
Abstract
Background. The diagnostic work-up and treatment of patients with neuroendocrine neoplasms (NENs) has undergone major recent advances and new methods are currently introduced into the clinic. An update of the WHO classification has resulted in a new nomenclature dividing NENs into neuroendocrine tumours (NETs) including G1 (Ki67 index ≤ 2%) and G2 (Ki67 index 3–20%) tumours and neuroendocrine carcinomas (NECs) with Ki67 index > 20%, G3. Aim. These Nordic guidelines summarise the Nordic Neuroendocrine Tumour Group's current view on how to diagnose and treat NEN-patients and are meant to be useful in the daily practice for clinicians handling these patients.
Classification
Gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs) are divided into neuroendocrine tumours (NETs, Grade 1 and 2 tumours) and neuroendocrine carcinomas (NECs, Grade 3 tumours) () and classified according to the WHO Classification of Tumours of the Digestive System 2010 and IUCC's TNM Classification of Malignant Tumours ().
Figure 1. Schematic drawing illustrating the classification of NENs.
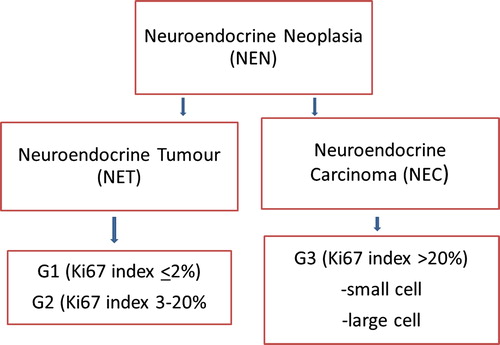
Table I. The WHO 2010 classification for neuroendocrine neoplasms (NEN) of the digestive system.
Epidemiology
NENs account for 1.0–1.5% of all GEP neoplasms [Citation1,Citation2]. Median age at diagnosis is approximately 65 years, except for appendicial and rectal NENs, which are diagnosed at a younger age [Citation1,Citation3,Citation4]. Data from USA show an increase in incidence of GEP-NENs from 1.0/100 000 per year in 1973–1977 to 3.65/100 000 per year in 2003–2007 [Citation3]. Similar incidence rates have been found in Europe [Citation4,Citation5].
The increased incidence may have several explanations: 1) increased awareness of NENs by clinicians and pathologists; 2) improved classifications; 3) improved diagnostic methods; 4) changes in population demography; and 5) a real increase in incidence.
The prevalence has been reported as high as 35/100 000 [Citation1], only exceeded by colorectal cancer among gastrointestinal (GI) cancers. The high prevalence is mainly explained by the long survival of NET patients.
At the time of diagnosis approximately 50% of all NENs are localised, 25% have regional disease and 25% have distant metastases [Citation1,Citation3,Citation4]. Approximately 25% of patients with GEP-NENs develop other primary cancers [Citation6].
Pathology
A core biopsy or surgical specimen should be used for histological diagnosis. Fine needle aspirate is insufficient for specific diagnosis and for calculation of Ki67 proliferation index.
The NET diagnosis is based on the growth pattern, the uniformity of tumour cells, and confirmed by immunohistochemical staining with the general neuroendocrine markers synaptophysin and chromogranin A (CgA). Neuron specific enolase (NSE) and CD56 are non-specific markers and should not be used for NET diagnosis. Calculation of Ki67 index is mandatory to establish tumour grade and should be assessed in areas with the highest number of tumour cells with nuclear labelling (hot spots). The percentage of stained cells is calculated with 500–2000 tumour cells as a reference. Other immunohistochemical markers (e.g. TTF-1, serotonin, CDX2) may give an indication of the site of the primary tumour.
Small-cell NECs are morphologically similar to small-cell lung cancer. Large-cell NECs usually have large pleomorphic cells and areas of necrosis but they can also be deceptively gland forming or insular. Pathologists should consider staining for synaptophysin and CgA in poorly differentiated GEP carcinomas. In the case of a NEC, synaptophysin staining is usually strongly positive whereas CgA may be negative.
Biochemical markers
Increased p-CgA is present in 60–80% of NET patients with residual tumour [Citation7] and high levels are associated with poor prognosis [Citation8]. CgA measurement is useful for diagnosis of NET, evaluation of treatment response and detection of progression and recurrence at an early stage [Citation9,Citation10]. Elevated levels of CgA are not specific for NETs. Chronic atrophic gastritis, treatment with proton pump inhibitors (PPIs), kidney, liver and heart failure as well as several non-neuroendocrine malignancies are associated with increased CgA [Citation11]. Several assays for measurement of CgA with different sensitivities and specificities exist [Citation12].
Depending on the primary tumour, laboratory findings and symptoms of the patient, measurement of specific markers, such as gastrin, insulin, c-peptide, pro-insulin, glucagon, VIP, pancreatic polypeptide (PP), somatostatin, ACTH and calcitonin should be performed. 5-hydroxyindoleacetic acid (5HIAA), a breakdown product of serotonin, is elevated in 70–75% of patients with SI-NETs and is traditionally measured in 24-hour urine collection. Measurement in the first voided urine in the morning seems to give comparable results [Citation13]. There are diet restrictions during the period of urine collection (avoiding serotonin rich food). Recently, a method for measurement of 5HIAA in blood has been developed and is presently validated for routine clinical use [Citation14].
NSE has only a sensitivity of 18–54% for NETs but may be a useful prognostic marker [Citation15].
Radiology
Computed tomography (CT) is the basic radiological modality for NEN imaging. However, magnetic resonance imaging (MRI) allows for better visualisation and characterisation of small liver and pancreatic lesions [Citation16]. Proper use of i.v. contrast media for CT (‘triple phase CT’), MRI and ultrasonography (US) is fundamental to visualise and characterise hyper- and hypovascular NET lesions. Therapy may render liver metastases clearly depicted in one phase at baseline examination to be completely disguised at follow-up and instead be visible in another phase [Citation17]. It is recommended to scan the thorax, abdomen and pelvis for primary tumour and/or metastases.
Diffusion-weighted MRI (DW-MRI) facilitates lesion detection [Citation18] and whole-body imaging (WBI) MRI for NENs have been implemented [Citation19]. By dynamic i.v. contrast-enhanced US it is possible to detect small (≤ 3 mm) liver metastases and to characterise previously equivocal tumour lesions [Citation20]. Alternate use of CT, MRI and US for follow-up may be advantageous in patients to visualise different tumour lesions, and to decrease the radiation dose especially in young patients with an expected long follow-up time with repeated imaging.
Endoscopic ultrasound (EUS) is highly sensitive for visualisation and loco-regional staging [Citation21] and for EUS-guided biopsy of the lesion. Intraoperative US is mandatory in hepato-biliary-pancreatic surgery for detection and localisation of small pancreatico-duodenal tumours and metastatic disease.
In the clinical daily routine, formal Response Evaluation Criteria in Solid Tumours (RECIST) evaluation may not always be feasible. Therefore, detailed information on lesion size should be included in the radiological report in order to facilitate monitoring of changes of the predefined target lesions over time. Since treatment usually result in tumour stabilisation rather than tumour shrinkage, other criteria such as decrease in tumour attenuation and contrast-enhancement need to be accounted for in the response evaluation [Citation22]. Due to the slow-growing nature of GEP-NETs it is important to compare lesion size over a longer period of time (6–12 months) to detect changes in tumour burden.
Somatostatin receptor imaging (SRI)
SRI is used for detection of the primary tumour, tumour staging, diagnosis of recurrent disease, and for evaluating eligibility for peptide receptor radionuclide therapy (PRRT).
Small tumours and tumours with no or low density of somatostatin receptors (SSTRs) may not be detected. There is no need to pause somatostatin analogue (SSA) treatment before SRI [Citation23].
SSTRs are also expressed in other tissues and differential diagnoses should be considered to avoid ‘false-positive’ findings, such as inflammation, scar healing and other non-neuroendocrine malignancies.
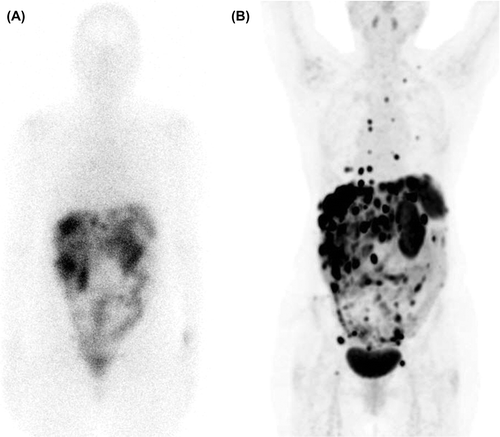
SRI is performed as a whole body investigation by scintigraphy (SRS) with tomography (SPECT) and recently also by positron emission tomography (PET) with 68Ga coupled to a somatostatin analogue. Both SPECT and PET should be performed together with a diagnostic CT for more precise localisation/interpretation. SRI with PET/CT is preferred to SPECT/CT because of higher spatial resolution and sensitivity of the former [Citation24] (). The PET/CT procedure takes only a few hours compared to 2–3 days with SPECT/CT and the radiation dose to the patient is lower with PET [Citation25].
Figure 2. Comparison of the resolution between SRS and PET/CT. A 56-year-old woman with a small intestinal NET G2 since 10 years with known liver metastases and peritoneal carcinomatosis. In picture 2A, SRS depicts only liver metastases, while PET/CT with 68Ga-DOTATATE performed one month later, displayed in picture 2B, shows extensive supra- and infra-diaphragmatic lymph node involvement as well as pleural and bone metastases. During the time period between the investigations the patient had radiologically, biochemically and clinically stable disease.
Other types of positron emission tomography (PET)
18F-FDG PET and SRI provide complementary information regarding the different biological characteristics of the lesions. Prognosis is worse if a tumour is 18F-FDG positive and SRI negative and vice versa [Citation26,Citation27]. 18F-FDG -PET should not be routinely performed in G1 NET, while it is especially recommended for staging of G3 tumours before surgery. In G2 tumours, and in case of tumour progression/aggressive clinical course in a G1 tumour, 18F-FDG PET/CT may be considered for staging and prognosis [Citation27].
SRI remains the gold standard for evaluating NETs using radionuclide imaging whereas PET/CT with 18F-L-DOPA or 11C-5-HTP are available as additional problem solving tools. Whether they add value in relation to PET/CT with 68Ga-DOTATATE or -DOTATOC is yet to be evaluated.
Imaging follow-up
The recommendations for follow-up of NEN- patients are based on current best practice since this is not a well-studied subject. The follow-up should generally be lifelong and the recommended imaging routines are detailed for each NEN type in the specific part of these guidelines. The imaging results need to be interpreted together with biochemical results and clinical symptoms. The recommended follow-up for G1–G2 patients after surgery or start of non-surgical therapy is initially every three months after which the interval can be prolonged to every 6–12 months in the case of stable disease. The surveillance may include CT/MRI/US which may be combined with SRI if progression is suspected or change in treatment is considered. NEC patients should be monitored by CT/MRI every 2–3 months.
Treatment overview
Surgery
All NEN patients should be considered for surgery. Patients with insulinomas, appendix and rectal NETs are often cured by surgery alone. However, curative surgery is probably possible in less than 30% of all NET patients and recurrence is common. When curative surgery is not possible, resection of the primary tumour and/or debulking surgery may be beneficial for control of local and endocrine symptoms [Citation28,Citation29].
There is no evidence or consensus for adjuvant or neo-adjuvant therapy for GEP-NET patients. However, chemotherapy may in few cases downstage the tumour obtaining curative resectability. To prevent excess hormone release during interventional procedures or surgery, peri-operative SSA treatment should be used, particularly in patients with SI-NETs or unknown primary.
As somatostatin-induced bile stones usually are asymptomatic, prophylactic cholecystectomy in NEN patients is generally not recommended. However, in patients with proven bile stones who are undergoing abdominal surgery, cholecystectomy may be considered [Citation30].
Chemotherapy in NET
Although well-designed, controlled studies are lacking, there are basically two indications established in clinical practice: advanced G1–G2 pancreatic NETs (P-NETs) and GEP-NEC (see separate section). Many studies on chemotherapy are difficult to interpret, as they report data from heterogeneous populations of NEN patients with a mixture of different primary tumour sites and the proliferation index is rarely reported. Many studies do not, for example, differentiate between SI-NET with a Ki67 index of < 2% and P-NETs with a Ki67 index of 5–10%. As chemotherapy mainly affects dividing cells, only a modest response would be expected when the proliferation index is low. Some have therefore advocated selecting NEN patients for chemotherapy based solely on the proliferation index, rather than the location of the primary tumour. In some patients, tumours with an initial low Ki67 index may develop a more aggressive phenotype over time, making re-biopsy advisable with reassessment of Ki67 index.
Streptozocin (STZ) in combination with 5-fluorouracil (5FU) is often used as a first-line treatment for advanced P-NET. It is usually given as a five-day induction course followed by one-day infusions every third week. STZ is nephrotoxic. Response rates (RRs) of 6–40% with progression-free survival (PFS) of 5–20 months and median survival of 16–24 months have been reported [Citation31]. In SI-NETs, however, results from a controlled randomised trial (CRT) comparing STZ to interferon seemed to favour the latter [Citation32].
Temozolomide may be used as first-line treatment for advanced P-NET. Retrospective data showed a response rate of 70% and PFS of 18 months of the convenient, oral combination of temozolomide and capecitabine [Citation33]. In a retrospective analysis of patients with P-NET receiving various temozolomide-based treatments, 34% experienced partial response with a median PFS of 13.6 months. By contrast, only 2% of non-P-NET patients had an objective response with a PFS of 9.6 months [Citation34]. Toxicity is usually mild with nausea and haematological toxicity. A number of other chemotherapy regimens have been used in NETs and reported as cases and small retrospective studies. None of these have sufficient power or quality to enable firm conclusions with respect to the effect in NETs.
Somatostatin analogues
More than 90% of NETs express SSTRs. The SSAs octreotide LAR (30 mg/4 weeks) and lanreotide autogel (120 mg/4 weeks) are used for symptomatic and anti-tumour treatment. It is unclear if the degree of uptake on SRI is predictive of the therapeutic effect of ¨cold¨ SSA treatment, and SSA may have a symptomatic effect even if the of uptake on SRI is low.
Similar symptomatic and biochemical response is observed in the majority of patients with SI-NET carcinoid syndrome and with functioning P-NET using either preparation (50–90%) [Citation35].
Two placebo-controlled CRTs of SSA treatment of NET-patients showing anti-tumour effect have been reported [Citation36,Citation37]. In the PROMID study including SI-NET patients, median time to tumour progression (TTP) was significantly longer with octreotide LAR (14.3 months) compared to placebo (6 months). In the CLARINET study including non-functioning NETs of different origins, median PFS was significantly longer on lanreotide (> 24 months) compared to placebo (18 months).
These studies show that SSA as a group has an antiproliferative effect in NETs, and treatment with SSA is therefore recommended in metastatic SI-NET disease and should be considered in P-NET with low Ki67 index (< 10%).
Common side effects of SSAs include abdominal pain, diarrhoea, flatulence, nausea, subcutaneous nodules at the injection site and development of bile stones. Steatorrhoea caused by reduced secretion of pancreatic enzymes should be treated with pancreatic enzyme replacement.
Pasireotide (SOM230) is a new SSA with broader and higher affinity for the SSTRs and reduces symptoms in octreotide LAR resistant patients [Citation38]. The side effects of pasireotide are similar to other SSAs except for hyperglycaemia, which is common and may be severe.
Interferon alpha
Interferon alpha (IFN) may reduce NET growth and hormone-induced symptoms. The dose should be titrated individually with doses between 3 and 5 MU s.c. 3–5 times weekly for recombinant IFN and 50–150 μg s.c. weekly for pegylated IFN. In SI-NET, symptomatic response is seen in up to 60%, biochemical response in 50%, tumour reduction in about 10% and tumour stabilisation in up to 65% lasting for long periods of time (> 36 months).
Common side effects include flu-like symptoms during the first few weeks of treatment and chronic fatigue. Bone-marrow depression is usually seen but related infections are extremely rare. Patients with autoimmune diseases or a history of mental disorder should not be treated with IFN [Citation39].
There are no placebo controlled trials of IFN and most studies are non-randomised. In the three CRTs published, the estimated PFS on IFN treatment was quite long. IFN should be considered for the treatment of G1 and G2 SI-NET (Ki67 < 10%) either as first-line treatment in combination with SSA or as second-line treatment when progression occurs during SSA mono-therapy.
Peptide receptor radionuclide therapy (PRRT)
Single-centre phase II studies of PRRT including almost 2000 patients have been published, giving evidence for the safety and efficacy of the treatment. Disease control rate ranges between 60% and 80%, PFS is usually between 2 and 3 years and symptoms and QoL improve significantly in the majority of patients after PRRT [Citation40–42]. Patients that respond to PRRT, but later progress, may be re-treated [Citation43].
The main indication for PRRT is inoperable and metastatic NETs overexpressing SSTR2. Patient selection is based on SRI; a high tumour uptake, relative to that of the normal liver, predicts a better treatment outcome [Citation44]. Treatment is more effective in G1 and G2 tumours than in G3 tumours [Citation40,Citation44]. Prerequisites for PRRT include a sufficient bone marrow, liver and kidney function.
177Lu-DOTATATE/DOTATOC or 90Y-DOTATOC are the preferred radiopharmaceuticals usually given as 2–5 cycles with 8–12 weeks intervals. Long-acting SSA treatment is interrupted 4–6 weeks before PRRT in order not to block the SSTRs. Short-acting analogues can be used until the day before PRRT. Treatment with chemotherapy should be stopped four weeks and IFN treatment at least two weeks before PRRT.
Acute side effects include nausea, vomiting and pain. Other side effects are bone marrow depression, which is usually reversible, and impaired renal function. Patients with extensive liver or bone metastases should be evaluated carefully prior to treatment because of the risk of severe hepatic failure or bone marrow toxicity. Myelodysplastic syndrome and leukaemia have been reported (1%) [Citation40–42].
There have been no published CRT of PRRT and the place for PRRT in the treatment algorithm is still not exactly defined although it is rarely used as a first-line treatment.
mTOR inhibitors
The mTOR-signalling pathway is often up-regulated in NENs. Everolimus, a specific inhibitor of the mTOR pathway, has been shown effective in NET treatment in two large phase III, multicentre CRTs. The RADIANT 2 study [Citation45] included 429 patients with different functioning NETs, patients who received everolimus + octreotide LAR tended to have longer PFS compared to those treated with placebo + octreotide LAR (16.4 vs. 11.3 months). The RADIANT 3 study [Citation46] including 410 patients with P-NET, showed a significant difference in favour of everolimus with PFS of 11.4 versus 5.4 months for placebo.
The main side effects of everolimus are stomatitis, rash, diarrhoea, hyperglycaemia, fatigue and infections. Dose reduction or temporary interruptions are frequently needed. A serious but uncommon side effect is non-infectious pneumonitis.
Everolimus is recommended for progressing P-NET. Its exact place in the treatment of SI-NETs and NETs from other primary sites than pancreas has still to be clarified.
Sunitinib
Sunitinib is an oral multi-tyrosine kinase inhibitor with its main affinity to the VEGF receptor.
In a multicentre phase III CRT including 173 patients with P-NETs, sunitinib treatment resulted in a significant improvement in PFS of 11.4 versus 5.5 months for placebo [Citation47].
Major side effects include rash, diarrhoea, nausea, vomiting, asthenia and fatigue. Dose reduction or temporary interruptions are frequently needed.
Sunitinib is recommended for progressing P-NET.
Specific part
This part refers mainly to G1 and G2 NET tumours while G3 (NEC) tumours are described in a common section at the end.
Inherited syndromes with associated NETs
Pancreatico-duodenal NETs can be part of a familial syndrome. Most common are MEN-1 and von Hippel Lindaus disease (VHL) but also patients with Carneys complex and neurofibromatosis type 1 (NF1) may develop NETs [Citation48].
In MEN-1, a mutation in the tumour suppressor gene encoding for the protein menin at 11q13 gives rise to multiple pancreatico-duodenal NETs, pituitary adenomas and parathyroid hyperplasia. Twenty-five percent of all gastrinoma patients have MEN-1. In these patients PTH, S-Ca and anterior pituitary hormones should be measured and family history recorded.
VHL has a mutation in 3p25–26 and gives rise to non-functioning P-NETs, pancreatic cysts and other tumour lesions. NF1 may be associated with somatostatinomas located at the ampulla of Vater [Citation48].
A reasonable approach is to perform genetic screening for MEN-1 in patients with P-NETs if there is a family history indicating MEN-1 lesions or if the patient presents with at least two lesions involved in the syndrome.
Oesophageal NETs
Oesophageal NENs are rare (< 1% of all NENs) and are usually NECs, see separate section.
Gastric NETs
1) Type I and II gastric NETs
Clinical presentation
Most type I and II gastric NETs are incidental findings at gastroscopy and comprise 85% of gastric NETs. In type I, the cause of hypergastrinaemia is destruction of parietal cells due to autoimmune chronic atrophic gastritis. The rare type II develops in patients with gastrin-producing tumours.
Diagnostic procedures
Gastric NETs are usually multiple, < 2 cm and with Ki67 index ≤ 5%. Multiple biopsies are recommended. P-CgA and s-gastrin are usually highly elevated but the clinical value of repeated measurements is questionable. In type I, gastric acid secretion is low or nil (high pH), while in type II acid secretion is elevated (low pH). EUS with evaluation of tumour invasion and regional lymph nodes is mandatory.
Treatment
Patients with tumours, single or multiple, < 1 cm should only undergo surveillance, since the lesions usually are benign. Tumours > 1 cm should be locally resected by endoscopy or surgery depending on the number of lesions, and whether or not there appears to be invasion into the muscularis propria.
Follow-up and prognosis
Repeated endoscopies, with 6–12 months interval should be performed. EUS, CT/MRI and SRI should only be performed when tumour growth or metastases are suspected. In type I gastric NETs local lymph node metastases are rarely found and liver metastases are almost never seen. In type II gastric NETs regional metastases are found in 5–10% and liver metastases in < 2%. The prognosis is good with a five-year survival of 100% in type I and 95% in type II gastric NETs.
2) Type III gastric NETs
Clinical presentation
Type III gastric NETs are sporadic, not associated with hypergastrinaemia and constitute 10–20% of all gastric NETs. Patients may present with symptoms of anaemia, GI-bleeding, dyspepsia and gastric obstruction. Local or liver metastases are seen in > 50% of the patients at diagnosis.
Diagnostic procedures
The tumour is usually solitary and larger than 2 cm in diameter with Ki67 index 3–20%. There are no specific biochemical markers but p-CgA may be elevated. EUS, CT of the thorax and abdomen and SRI should be performed for staging of the tumour.
Treatment
Surgical resection with lymph node dissection should be performed when possible. In histamine producing tumours symptoms may be reduced by SSA and H2-receptor blockers. Although firm clinical data is missing, for disseminated cases, IFN and/or SSA may be used if Ki67 < 10%, while chemotherapy is an option if Ki67 > 10%.
Follow-up and prognosis
Measurement of p-CgA, if elevated initially, gastroscopy and imaging with CT/MRI should be performed every 3–6 months. Five-year survival is 50%.
Duodenal NETs
Clinical presentation
The most common tumours are gastrinomas (40%) followed by somatostatinomas (30%) while other diagnoses, such as gangliocytic paragangliomas and serotonin-producing tumours are rare.
Diagnostic procedures
Histological examination is crucial to distinguish between the different types of duodenal NETs. S-gastrin and p-CgA should be measured. If indicated by symptoms, measurement of U-5HIAA and p-calcitonin should also be considered. Pancreatico-duodenal CT/MRI, EUS, and SRI are recommended for staging.
Treatment
Endoscopic resection may be possible in selected NETs < 1 cm. Otherwise, local resection(s), pancreaticoduodenectomy or pancreatic sparing duodenectomy with reimplantation of the ampulla of Vater may be performed. Intraoperative US of the liver and pancreas is mandatory. Surgical treatment of gastrinomas in patients with MEN-1 is controversial, since these tumours are generally multiple and rarely metastasise to the liver. If operated, duodenotomy with a thorough search for multiple tumours in the entire duodenum and pancreas is preferred and intraoperative endoscopy or EUS may be performed if required.
Unless radically resected, patients with gastrinomas should be treated with PPI, often in high doses, since even very small duodenal gastrinomas may give profound acid-related symptoms. Other functional NETs may benefit of SSA treatment. Metastatic disease should be treated as P-NETs.
Follow-up and prognosis
Measurement of p-CgA, and tumour specific hormones, endoscopy, EUS and CT/MRI should be done every 3–12 months dependent on the malignant potential of the tumour. The prognosis is good with a median survival of > 100 months for local and regionally metastasised tumours. For tumours with distant metastases the five-year survival is around 60% [Citation1].
Pancreatic NETs
Clinical presentation
P-NETs account for 1–2% of all pancreatic neoplasms. Apart from insulinomas, where less than 10% are malignant, P-NETs have a significant malignant potential. Non-functioning tumours constitute approximately 65% of P-NETs and may present with symptoms similar to pancreatic adenocarcinomas. However, in many cases the patient may be remarkably devoid of symptoms. The remaining 35% are functioning tumours, causing a specific syndrome of which 50% produce insulin, 40% gastrin, and the rest produce a wide variety of hormones, such as glucagon, somatostatin and VIP. Even small tumours of a few mm may give rise to fulminant symptoms. Metastases are usually seen in the liver and regional lymph nodes.
Diagnostic procedures
Expression of specific hormones may be tested on tissue sections by immunohistochemistry (IHC). Ki67 is generally 5–15% [Citation49]. Measurement of p-CgA, s-insulin, s-C-peptide, s-proinsulin, s-gastrin, p-VIP, p-glucagon, s-calcitonin, s-pancreatic polypeptide and p-somatostatin should be considered. For the diagnosis of insulinoma a 72-hour fasting test is strongly recommended. For the diagnosis of gastrinoma, measurement of pH or basal and maximal gastric acid output is recommended to distinguish gastrinomas from secondary hypergastrinaemia.
Pancreatico-duodenal CT/MRI, EUS and SRI should be performed before the surgical decision is taken [Citation50]. Benign insulinomas are in contrast to the malignant ones often negative on SRI. If conventional imaging modalities are negative, angiography with arterial stimulation by calcium or secretin with venous sampling or PET with specific tracers may localise the tumour. Intra-operative US of the pancreas and liver is mandatory.
Treatment
Surgery. Since 90% of the insulinomas are benign, enucleation or resection is feasible and can often be performed laparoscopically.
For other P-NETs a pancreaticoduodenectomy or distal/left pancreatectomy, including a splenectomy may be combined with regional lymph node dissection, hepatic surgery and/or radiofrequency ablation of liver metastases. Total pancreatectomy may be performed to achieve radical resection of NETs infiltrating major parts of the pancreas or in the case of multiple tumours in the gland. Involvement of the portal and mesenteric veins or adjacent organs should not be a contraindication for surgery. If radical surgery is not possible, debulking procedures may be considered in order to reduce severe endocrine symptoms.
Pancreatic neuroendocrine incidentalomas less than 2 cm in size usually have no or minimal growth for many years and may therefore be managed non-operatively with close surveillance [Citation51].
Surgery is recommended for patients with MEN-1 with functional NETs and non-functional NETs with a size > 2 cm or if the tumours show progression.
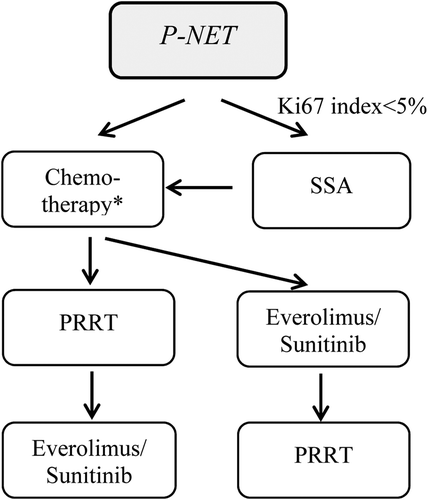
Systemic treatment. Palliative chemotherapy with STZ + 5FU or temozolomide+ capecitabine is used as first-line treatment for metastatic or locally advanced disease when Ki67 is < 20%. In patients with Ki67 < 10% and high SSTR expression, SSA treatment may be considered [Citation37]. Everolimus and sunitinib are often used as second line treatment. PRRT should be considered in patients with tumours with high SSTR expression, usually as second or third line treatment. Data on the use of IFN in P-NET is limited. See a suggested algorithm for systemic treatment of P-NETs in .
Figure 3. Treatment algorithm for the systemic treatment of pancreatic NETs. *indicates treatment regimens with either streptozotocin + 5-fluourouracil or temozolomide +/2 capecitabine. Debulking treatment, such as surgery, radiofrequency ablation and liver embolisation are not included in the algorithm.
Symptomatic treatment. In patients with gastrinomas, PPIs can control the symptoms of increased acid secretion. For patients with insulinomas frequent small meals may temporarily reduce the hypoglycaemic attacks. Everolimus has proven to be very effective in treating hypoglycaemia by inhibiting insulin secretion and increasing insulin resistance in patients with malignant tumours [Citation52]. Diazoxide, SSA and continuous glucose infusion are other options which may be tried. In some cases with severe and frequent attacks of hypoglycaemia constant infusion of glucagon may be necessary. Glucocorticoids are not recommended unless other treatments have failed, since the secretion of endogenous corticoids from the adrenals is suppressed and the protective counter-regulatory mechanism is blocked in case of hypoglycaemic attacks. SSAs are efficient in reducing VIP-induced diarrhoea and skin lesions in glucagonomas.
Follow-up and prognosis
Patients with radically resected benign insulinomas should have a clinical and biochemical follow-up but do not need further controls. The prognosis is excellent and similar to that of the normal population. Other radically operated patients should be followed with biochemical markers and CT/MRI every 6–12 months for at least 10 years.
Patients with residual disease should be monitored closely during treatment, initially every three months, with biochemical markers, CT/MRI and when indicated by SRI. If the patient reports new endocrine symptoms, relevant pancreatic hormones and tumour markers should be measured, since the tumours may switch profile and consequently the patient may need a change of treatment. The median overall survival in a large cohort of 324 patients was 99 months [Citation53]. The five- and 10-year survival was 64% and 44%. For patients with gastrinoma the five-year survival is close to 100% in the absence of liver metastases and 50% when liver metastases are present. For patients with malignant insulinomas the median survival is only two years.
Small intestinal NETs (classical carcinoid tumours)
Clinical presentation
Approximately 25–30% of GEP-NENs arise in the small bowel, mainly in the distal part of the ileum. There may be multiple tumours in the intestinal wall. Most primary tumours are 1–2 cm in diameter and approximately 60% are metastatic at diagnosis. Metastases are usually located in the mesenteric and para-aortal lymph nodes and in the liver, whereas peritoneal spread is less common.
The primary tumour, mesenteric lymph node metastases and tumour-induced fibrosis may induce kinking of the bowel, partial bowel obstruction and vascular encasement, which may cause abdominal pain, diarrhoea and weight loss. Tumours are often found incidentally during operation for another abdominal malignancy, in particular colorectal cancer. The hormone-induced carcinoid syndrome with flushing and diarrhoea is seen in the presence of liver metastases or large retroperitoneal masses draining into the caval vein, and is present in less than 30% of the patients at diagnosis. Right-sided heart failure due to tricuspid valve insufficiency and pulmonary valve stenosis (carcinoid heart disease) provoked by serotonin-induced fibrosis is seen in 10–30% of patients.
Hereditary SI-NETs
SI-NETs are usually sporadic tumours but a Swedish series of 10 families with hereditary SI-NETs has been published [Citation54]. The genetic changes behind the development of SI-NETs are still unknown.
Diagnostic procedures
The growth pattern is typically insular, the tumours stain positive for serotonin and usually have a Ki67 index of 1–5%. P-CgA and U-5HIAA should be monitored. Measurement of s-Pro-BNP may be useful to detect and follow carcinoid heart disease. Staging with SRI and diagnostic CT-can should always be performed, which also provides information about possible metastatic encasement of the superior mesenteric vessels for preoperative planning. Echocardiography should be performed initially in all patients with metastatic disease to evaluate possible carcinoid heart disease, and should be repeated at least yearly in those with proven abnormalities.
Treatment
Surgery. The primary tumour should be resected together with any central mesenteric lymph node(s). Intra-operative palpation of the entire small intestine should be performed to identify additional tumours. However, in case of acute surgery extensive central mesenteric dissection should be avoided due the high risk of damage to the mesenteric vessels resulting in short bowel syndrome. Intestinal bypass should be avoided as it may complicate further surgery.
In patients with limited metastatic disease R0 resection should be attempted (resection of the primary tumour, lymph node metastases and the few liver metastases). In patients with disseminated disease the primary tumour and regional metastases should be resected if the patient has symptoms of imminent bowel obstruction or ischemia [Citation55]. In patients with disseminated disease without symptoms, resection of the primary and regional metastases is controversial, as there is no evidence that resection of the primary will prolong survival [Citation55].
Surgery of liver metastases should always be considered, also for debulking to reduce endocrine symptoms. If surgery is not possible, local ablation in cases with few liver metastases and tumour size < 3–5 cm may be considered.
In cases of manifest carcinoid heart disease, valve replacement using biological valves should be considered to improve cardiac function and should be performed before abdominal surgery to reduce the blood pressure in the hepatic veins.
Systemic treatment. SSA is the primary treatment for most patients with residual disease, regardless of the presence of endocrine symptoms [Citation36,Citation37]. The symptomatic effect of SSA may be achieved also in patients with a negative SRI.
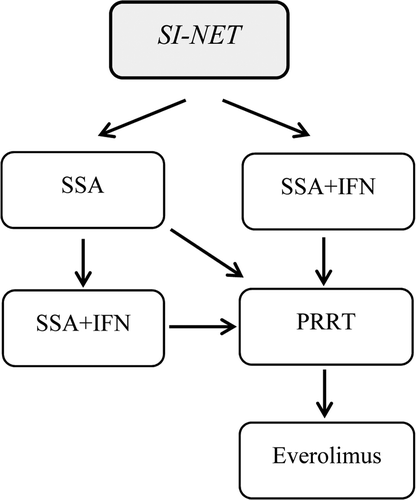
IFN treatment alone or combined with SSA may be used in low proliferating tumours [Citation56–58]. PRRT should be considered in patients who progress on SSA and IFN therapy. Everolimus may be considered in SI-NETs progressing on standard therapy, although definite evidence of benefit is lacking [Citation45]. Chemotherapy is generally not recommended in low proliferating SI-NET. See a suggested algorithm for systemic treatment of SI-NETs in .
Figure 4. Treatment algorithm for the systemic treatment of small intestinal NETs. Debulking treatment, such as surgery, radiofrequency ablation and liver embolisation are not included in the algorithm.
Follow-up and prognosis
After radical surgery, follow up is recommended every 3–12 months for at least 10 years with measurements of p-CgA and imaging. Recurrence is seen in up to 75% after 15 years [Citation28]. A rise in p-CgA is often the first evidence of recurrence [Citation9]. Patients with disseminated disease should be examined every 3–12 months with measurements of p-CgA, U-5HIAA and CT/MRI. SRI should be performed when whole body imaging is required or PRRT considered. Five-year survival for localised or regional disease is 75%. Median survival is > 100 months for patients with local and regional disease and 60 months for distant disease [Citation8].
Appendix NETs
Clinical presentation
These tumours are almost always found by the pathologist in the tissue specimen after appendectomy for acute appendicitis. More than 60% are < 1 cm and belong to the WHO G1 class of tumours.
Diagnostic procedures
Most appendix NETs (85%) are morphologically, and by IHC, similar to SI-NETs, being positive for serotonin. Less than 5% are small benign tubular carcinoids, which are negative for serotonin but positive for glucagon. Biochemical markers (P-CgA and U-5HIAA) are usually normal postoperatively. Follow-up by CT/MRI and SRI are only recommended after surgery if unclear resection margins or in tumours > 2 cm.
Treatment
Appendectomy is sufficient treatment in > 90% of cases and right-sided hemicolectomy (RSH) is very rarely indicated if the tumour is < 1 cm. If the tumour is > 2 cm, RSH with lymph node dissection should be performed. However, it has recently been suggested that all patients with tumours > 1 cm should be offered a RSH [Citation59]. Irrespective of size, a RSH should be performed if the tumour involves resection margins, has a location at the base of the appendix, infiltrates into the mesentery, show vascular invasion or spread to regional lymph nodes. In tumours with a Ki67 index > 10%, RSH should be performed.
In metastatic disease, medical treatment is similar to that for SI-NETs.
Follow-up and prognosis
In patients where appendectomy is considered sufficient p-CgA may be measured after 3–6 months. If normal no further follow-up is required. The five-year survival is close to 100%. However, follow-up is required in patients who have undergone hemicolectomy because of high-risk features, and should be identical to that of SI-NETs. For these patients the five-year survival is < 70%.
Goblet cell carcinoids
Clinical presentation
Approximately 10% of appendiceal NENs are goblet cell carcinoids. In localised tumours the presentation is similar to that of other appendiceal carcinoids. In disseminated cases, the disease may induce diffuse peritoneal carcinomatosis and ovarian metastases with ascites, but liver metastases are rare.
Diagnostic procedures
P-CgA and U-5HIAA are normal and should not be used in follow-up as a routine. S-CEA, s-CA19-9 and s-CA125 may be elevated and can serve as tumour markers. All patients should have CT/ MRI for staging. SRI is usually negative but FDG-PET/CT may be positive.
Treatment
Localised disease should be treated with RSH with lymph node dissection. Hystero- salpingo-oophorectomy should not be performed unless ovarian metastases are present. Peritoneal metastatic disease may be treated aggressively, including peritonectomy, heat-induced peritoneal chemotherapy (HIPEC) and systemic chemotherapy. Since there are no RCTs, use of chemotherapy usually follows colon cancer guidelines.
Follow-up and prognosis
CT/MRI should be performed as for colon cancer. Measurements of tumour markers may be useful for localised and regional disease. The five-year survival is 80%, which drops to below 20% in disseminated tumours [Citation60].
Colon NETs
Clinical presentation
Most colon NENs originating in the coecum, are G1/G2 tumours and resemble SI-NETs. Tumours in the remaining part of the colon are usually NECs. NENs in the colon comprise < 1% of colonic cancers.
Diagnostic procedures
CgA should be measured in all patients while U-5HIAA should be measured in NETs originating in the right colon. Colonoscopy with biopsy is mandatory. CT of the thorax and abdomen and SRI are performed for staging of regional and distant metastases.
Treatment
Surgical treatment includes bowel resection with lymph node dissection. The medical treatment for advanced disease depends on the Ki67 index of the tumour, SSA with or without IFN should be considered if the Ki67 index is below 10% and chemotherapy when Ki67 index is above 10%.
Follow-up and prognosis
For G1 and G2 NETs, see SI-NETs. For G3 NECs see below.
Rectal NETs
Clinical presentation
Rectal NETs comprise approximately 20% of GEP-NENs and are usually small, benign polyps that are found incidentally at routine sigmoideoscopy. Larger NET polyps may cause rectal bleeding. Hormone-related symptoms are never seen.
Diagnostic procedures
Tumours show a trabecular growth pattern with synaptophysin, CgA and glucagon positivity and serotonin negative tumour cells. P-CgA and U-5HIAA are usually normal. Endoscopy with biopsies/resection is recommended. In tumours > 2 cm, rectal US or MRI is recommended to visualise the extent of the primary tumour and regional lymph node metastases. CT thorax-abdomen and SRI for additional staging are also recommended.
Treatment
Rectal NET polyps < 1 cm can be radically resected by endoscopy. Tumours between 1 and 2 cm are treated with endoscopic submucosal dissection or transendoscopic mucosectomy (TEM) if radical excision can be assured. Tumours > 2 cm may be resected by TEM or by a low anterior or abdomino-peritoneal resection depending on size, Ki67 index, muscular invasion and presence of regional lymph node metastases.
Patients with metastatic disease with a positive SRI may respond well to PRRT. SSA and IFN may stabilise tumours with low proliferation index and chemotherapy can be used in more aggressive tumours.
Follow-up and prognosis
Patients with radically resected G1 polyps < 2 cm do not need further surveillance except for one endoscopic follow-up within the first year. Those with radically resected larger or higher grade lesions, or lymph node metastases should be followed by endoscopy, transrectal US and CT/MRI at least annually for at least 10 years. Five-year survival of patients with localised disease is > 90%, regional disease 50% and distant disease 30–40%.
Management of neuroendocrine liver metastases
Single or few hepatic metastases may be resected with curative intent, which is possible in < 10%, as most NEN liver metastases are multiple at diagnosis. Palliative debulking surgery and/or RF/microwave ablation is indicated to minimise severe hormone or local symptoms, but the impact on survival is uncertain [Citation61,Citation62]. However, RF/microwave ablation should not be performed if surgical resection is possible.
When surgery or RF/microwave ablation is impossible and the majority of the tumour burden is confined to the liver, liver metastases may be treated by embolisation of the hepatic artery or its branches. Beneficial effects on hormone-induced symptoms are seen in 50–90%, with duration of 14–17 months. Reduction in tumour size is seen in 55% of the patients with duration of 10–24 months, and median survival of 50 months. In a randomised study there was no significant difference in two-year PFS between bland- and chemoembolisation [Citation63] and for SI-NET bland embolisation may be superior to chemoembolisation [Citation64].
Radio-embolisation with intrahepatic infusion of small resin or 90Y coated glass particles has shown promising results. However, only single-centre studies without comparison to bland- or chemoembolisation have been performed [Citation65,Citation66]. Orthotopic liver transplantation is rarely used, but may be considered in carefully selected young patients without extrahepatic disease, severe uncontrollable endocrine symptoms and a Ki67 index < 10%. Approximately 20% are recurrence free after five years and the five-year survival rate may be as high as 90% [Citation67].
Gastroenteropancreatic neuroendocrine carcinoma (GEP-NEC)
Clinical presentation
GEP-NECs account for approximately 10–20% of GEP-NEN. The primary site is usually in the oesophagus, stomach, pancreas or colon [Citation68] with metastatic disease at diagnosis in 50–70% [Citation4]. Symptoms are related to the organ from which the carcinoma develops as well as from metastatic sites. Weight loss is frequent and hormone- induced endocrine symptoms are rare.
Diagnostic procedures
GEP-NECs are characterised by necrosis, abundant mitoses and positive immunohistochemical staining for synaptophysin and less frequent CgA [Citation68]. Ki67 index is by definition > 20%, but usually > 50%. If the neuroendocrine part in a tumour with both adenocarcinoma and neuroendocrine components is < 30%, the tumour is classified as an adenocarcinoma with neuroendocrine features and these patients should be treated as patients with adenocarcinomas. If both the carcinoma (usually adenocarcinoma) and the neuroendocrine component exceed 30% of the tumour, it is classified as mixed adenoneuroendocrine carcinoma (MANEC). Tumours with an adenocarcinoma component of < 30% are depicted as NECs. P-CgA may be elevated. CT of the thorax-abdomen should be performed for staging. FDG-PET is usually positive and should be performed before curatively intended treatment of localised disease [Citation69]. The relevance of SRI in GEP-NEC patients is uncertain [Citation68,Citation69]. Common metastatic sites are liver, lymph nodes, lung and bone. CT/MRI of the brain should be performed if there are signs of cerebral involvement.
Treatment
The treatment approach for localised disease is at present neither consistent nor uniform. If radical resection of the primary tumour and metastases is possible surgery may be considered as first line treatment. However, surgery alone is rarely curative, even for apparently localised disease [Citation70]. Surgery for distant metastases or debulking surgery is generally not recommended.
Adjuvant treatment. The aggressive behaviour of GEP-NECs warrants consideration of adjuvant therapy after surgery. Therefore, although definite data are missing, 4–6 cycles of cis/carboplatin and etoposide should be strongly considered [Citation70,Citation71]. Prophylactic cerebral irradiation is not recommended.
Palliative treatment. For patients with metastatic GEP-NECs rapid consideration of chemotherapy is essential. Median survival is only one month for patients who are not given chemotherapy [Citation68]. In a large, retrospective Nordic study, treatment with cis/carboplatin and etoposide showed a RR of 31%, PFS of four months and median survival of 11 months with similar effect of cis- and carboplatin-based regimes [Citation68]. Tumours with a Ki67 index < 55% were less responsive to platinum-based chemotherapy, but the patients had a significantly longer survival compared to those with higher Ki67 levels. Consequently, patients with a Ki67 index of > 55% should be treated with platinum-based chemotherapy while patients with a Ki67 index < 55%, could be considered for other regimens, such as temozolomide-based therapy.
There is no established second-line therapy for GEP-NEC. Temozolomide-based therapy given after platinum-based chemotherapy resulted in a RR of 33% and PFS of six months [Citation72] and it is frequently used. Irinotecan and 5FU (FOLFIRI) given as second-line treatment resulted in a RR of 31% and PFS of four months [Citation73]. PRRT may be an option for patients with a high uptake on SRI and a Ki67 index in the lower range [Citation44]. The optimal treatment of MANEC is controversial.
Follow-up and prognosis
Radically operated patients should be monitored with CT of thorax/ abdomen every three months initially as patients often have a rapid recurrence. If there is no recurrence within five years the patient can be considered as cured. Median survival was 34 months for patients with localised disease, 14 months with regional disease and five months with distant disease [Citation1]. Good PS, pancreatic primary, normal serum LDH, normal platelet count, large cell morphology, presence of both neuroendocrine immunohistochemical markers, and Ki67 index < 55% are favourable prognostic parameters [Citation4,Citation68,Citation72,Citation74].
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
This work was supported by an unrestricted grant from Novartis Oncology and Ipsen AB.
References
- Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after ‘carcinoid’: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008;26:3063–72.
- Lepage C, Bouvier AM, Faivre J. Endocrine tumours: Epidemiology of malignant digestive neuroendocrine tumours. Eur J Endocrinol 2013;168:R77–83.
- Lawrence B, Gustafsson BI, Chan A, Svejda B, Kidd M, Modlin IM. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am 2011;40:1–18, vii.
- Korse CM, Taal BG, van Velthuysen ML, Visser O. Incidence and survival of neuroendocrine tumours in the Netherlands according to histological grade: Experience of two decades of cancer registry. Eur J Cancer Care 2013; 49:1975–83.
- van der Zwan JM, Trama A, Otter R, Larranaga N, Tavilla A, Marcos-Gragera R, et al. Rare neuroendocrine tumours: Results of the surveillance of rare cancers in Europe project. Eur J Cancer Care 2013;49:2565–78.
- Mocellin S, Nitti D. Gastrointestinal carcinoid: Epidemiological and survival evidence from a large population-based study (n = 25 531). Ann Oncol 2013;24:3040–4.
- Kanakis G, Kaltsas G. Biochemical markers for gastroenteropancreatic neuroendocrine tumours (GEP-NETs). Best Pract Res Clin Gastroenterol 2012;26:791–802.
- Janson ET, Holmberg L, Stridsberg M, Eriksson B, Theodorsson E, Wilander E, et al. Carcinoid tumors: Analysis of prognostic factors and survival in 301 patients from a referral center. Ann Oncol 1997;8:685–90.
- Welin S, Stridsberg M, Cunningham J, Granberg D, Skogseid B, Oberg K, et al. Elevated plasma chromogranin A is the first indication of recurrence in radically operated midgut carcinoid tumors. Neuroendocrinology 2009;89:302–7.
- Jensen KH, Hilsted L, Jensen C, Mynster T, Rehfeld JF, Knigge U. Chromogranin A is a sensitive marker of progression or regression in ileo-cecal neuroendocrine tumors. Scand J Gastroenterol 2013;48:70–7.
- Tropea F, Baldari S, Restifo G, Fiorillo MT, Surace P, Herberg A. Evaluation of chromogranin A expression in patients with non-neuroendocrine tumours. Clin Drug Investig 2006;26:715–22.
- Stridsberg M, Eriksson B, Oberg K, Janson ET. A comparison between three commercial kits for chromogranin A measurements. J Endocrinol 2003;177:337–41.
- Gedde-Dahl M, Thiis-Evensen E, Tjolsen AM, Mordal KS, Vatn M, Bergestuen DS. Comparison of 24-h and overnight samples of urinary 5-hydroxyindoleacetic acid in patients with intestinal neuroendocrine tumors. Endocr Connect 2013;2:50–4.
- Tohmola N, Itkonen O, Sane T, Markkanen H, Joenvaara S, Renkonen R, et al. Analytical and preanalytical validation of a new mass spectrometric serum 5-hydroxyindoleacetic acid assay as neuroendocrine tumor marker. Clin Chim Acta 2014;428:38–43.
- Yao JC, Pavel M, Phan AT, Kulke MH, Hoosen S, St Peter J, et al. Chromogranin A and neuron-specific enolase as prognostic markers in patients with advanced pNET treated with everolimus. J Clin Endocrinol Metab 2011;96:3741–9.
- Elias D, Lefevre JH, Duvillard P, Goere D, Dromain C, Dumont F, et al. Hepatic metastases from neuroendocrine tumors with a ‘thin slice’ pathological examination: They are many more than you think. Ann Surg 2010;251:307–10.
- Sundin A. Radiological and nuclear medicine imaging of gastroenteropancreatic neuroendocrine tumours. Best Pract Res Clin Gastroenterol 2012;26:803–18.
- Brenner R, Metens T, Bali M, Demetter P, Matos C. Pancreatic neuroendocrine tumor: Added value of fusion of T2-weighted imaging and high b-value diffusion-weighted imaging for tumor detection. Eur Radiol 2012;81:e746–9.
- Schraml C, Schwenzer NF, Sperling O, Aschoff P, Lichy MP, Muller M, et al. Staging of neuroendocrine tumours: Comparison of [(6)(8)Ga]DOTATOC multiphase PET/CT and whole-body MRI. Cancer Imaging 2013;13:63–72.
- Massironi S, Conte D, Sciola V, Pirola L, Paggi S, Fraquelli M, et al. Contrast-enhanced ultrasonography in evaluating hepatic metastases from neuroendocrine tumours. Dig Liver Dis 2010;42:635–41.
- Atiq M, Bhutani MS, Bektas M, Lee JE, Gong Y, Tamm EP, et al. EUS-FNA for pancreatic neuroendocrine tumors: A tertiary cancer center experience. Dig Dis Sci 2012;57: 791–800.
- Sundin A, Rockall A. Therapeutic monitoring of gastroenteropancreatic neuroendocrine tumors: The challenges ahead. Neuroendocrinology 2012;96:261–71.
- Velikyan I, Sundin A, Eriksson B, Lundqvist H, Sorensen J, Bergstrom M, et al. In vivo binding of [68Ga]-DOTATOC to somatostatin receptors in neuroendocrine tumours – impact of peptide mass. Nucl Med Biol 2010;37:265–75.
- Pfeifer A, Knigge U, Mortensen J, Oturai P, Berthelsen AK, Loft A, et al. Clinical PET of neuroendocrine tumors using 64Cu-DOTATATE: First-in-humans study. J Nucl Med 2012;53:1207–15.
- Sandstrom M, Velikyan I, Garske-Roman U, Sorensen J, Eriksson B, Granberg D, et al. Comparative biodistribution and radiation dosimetry of 68Ga-DOTATOC and 68Ga-DOTATATE in patients with neuroendocrine tumors. J Nucl Med 2013;54:1755–9.
- Garin E, Le Jeune F, Devillers A, Cuggia M, de Lajarte-Thirouard AS, Bouriel C, et al. Predictive value of 18F-FDG PET and somatostatin receptor scintigraphy in patients with metastatic endocrine tumors. J Nucl Med 2009;50:858–64.
- Binderup T, Knigge U, Loft A, Federspiel B, Kjaer A. 18F-fluorodeoxyglucose positron emission tomography predicts survival of patients with neuroendocrine tumors. Clin Cancer Res 2010;16:978–85.
- Norlen O, Stalberg P, Oberg K, Eriksson J, Hedberg J, Hessman O, et al. Long-term results of surgery for small intestinal neuroendocrine tumors at a tertiary referral center. World J Surg 2012;36:1419–31.
- Capurso G, Bettini R, Rinzivillo M, Boninsegna L, Delle Fave G, Falconi M. Role of resection of the primary pancreatic neuroendocrine tumour only in patients with unresectable metastatic liver disease: A systematic review. Neuroendocrinology 2011;93:223–9.
- Norlen O, Hessman O, Stalberg P, Akerstrom G, Hellman P. Prophylactic cholecystectomy in midgut carcinoid patients. World J Surg 2010;34:1361–7.
- Moertel CG, Hanley JA, Johnson LA. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med 1980;303:1189–94.
- Dahan L, Bonnetain F, Rougier P, Raoul JL, Gamelin E, Etienne PL, et al. Phase III trial of chemotherapy using 5-fluorouracil and streptozotocin compared with interferon alpha for advanced carcinoid tumors: FNCLCC-FFCD 9710. Endocr Relat Cancer 2009;16:1351–61.
- Strosberg JR, Fine RL, Choi J, Nasir A, Coppola D, Chen DT, et al. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer 2011;117:268–75.
- Kulke MH, Hornick JL, Frauenhoffer C, Hooshmand S, Ryan DP, Enzinger PC, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res 2009;15:338–45.
- Modlin IM, Pavel M, Kidd M, Gustafsson BI. Review article: Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther 2010;31:169–88.
- Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J Clin Oncol 2009;27:4656–63.
- Caplin M, Pavel M, Cwikla JB, Phan AT, Raderer M, Sedlackova E et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014;371: 224–33.
- Kvols LK, Oberg KE, O’Dorisio TM, Mohideen P, de Herder WW, Arnold R, et al. Pasireotide (SOM230) shows efficacy and tolerability in the treatment of patients with advanced neuroendocrine tumors refractory or resistant to octreotide LAR: Results from a phase II study. Endocr Relat Cancer 2012;19:657–66.
- Oberg K. Biotherapies for GEP-NETs. Best Pract Res Clin Gastroenterol 2012;26:833–41.
- Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: Toxicity, efficacy, and survival. J Clin Oncol 2008; 26:2124–30.
- Imhof A, Brunner P, Marincek N, Briel M, Schindler C, Rasch H, et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol 2011;29:2416–23.
- Villard L, Romer A, Marincek N, Brunner P, Koller MT, Schindler C, et al. Cohort study of somatostatin-based radiopeptide therapy with [(90)Y-DOTA]-TOC versus [(90)Y-DOTA]-TOC plus [(177)Lu-DOTA]-TOC in neuroendocrine cancers. J Clin Oncol 2012;30:1100–6.
- van Essen M, Krenning EP, Kam BL, de Herder WW, Feelders RA, Kwekkeboom DJ. Salvage therapy with (177)Lu-octreotate in patients with bronchial and gastroenteropancreatic neuroendocrine tumors. J Nucl Med 2010;51:383–90.
- Ezziddin S, Opitz M, Attassi M, Biermann K, Sabet A, Guhlke S, et al. Impact of the Ki-67 proliferation index on response to peptide receptor radionuclide therapy. Eur J Nucl Med Mol 2011;38:459–66.
- Pavel ME, Hainsworth JD, Baudin E, Peeters M, Horsch D, Winkler RE, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011;378:2005–12.
- Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011;364:514–23.
- Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011;364:501–13.
- Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 2008;113:1807–43.
- Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): Incidence, prognosis and recent trend toward improved survival. Ann Oncol 2008;19:1727–33.
- Anderson MA, Carpenter S, Thompson NW, Nostrant TT, Elta GH, Scheiman JM. Endoscopic ultrasound is highly accurate and directs management in patients with neuroendocrine tumors of the pancreas. Am J Gastroenterol 2000;95:2271–7.
- Birnbaum DJ, Gaujoux S, Cherif R, Dokmak S, Fuks D, Couvelard A, et al. Sporadic nonfunctioning pancreatic neuroendocrine tumors: Prognostic significance of incidental diagnosis. Surgery 2014;155:13–21.
- Kulke MH, Bergsland EK, Yao JC. Glycemic control in patients with insulinoma treated with everolimus. N Engl J Med 2009;360:195–7.
- Ekeblad S, Sundin A, Janson ET, Welin S, Granberg D, Kindmark H, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res 2007;13:2986–91.
- Cunningham JL, Diaz de Stahl T, Sjoblom T, Westin G, Dumanski JP, Janson ET. Common pathogenetic mechanism involving human chromosome 18 in familial and sporadic ileal carcinoid tumors. Genes Chromosomes Cancer 2011;50:82–94.
- Capurso G, Rinzivillo M, Bettini R, Boninsegna L, Delle Fave G, Falconi M. Systematic review of resection of primary midgut carcinoid tumour in patients with unresectable liver metastases. Br J Surg 2012;99:1480–6.
- Kolby L, Persson G, Franzen S, Ahren B. Randomized clinical trial of the effect of interferon alpha on survival in patients with disseminated midgut carcinoid tumours. Br J Surg 2003;90:687–93.
- Arnold R, Rinke A, Klose KJ, Muller HH, Wied M, Zamzow K, et al. Octreotide versus octreotide plus interferon-alpha in endocrine gastroenteropancreatic tumors: A randomized trial. Clin Gastroenterol Hepatol 2005;3: 761–71.
- Faiss S, Pape UF, Bohmig M, Dorffel Y, Mansmann U, Golder W, et al. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors – the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol 2003;21:2689–96.
- Grozinsky-Glasberg S, Alexandraki KI, Barak D, Doviner V, Reissman P, Kaltsas GA, et al. Current size criteria for the management of neuroendocrine tumors of the appendix: Are they valid? Clinical experience and review of the literature. Neuroendocrinology 2013;98:31–7.
- McGory ML, Maggard MA, Kang H, O’Connell JB, Ko CY. Malignancies of the appendix: Beyond case series reports. Dis Colon Rectum 2005;48:2264–71.
- Eriksson J, Stalberg P, Nilsson A, Krause J, Lundberg C, Skogseid B, et al. Surgery and radiofrequency ablation for treatment of liver metastases from midgut and foregut carcinoids and endocrine pancreatic tumors. World J Surgery 2008;32:930–8.
- Knigge U, Hansen CP. Surgery for GEP-NETs. Best Pract Res Clin Gastroenterol 2012;26:819–31.
- Maire F, Lombard-Bohas C, O’Toole D, Vullierme MP, Rebours V, Couvelard A, et al. Hepatic arterial embolization versus chemoembolization in the treatment of liver metastases from well-differentiated midgut endocrine tumors: A prospective randomized study. Neuroendocrinology 2012; 96:294–300.
- Gupta S, Johnson MM, Murthy R, Ahrar K, Wallace MJ, Madoff DC, et al. Hepatic arterial embolization and chemoembolization for the treatment of patients with metastatic neuroendocrine tumors: Variables affecting response rates and survival. Cancer 2005;104:1590–602.
- Kalinowski M, Dressler M, Konig A, El-Sheik M, Rinke A, Hoffken H, et al. Selective internal radiotherapy with Yttrium-90 microspheres for hepatic metastatic neuroendocrine tumors: A prospective single center study. Digestion 2009;79:137–42.
- Paprottka PM, Hoffmann RT, Haug A, Sommer WH, Raessler F, Trumm CG, et al. Radioembolization of symptomatic, unresectable neuroendocrine hepatic metastases using yttrium-90 microspheres. J Vasc Interv Radiol 2012;35:334–42.
- Le Treut YP, Gregoire E, Klempnauer J, Belghiti J, Jouve E, Lerut J, et al. Liver transplantation for neuroendocrine tumors in Europe – results and trends in patient selection: A 213-case European liver transplant registry study. Ann Surg 2013;257:807–15.
- Sorbye H, Welin S, Langer SW, Vestermark LW, Holt N, Osterlund P, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann Oncol 2013;24: 152–60.
- Binderup T, Knigge U, Loft A, Mortensen J, Pfeifer A, Federspiel B, et al. Functional imaging of neuroendocrine tumors: A head-to-head comparison of somatostatin receptor scintigraphy, 123I-MIBG scintigraphy, and 18F-FDG PET. J Nucl Med 2010;51:704–12.
- Brenner B, Shah MA, Gonen M, Klimstra DS, Shia J, Kelsen DP. Small-cell carcinoma of the gastrointestinal tract: A retrospective study of 64 cases. Br J Cancer 2004;90:1720–6.
- Ku GY, Minsky BD, Rusch VW, Bains M, Kelsen DP, Ilson DH. Small-cell carcinoma of the esophagus and gastroesophageal junction: Review of the Memorial Sloan-Kettering experience. Ann Oncol 2008;19:533–7.
- Welin S, Sorbye H, Sebjornsen S, Knappskog S, Busch C, Oberg K. Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer 2011;117:4617–22.
- Hentic O, Hammel P, Couvelard A, Rebours V, Zappa M, Palazzo M, et al. FOLFIRI regimen: An effective second-line chemotherapy after failure of etoposide-platinum combination in patients with neuroendocrine carcinomas grade 3. Endocr Relat Cancer 2012;19:751–7.
- Strosberg JR, Coppola D, Klimstra DS, Phan AT, Kulke MH, Wiseman GA, et al. The NANETS consensus guidelines for the diagnosis and management of poorly differentiated (high-grade) extrapulmonary neuroendocrine carcinomas. Pancreas 2010;39:799–800.