but plasma KRAS quantification is a strong predictor of outcome.
A significant proportion of patients with chemotherapy resistant metastatic colorectal cancer (mCRC) are in good performance and third-line treatment is therefore relevant. Therapy with monoclonal antibodies (MoAbs) targeting the epidermal growth factor receptor (EGFR) is confined to patients with KRAS wild type tumors [Citation1,Citation2]. Consequently, patients with KRAS mutant disease who develop resistance to standard cytotoxic agents have no effective treatment options.
Temsirolimus is a novel inhibitor of mammalian target of rapamycin (mTOR), a central intracellular signaling molecule and a member of the phosphoinositide-kinase-related kinase family. It acts as a component of the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway that mediates eukaryotic cell growth and proliferation [Citation3]. Preclinical studies have demonstrated that temsirolimus inhibits the proliferation of different malignant tumors, including colorectal cancer cell lines [Citation4–6]. Enhanced antitumor activity has been reported when combined with different cytostatic drugs and recent preclinical data have demonstrated that the combination of rapamycin with irinotecan show potent inhibition of colorectal xenografted tumors [Citation7]. Temsirolimus is presently approved for treatment of renal cell carcinomas and has a favorable toxicity profile [Citation8,Citation9]. A low-dose regimen has shown clinical activity in patients with mantle cell lymphoma [Citation10] and a phase II study in heavily pre-treated metastatic breast cancer reported an objective response rate of 9.2% [Citation11]. mTOR inhibitor everolimus was evaluated in patients with different advanced tumors and clinical benefit was observed in 4/55 patients including one with mCRC [Citation12]. Taken together the current data hold promise of activity alone or in combination with chemotherapy in patients with CRC, including KRAS mutant disease justifying clinical trials.
The identification of KRAS mutation as a marker for non-responsiveness to anti-EGFR MoAbs has underlined the need for translational research studies being conducted in parallel to clinical trials. We have developed a highly sensitive method for mutational analysis which enables us to quantify the amount of KRAS mutation alleles in plasma samples.
The present study aimed to evaluate the efficacy of temsirolimus in chemotherapy refractory mCRC with KRAS mutation. Furthermore, we wanted to investigate if resistance to temsirolimus could be reversed by the addition of irinotecan. Finally, we analyzed pre-treatment blood samples for KRAS mutations to investigate the association between quantitative measures of KRAS mutated alleles and clinical outcome.
Material and methods
Trial design
The study was conducted as two consecutive non-randomized phase II trials (TIRASMUS, ClinicalTrials.gov identifier; NCT00827684). Patients were treated with monotherapy temsirolimus until disease progression or unacceptable toxicity. Thereafter the patients received combination therapy, which continued until progression or unacceptable toxicity.
The primary endpoints were response to monotherapy and combination therapy according to RECIST. Secondary endpoints included PFS, OS, toxicity and evaluation of the predictive and prognostic value KRAS mutation alleles in plasma.
Written informed consent was obtained from each patient. The study followed the Declaration of Helsinki and good clinical practice and was approved by ethics committees and the Danish Medicines Agency.
Patients
Patients met the following criteria: age ≥ 18 and ≤ 70, histologically confirmed metastatic colorectal adenocarcinoma with KRAS mutation and measurable disease as per RECIST. KRAS status was determined on archival tumor tissue. All patients had progressed on treatment with fluoropyrimidines, oxaliplatin and irinotecan. Adequate renal, hepatic and bone marrow function was required, in addition to PS ≤ 2, cholesterol and triglyceride within normal range.
Treatment schedules
Patients initially received weekly doses of temsirolimus 25 mg IV, followed by response evaluation every six weeks. Combination therapy comprised irinotecan 180 mg/m2 IV every second week and weekly temsirolimus. In this step the initial dose of temsirolimus was 15 mg, and if tolerated increased to 25 mg. The initially low dose was chosen to avoid any unexpected toxicity from the combination regimen. Dose reduction to less than 15 mg per week was not allowed and further non-tolerance excluded the patients from therapy. A treatment pause for more than three weeks, regardless of reason also excluded the patient. Similarly, combination therapy was given until progression, unacceptable toxicity, treatment pause or patient’s withdrawal of consent.
Evaluation criteria
Computed tomography (CT) scans of chest and abdomen were used for evaluation of response according to RECIST (version 1.0) and performed less than four weeks prior to first treatment given and every six weeks during treatment. Follow-up scans were performed every three months in patients who stopped treatment on stable disease and continued until first sign of progression.
Safety assessment
The NCI-CTCAE version 3.0 was used to assess toxicity and recorded at every visit for treatment until resolved. On combination therapy any dose reduction was applied to both drugs regardless of the type of toxicity.
Biomarker collection and analysis
Archived tumor tissue was obtained and blood samples for translational research drawn prior to therapy. KRAS tumor status was determined prior to treatment. Plasma samples were analyzed for KRAS mutations.
DNA purification and KRAS mutational analysis in plasma
DNA was purified from 1.0 ml EDTA-plasma samples using a QIAsymphony virus/bacteria midi-kit on a QIAsymphony robot (Qiagen, Düsseldorf, Germany) according to the manufacturer's instructions. Primers and probe for the KRAS assay were developed using the OLIGO 7 software (Molecular Biology Insights Inc., Cascade, CO, USA). The assay utilize an Amplification Refractory Mutation System-Quantitative PCR methodology which detects six mutations in KRAS codon 12 (Gly12Ala, Gly12Arg, Gly12Asp, Gly12Cys, Gly12Ser, Gly12Val) and one in codon 13 (Gly13Asp).
A wild type blocking oligo was added to increase the specificity. The oligos were modified by including HyNA nucleotides (Pentabase Aps, Soendersoe, Denmark), which increased the melting temperature and blocked extension. An in-house cyclophilin (gCYC) qPCR was included as positive control and reference. Final primer mixtures are shown in Supplementary Table I (to be found online at http://www.informahealthcare.com/doi/abs/10.3109/0284186X.2013.776175). All qPCR reactions were performed in a volume of 25 μl (duplicates) on an ABI Prism7900HT using ABI Universal Mastermix with UNG. A mixture of patient DNA representing all seven KRAS mutations was included as positive controls as well as (negative) water controls and wild type donor DNA controls. The qPCR reaction conditions were: 2 min at 50°C and 10 min at 95°C, followed by 50 cycles of 15 s at 95°C and 60 s at 60°C.
Table I. Patients characteristics.
Quantification of KRAS in plasma
From standard curves the slopes were calculated for the KRAS (3.6) primer sets. The Y-intercept (cycle threshold (Ct) value) corresponding to one DNA copy of the target DNA were set to 43. Quantification of KRAS was done by calculating the copy number of mutated KRAS alleles as 10((Y-intercept(KRAS)-meanCt(KRAS))/slope(KRAS)) and normalization to plasma volume.
Statistics
The study was conducted according to Simon's two-stage design. The initial planned sample size for the first stage was 15 patients. Due to the unknown response rate of mCRC patients to mTOR inhibition a stabilization of disease on monotherapy was considered relevant, whereas response was required for acceptance of the combination regimen. A success rate of 10% was deemed unacceptable and considered interesting if ≥ 30%. α was set to 0.05 and β = 0.2. Consequently, if one or more of the first 15 patients achieved stable disease on monotherapy, enrolment was extended by another 25 patients. Similar conditions were applied to combination therapy, but as no objective response was recorded according to RECIST in the first 15 evaluable patients, further enrolment in the second stage was suspended. Since not all patients were expected to precede to combination therapy an estimated total sample size of 50 patients was set.
The association between marker status and objective response, baseline characteristics and toxicity rates was determined by two-sided t-tests or χ²-test. The Kaplan-Meier method was used to estimate time to progression (TTP) and overall survival (OS). A multivariate Cox regression analysis was performed to examine whether the different variables were associated with reduced survival. P-values referred to two-tailed tests and were considered significant when p ≤ 0.05. Statistics were carried out using the NCSS statistical software 2007 v.07.1.5 (NCSS Statistical Software, Utah 84037, USA, www.ncss.com).
Results
Patient characteristics
Sixty-four patients were included in two Danish oncology centers. All patients had progressed on fluoropyrimidines, oxaliplatin and irinotecan and 86% had received prior anti-angiogenic therapy. Patient characteristics are listed in . Two patients experienced an anaphylactic reaction at the first infusion and one patient died before commencing treatment, leaving 61 patients having at least one cycle of monotherapy, and a total of 35 patients proceeding to the combination regimen. The reasons for discontinuation of treatment were toxicity (two), patient’s wish (five), postponed treatment (two), progression (41) and death (three), other (seven, including one myocardial infarct, and one discovered neuro-endocrine component). At time of this report 52 patients were dead and 12 still alive, including three still on treatment. The cause of death was recorded as mCRC in all deceased patients.
Toxicity
The serious adverse events are presented in . There were no grade 4 events, but two patients discontinued treatment after anaphylactic reaction and three had a mild allergic reaction to temsirolimus. The majority of grade 2–3 GI-related toxicity occurred during combination therapy as expected. Grade 2–3 hematological toxicity from monotherapy was primarily reversible thrombocytopenia, and grade 3 leukopenia occurred only during the combination treatment.
Table II. Reported toxicities possibly or definitely related to treatment in TIRASMUS.
Efficacy monotherapy
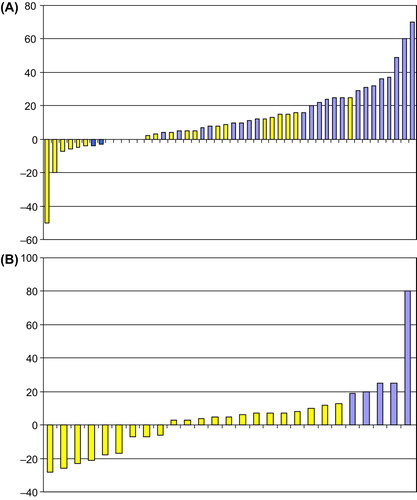
The median number of cycles was 3 (range 0–17). None of the patients achieved an objective response according to RECIST but 38% (24/64) of patients achieved SD on monotherapy, when analyzing the overall best responses. Of note, the results showed that four patients achieved a minor response (defined more than 5% tumor shrinkage) including two with reduction ≥ 20% as illustrated in .
Figure 1. Waterfall plot of changes in target lesions during monotherapy (A) and combination treatment (B). The plot illustrates the best response by changes in target lesions (only) from baseline presented in percentages on the Y-axis. Each patient is represented on the X-axis. Forty-eight patients were eligible for tumor evaluation during monotherapy, whereas 27 patients had evaluation scans performed during combination treatment. The colours illustrate the best response according to RECIST evaluation. Yellow represents the patients with SD during treatment and blue the patients who progressed.
Efficacy combination therapy
The median number of cycles was three (range 0–19). Sixty-three percent (22/35) of patients who commenced combination therapy obtained SD, including eight with a minor response, (three with reduction ≥ 10% and three ≥ 20%.). The maximum changes in target lesions are illustrated in . Three of the patients who achieved tumor reduction during combination therapy had also shown reduction from monotherapy.
Progression and survival data
The median TTP of monotherapy and combination therapy was 45 days (95% CI 41–67) and 84 days (95% CI 48–120), respectively. The median OS was 160 days in the total cohort (95% CI 103–232).
Biomarker study
Fifty-five patients had available plasma and tumor DNA for comparison. The remaining nine included seven patients who had no baseline sample available, and two which revealed inconclusive results (Supplementary Figure 1, to be found online at http://www.informahealthcare.com/doi/abs/10.3109/0284186X.2013.776175). The overall concordance between tumor and plasma analysis was 82%.
A high level of pKRAS was associated with early progression (). In patients with a high concentration of baseline pKRAS (above the 75% quartile) only 23% had temporary disease control, and 77% of the patient in this group progressed at time of first tumor evaluation compared to 43% of patients with lower levels (p = 0.036). All patients who achieved tumor reduction had low levels of baseline pKRAS.
Table III. Correlation between disease control and plasma KRAS levels.
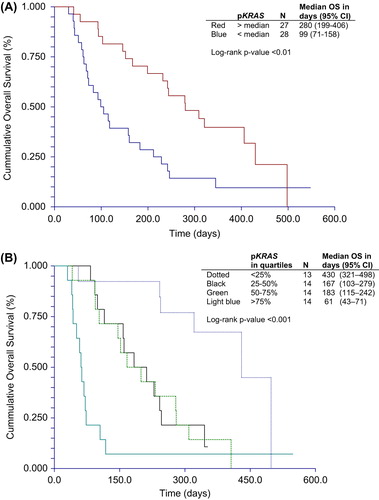
The median overall survival was significantly longer in patients with pKRAS values below the median (). The median survival in patients with levels below the median was 280 days (95% CI 199–406) compared to 99 days (95% CI 71–158) in those with higher levels, HR = 2.3 (95% CI 1.27–4.34, p = 0.0032). The overall survival according to the quartiles of pKRAS is illustrated in , which shows that patients in the highest quartile had a significantly worse prognosis than those in the lower quartiles.
Figure 2. Overall survival and patients divided into groups with pKRAS above and below the median level (A) and by pKRAS in Quartiles (B).
A Cox regression multivariate survival analysis including PS at inclusion, number of metastatic sites, and pKRAS confirmed the independent prognostic value of pKRAS in these patients.
Discussion
In this phase II study we were unable to demonstrate a clinically meaningful anti-tumor activity of temsirolimus alone. No objective responses according to RECIST were seen, but we did observe stabilization of disease in a proportion of the patients during both treatment regimens (38% and 63%) and tumor reduction in 7% and 23% of patients treated.
It is possible that the minimal responses observed were indicators of potential clinical effect and that therapy was given in insufficient doses. We chose the initial 15 mg and then increased it to 25 mg dose to avoid unexpected severe toxicity during the combination regimen, (which is of high priority in this specific group of incurable patients to whom quality of life is very important). Thus, for safety reasons a low dose approach was chosen and appeared feasible. Data have suggested that, e.g. breast cancer patients seem to benefit from a higher dose [Citation13], and it is also possible that the low-dose regimen used here may be adequate in other tumor types, but not in adenocarcinomas. However, an established dose escalation study was not the aim of this investigation.
The TTP was longer and the proportion of patients with tumor reduction was higher during combination therapy compared to single agent therapy. It is worth noticing that these heavily pre-treated patients after progression on temsirolimus alone achieved a median TTP of nearly three months on the combination regimen. This could indicate a potential effect and it is possible that with proper selection of patients a subgroup of patients would benefit from treatment.
The present study included patients with chemotherapy refractory KRAS mutant diseases, which clearly constitutes a major therapeutic challenge. The expected time to progression and clinical deterioration is unfortunately short, and identification of reliable criteria for selection of patients with a good chance of benefit is therefore of major importance.
It is possible that improvement of outcome could be achieved by selection based on mutational status. Nicolantonio et al., demonstrated that cell lines with PIK3CA alterations are more sensitive to mTOR inhibition in contrast to KRAS mutations, which interestingly seemed to be associated with resistance [Citation14], but another study presented two cases of ovarian cancer with RAS/RAF mutations who responded to mTOR inhibition. Further data suggested that mTOR inhibition may reverse colorectal cancer cell lines resistant to EGFR inhibitors [Citation15,Citation16]. Translational research studies of patients treated with mTOR inhibitors are therefore highly relevant.
Many aspects of KRAS mutant colorectal cancer need further investigations, including methodological issues, tumor heterogeneity, and different mutational status in primary and metastatic disease [Citation17–19]. Furthermore, results have indicated that clinical outcome could be influenced by the different types of KRAS mutations [Citation20]. The present study allowed us to demonstrate that KRAS analysis can be performed in the peripheral blood and we confirmed a high concordance between KRAS status in the primary tumor and plasma [Citation21,Citation22]. More importantly, we were able to quantify the amount of KRAS mutation alleles in plasma and revealed impaired outcome with increasing concentrations. To our best knowledge there is no available literature suitable for comparison, but we have recently reported a similar correlation in patients treated with cetuximab and irinotecan [Citation23]. Patients with a high level of pKRAS had a high risk of early progression and poor survival and may therefore not be candidates for third line treatment. We suggest that not the KRAS status itself, but rather the quantitative amount of KRAS mutational alleles is associated with aggressive disease behavior and could be used for tailoring treatment in mCRC. The obvious limitations of a small sample size and non- randomized study does not allow for a distinction between a potential predictive or prognostic value. Consequently, verification in larger samples sizes and randomized trials are needed to establish the true clinical impact of pKRAS as a marker.
In conclusion, temsirolimus alone or in combination with irinotecan showed limited efficacy in patients with KRAS mutant chemotherapy resistant mCRC. This study did not, however, investigate the potential efficacy in higher doses or less advanced disease with combination regimens. The measurement of KRAS mutations in the plasma is a relevant alternative to tissue analysis, and baseline quantification may potentially help to identify patients with a reasonable chance of benefit from third line treatment.
Supplementary Table I and Figure 1.
Download PDF (185.4 KB)Acknowledgements
We thank Lone Frischknecht, Lone Hartmann Hansen and Pia Nielsen for technical assistance. Funding was kindly received from Research Council Vejle Hospital. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol 2010;11:753–62.
- Qiu LX, Mao C, Zhang J, Zhu XD, Liao RY, Xue K, et al. Predictive and prognostic value of KRAS mutations in metastatic colorectal cancer patients treated with cetuximab: A meta-analysis of 22 studies. Eur J Cancer 2010;46:2781–7.
- Dancey JE, Curiel R, Purvis J. Evaluating temsirolimus activity in multiple tumors: A review of clinical trials. Semin Oncol 2009;36(Suppl 3):S46–58.
- Zhang YJ, Dai Q, Sun DF, Xiong H, Tian XQ, Gao FH, et al. mTOR signaling pathway is a target for the treatment of colorectal cancer. Ann Surg Oncol 2009;16:2617–28.
- Pencreach E, Guérin E, Nicolet C, Lelong-Rebel I, Voegeli AC, Oudet P, et al. Marked activity of irinotecan and rapamycin combination toward colon cancer cells in vivo and in vitro is mediated through cooperative modulation of the mammalian target of rapamycin/hypoxia-inducible factor- 1alpha axis. Clin Cancer Res 2009;15:1297–307.
- Gulhati P, Cai Q, Li J, Liu J, Rychahou PG, Qiu S, et al. Targeted inhibition of mammalian target of rapamycin signaling inhibits tumorigenesis of colorectal cancer. Clin Cancer Res 2009;15:7207–16.
- Pencreach E, Guérin E, Nicolet C, Lelong-Rebel I, Voegeli AC, Oudet P, et al. Marked activity of irinotecan and rapamycin combination toward colon cancer cells in vivo and in vitro is mediated through cooperative modulation of the mammalian target of rapamycin/hypoxia-inducible factor- 1alpha axis. Clin Cancer Res 2009;15:1297–307.
- Gerullis H, Ecke TH, Eimer C, Heuck CJ, Otto T, et al. mTOR-inhibition in metastatic renal cell carcinoma. Focus on temsirolimus: A review. Minerva Urol Nefrol 2010;62: 411–23.
- Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, et al. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol 2004;22: 2336–47.
- Farag SS, Zhang S, Jansak BS, Wang X, Kraut E, Chan K, et al. Phase II trial of temsirolimus in patients with relapsed or refractory multiple myeloma. Leuk Res 2009;33:1475–80.
- Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 2005;23:5314–22.
- Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: A phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 2008;26:1603–10.
- Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 2005;23:5314–22.
- Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest 2010;120:2858–66.
- Bianco R, Garofalo S, Rosa R, Damiano V, Gelardi T, Daniele G, et al. Inhibition of mTOR pathway by everolimus cooperates with EGFR inhibitors in human tumours sensitive and resistant to anti-EGFR drugs. Br J Cancer 2008; 98:923–30.
- Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR Axis Inhibitor. Mol Cancer Ther 2011;10:558–65.
- Bouchahda M, Karaboué A, Saffroy R, Innominato P, Gorden L, Guettier C, et al. Acquired KRAS mutations during progression of colorectal cancer metastases: Possible implications for therapy and prognosis. Cancer Chemother Pharmacol 2010;66:605–9.
- Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 2010;16:790–9.
- Linardou H, Briasoulis E, Dahabreh IJ, Mountzios G, Papadimitriou C, Papadopoulos S, et al. All about KRAS for clinical oncology practice: Gene profile, clinical implications and laboratory recommendations for somatic mutational testing in colorectal cancer. Cancer Treat Rev 2011;37:221–33.
- De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010;304:1812–20.
- Lecomte T, Ceze N, Dorval E, Laurent-Puig P. Circulating free tumor DNA and colorectal cancer. Gastroenterol Clin Biol 2010;34:662–81.
- Yen LC, Yeh YS, Chen CW, Wang HM, Tsai HL, Lu CY, et al. Detection of KRAS oncogene in peripheral blood as a predictor of the response to cetuximab plus chemotherapy in patients with metastatic colorectal cancer. Clin Cancer Res 2009;15:4508–13.
- Spindler KL, Pallisgaard N, Vogelius I, Jakobsen A. Quantitative cell free DNA, KRAS and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res 2012;18:1177–85.