Abstract
Objective. This study investigated the effects of short-term intermittent hypoxia (IH) preconditioning on cardiac structure and function in rats and the influence of ischemia reperfusion (I/R) injury. Special attention was then paid to the involvement of hypoxia-inducible factor-1α (HIF-1α) and vascular endothelial growth factor (VEGF).
Methods. Wistar rats were given IH treatment for 1, 7, 14, or 28 days. Some of them were thereafter subject to myocardial infarction surgery. Right ventricle systolic pressure (RVSP), myocardial capillary density (CD), and mRNA/protein expression of HIF-1α, VEGF, and Bcl-2 in rat myocardial tissue were determined. Apoptotic cell number was determined by TUNEL staining, and concentrations of malondialdehyde (MDA) and superoxide dismutase (SOD) were measured.
Results. IH treatment for 1, 7, 14, and 28 days reduced the myocardial infarction size, whereas IH for 28 days increased the RVSP, ratio of right to left ventricle weight (RV/LV+S), and CD. IH up-regulated the mRNA and protein levels of HIF-1α, VEGF, and Bcl-2 both under normal and I/R conditions. The induced expression of HIF-1α and VEGF by IH reached a peak after 7 days of treatment. Moreover, IH for 28 days induced cardiomyocyte apoptosis, whereas prior treatment with IH for 1, 7, 14, and 28 days all markedly attenuated the apoptosis effected by the subsequent I/R injury. IH also decreased the concentrations of MDA but increased those of SOD in myocardial tissue of both in normal rats and following I/R.
Conclusions. The present study demonstrates that short-term IH protects the heart from I/R injury through inhibiting apoptosis and oxidative stress. The up-regulation of HIF-1α and VEGF by short-term IH may participate in the cardioprotective effect of IH.
Introduction
Acute hypoxia is characterized by pronounced and selective redistribution of the blood-flow that ensures adequate O2 supply to vital organs. The pattern of regional blood-flow distribution later returns towards that of normoxia, and organ O2 supply is maintained via an increase in blood hemoglobin concentration secondary to chronic hypoxia (Citation1). Meanwhile, vital organs develop resistance to ischemic stress. Since the first report about beneficial effects of preconditioning on ischemia/reperfusion (I/R) injuries by Murry et al. (Citation2), the underlying mechanisms of preconditioning have been studied extensively. In fact, this principle has been applied in the development of therapeutic strategies for prevention and treatment of ischemia in brain (Citation3,4), heart (Citation5,6), and other organs (Citation7,8). While scientific and practical reasons may explain these findings, the unpredictability of acute ischemic syndromes remains a major obstacle to the timely application of novel treatments. Thus, there is a need for more effective and simple methods of preconditioning.
Intermittent hypoxia (IH) conditioning has served as a non-invasive, drug-free technique aiming to improve physiological performance and well-being using the phenomena of adaptation to reduced oxygen (Citation9). Some previous reports have indicated that IH conditioning results in protective effects in brain (Citation10,11), heart (Citation12,13), and other organs (Citation14). In contrast, there is evidence to suggest that there are harmful effects of IH in obstructive sleep apnea (OSA). Studies of IH in OSA have been characterized by brief, recurrent cycles of hypoxia–reoxygenation, typically less than 60 s in duration (Citation10,15,16), whereas IH conditioning programs, which have proven effective, generally use hypoxia periods of several minutes–hours (Citation12,13). The differences of intensity, frequency, and duration of IH cycles are thought to be the major determinants of the effects (Citation17). Our hypothesis is that IH preconditioning with less than 60 s in duration, commonly used in studies of OSA, also may be protective while this intervention is limited by short-term administration.
To test this hypothesis, we determined the effects of short-term IH on myocardial infarction size in rats and investigated changes in hypoxia-inducible factor-1α (HIF-1α) and vascular endothelial growth factor (VEGF) expression after IH and IH + I/R. Moreover, we determined apoptotic cell numbers, expression of the anti-apoptosis protein Bcl-2, and the oxidative stress markers malondialdehyde (MDA) and superoxide dismutase (SOD) after IH and IH + I/R in rat myocardial tissue.
Materials and methods
Animals
Male Wistar rats (250 ± 30 g) were supplied by the Animal Center of The Third Military Medical University. They were housed under controlled temperature (23–25°C) and lighting (08.00–20.00) conditions and with free access to tap water and rat chow. All procedures were performed in accordance with institutional guidelines for animal care and the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996). Three groups of animals were studied: 1) untreated controls that remained in their usual cages throughout the 28-day period (normoxia control, n = 14); 2) animals that underwent 1, 7, 14, and 28 days of IH exposure as described below (n = 7 at each time point); 3) animals that underwent 1, 7, 14, and 28 days of IH exposure plus a myocardial ischemia/reperfusion intervention (n = 7 at each time point).
IH protocol
IH exposure was performed as previously described (Citation18,19). Briefly, during the daily 4-h exposure protocol, both experimental and sham control animals were housed in identical cylindrical plastic chambers (length 25 cm, diameter 15 cm, height 15 cm) placed side by side. Animals were unrestrained and freely mobile within the exposure chambers. Continuous measurement of chamber fractional O2 (FcO2) concentration was performed using an O2 analyzer. With the use of timed solenoid valves, the exposure chamber was flushed with 90% N2 + 10% O2 for 30 s of each minute. Gas flows were adjusted so that the chamber FcO2 declined over the first 30 s of each minute to 7.4%–7.8%, stabilizing at this level for 5–7 s and then gradually increasing to 21% over the rest of each minute. This cycle was repeated for 4 h/day during the animals' sleep period (08.00–12.00). Meanwhile, normoxia control rats were placed in a chamber flushed with compressed air instead of nitrogen.
Myocardial infarction model
At the end of the IH treatment period myocardial infarction surgery was performed as previously described (Citation20). Briefly, rats were anesthetized with ketamine and xylazine (100 and 15 mg/kg, respectively) before undergoing a thoracotomy. A single 4-0 nylon suture was tied around the proximal left anterior descending coronary artery. Approximately 30 minutes later, the suture was removed to produce reperfusion. During these surgical procedures, the rats were maintained on a heating pad. Sham-operated controls were treated in an identical way, including administration of anesthesia and thoracotomy, but did not undergo occlusion with the ligature. Four hours after reperfusion (or sham procedures), the rats were re-anesthetized and hearts rapidly isolated. Left and right ventricles were dissected and weighed. Left ventricles were fixed in 4% paraformaldehyde, 0.1 M phosphate buffer (pH 7.4) at 4°C for 8 hours. Tissues were embedded in paraffin, and 5-μm thick sections were cut for histological analysis.
In-vivo hemodynamic measurements
Right ventricle systolic pressure (RVSP) was measured as previously described (Citation18). Briefly, rats were anesthetized with ketamine and xylazine (100 and 15 mg/kg, respectively) and placed supine while spontaneously breathing room air. Cardiac catheters were introduced from the right external jugular vein into the right ventricle. RVSP was measured in real time and recorded. Animals were then killed, and the left ventricle including the interventricular septum and right ventricle were removed and weighed. The ratio of right to left ventricular weight was then calculated.
Myocardial capillary density measurement
Alkaline phosphatase staining was used as a specific marker of endothelial cells in paraffin-embedded sections (Citation21,22) and capillary density of hearts assessed as described previously (Citation21,22). In each heart section, four sections were selected and analyzed with Image-pro Plus software (Image-Pro Plus version 6.0, Media Cybernetics, Rockville, Maryland, USA). In every section, we selected three visual fields to measure the capillary density and capillary/myocyte ratio (C/M).
Western blot analysis
Tissue from myocardium was homogenized in RIPA lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 2 mM EDTA, supplemented with a cocktail of protease and phosphatase inhibitors (Citation23,24). Protein concentrations were determined by Bradford reagent according to the manufacturer's instructions. Samples containing equal amounts of total protein (30 μg) were resolved by SDS-PAGE under reducing conditions. After electrophoresis, proteins were transferred to a PVDF membrane and probed with various primary antibodies to Bcl-2, VEGF, and HIF-1α (Santa Cruz Biotechnology, Santa Cruz, CA). The optical density of the specific protein bands was quantified by using a densitometer (GE Healthcare Bio-Sciences, Piscataway, NJ, USA).
Reverse transcription PCR (RT-PCR)
Total RNA was extracted from tissues using Trizol (Invitrogen, Carlsbad, CA, USA) according to the instruction of the manufacturer (Citation25). RNA was then transcribed into cDNA using reverse transcriptase (Tokyo, Japan). To determine mRNA expression, the following primers were used: 1) HIF-1α: forward, 5′- CAGCAGACCCAGTTACAGAA -3′, reverse, 5′- CAGCAGACCCAGTTACAGAA -3′; 2) VEGF: forward, 5′- ATGAACTTTCTGCTCTCTTG -3′; reverse, 5′- CTTTCTTTGGTCTGCATTCA -3′; 3) β-actin: forward, 5′- GTGGGGCGCCCCGGC ACCA -3′, reverse, 5′- CTCCTTAATGTCACGC ACGATTT -3′. PCR was performed at 94°C for 30 s, then 30 cycles at 94°C for 10 s, at 60°C for 1 min, and at 68°C for 1 min; extension was carried out at 68°C for 7 min. PCR products were electrophoretically separated on 1.5% agarose gels.
Terminal deoxyribonucleotidyl transferase (TDT)-mediated dUTP-digoxigenin nick end labeling (TUNEL) staining
TUNEL staining was performed as described previously (Citation26,27) to evaluate myocyte apoptosis. Paraffin was removed with xylene, and then the sections were washed with PBS. After washing, sections were equilibrated at room temperature for 5–10 minutes in equilibration buffer (200 mmol/L potassium cacodylate, 0.2 mmol/L dithiothreitol, 0.25 g/L bovine serum albumin, and 2.5 mmol/L cobalt chloride in 25 mmol/L Tris-HCl, pH 6.6) and then incubated in the presence of biotinylated nucleotide mix and terminal deoxynucleotidyl transferase at 37°C for 1 hour in a humidified chamber. The reaction was terminated by standard saline citrate and 3% H2O2 added to block endogenous peroxidase. After washing (×3) in PBS, streptavidin horseradish peroxidase was added for 1 hour at room temperature and again washed with PBS. After subsequent diaminobenzidine staining, positive cells were counted. Apoptotic-positive cells were counted in five fields of every section, and for each rat six sections were examined.
Measurements of MDA and SOD
MDA content and SOD activity were determined according to the manufacturers' instructions using commercially available kits (Citation28). Briefly, tissues were homogenized on ice in normal saline, frozen in a refrigerator at –80°C for 5 min, and centrifuged for 15 min at 4,000 g. Supernatants were transferred into fresh tubes for evaluation. MDA concentrations were measured by chemical analysis (kits supplied by Nanjing Jiancheng Biological Product, Nanjing, China) as previously described (Citation28). SOD activity was evaluated by inhibition of nitroblue tetrazolium reduction by superoxide anion generated by the xanthine/xanthine oxidase system using a commercial assay kit (Nanjing Jiancheng Biological Product, Nanjing, China) as described (Citation28).
Assessment of infarct size
Assessment of infarct size was performed as reported (Citation29). The ascending aorta was cannulated with a 22-gauge tubing adapter, and 1% Evans blue was perfused into the aorta and coronary arteries. Evans blue was distributed to the areas of the myocardium proximal to the ligature. Areas of the myocardium not stained with Evans blue were defined as ‘area at risk'. Hearts were weighed and sliced into four separate sections. Each section was weighed separately and then incubated in 1.5% triphenyltetrazolium chloride for 30 min. The area of infarction was white, whereas viable myocardium was red. Infarct size was then expressed as a percentage of the area at risk by using the following equation: (weight of the area of infarction/weight of the area at risk) ×100%.
Statistical analysis
Statistics were analyzed with SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Data are expressed as mean ± SD. One-way analysis of variance (ANOVA) followed by Tukey post hoc was used for multiple comparisons. Correlation between different variables was assessed by Pearson's analysis, and P < 0.05 was considered statistically significant.
Results
Effects of IH on cardiovascular parameters
Wistar rats were subject to IH for 4 h/day for 1, 7, 14, and 28 days. A slight increase of the RVSP was observed after 28 days of IH treatment, while there were no changes of RVSP at 1, 7, and 14 days of IH (). Consistent with these results, the RV/LV+S ratio (the ratio between weights of right ventricle and left ventricle plus ventricular septum) was significantly increased by IH after 28 days but not at any other time point. Moreover, although there was a tendency towards an increase in capillary density (CD), a statistically significant change was only observed after 28 days of IH (). Similarly, C/M also gradually increased during the IH treatment. At 14 and 28 days of IH treatment, C/M was significantly greater than that in the control group ().
Table I. Hemodynamic parameters and capillary density in rats treated with IH.
Effects of IH on myocardial I/R injury
As IH was reported to be involved in I/R injuries, we hypothesized that treatment of rats with IH might affect myocardial I/R injury. After IH treatment, myocardial infarction size was reduced in all groups compared with the I/R control group (). The greatest reduction in infarction size was observed in the IH 7 d + I/R group, yielding an approximate 55% reduction. There were no statistically significant differences between groups of animals subjected to different time periods of IH.
Figure 1. Effect of IH on I/R-induced myocardial infarction size in rats. **P < 0.01 versus I/R group.
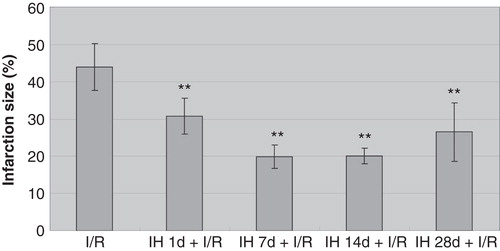
Effects of IH on mRNA and protein levels of myocardial VEGF and HIF-1α
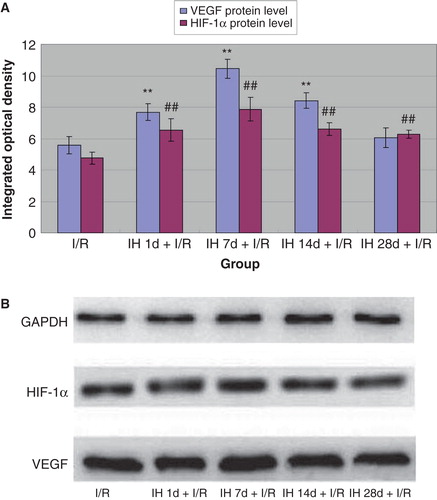
Because VEGF and HIF-1α have been shown to play important roles in myocardial infarction and associated prognosis (Citation30,31), we further determined their expression in myocardial tissue. Compared with controls, all IH animals displayed pronounced up-regulation of myocardial VEGF both at the mRNA and protein levels ( and ). Likewise, mRNA and protein levels of HIF-1α were increased, reaching the highest level at 7 days of IH treatment ( and ). Moreover, we examined the expression of VEGF and HIF-1α in myocardial tissue of animals subjected to I/R. I/R injury induced an up-regulation of these two factors, and IH treatment further increased this increase. Compared with the I/R group, mRNA () and protein levels () of VEGF and HIF-1α in IH + I/R animals increased and particularly so at 7 days after IH.
Figure 2. A: Effects of IH on mRNA levels of VEGF (shaded bars) and HIF-1α (open bars). **P < 0.01 versus control group, ## P < 0.01 versus control group. B: Effects of IH on mRNA levels of VEGF. C: Effects of IH on mRNA levels of HIF-1α.
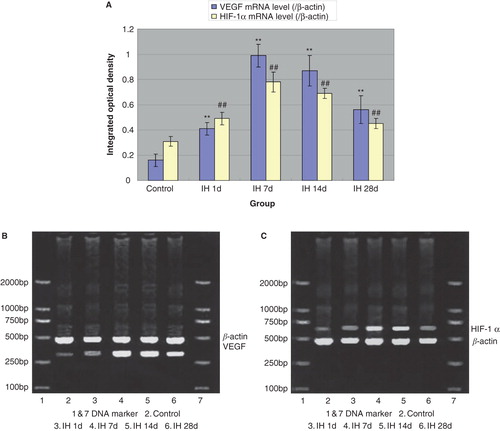
Figure 3. A: Effects of IH on protein levels of VEGF (gray bars) and HIF-1α (black bars). **P < 0.01 versus control group, # P < 0.05, ## P < 0.01 versus control group. B: Effects of IH on protein levels of VEGF and HIF-1α.
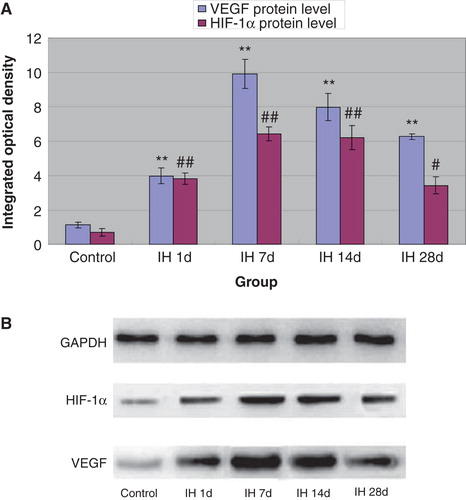
Figure 4. A: Effects of IH on I/R-induced expression of VEGF (gray bars) and HIF-1α (black bars) mRNA. **P < 0.01 versus I/R controls, ## P < 0.01 versus I/R controls. B: Effects of IH on I/R-induced expression of VEGF mRNA. C: Effects of IH on I/R-induced expression of HIF-1α mRNA.
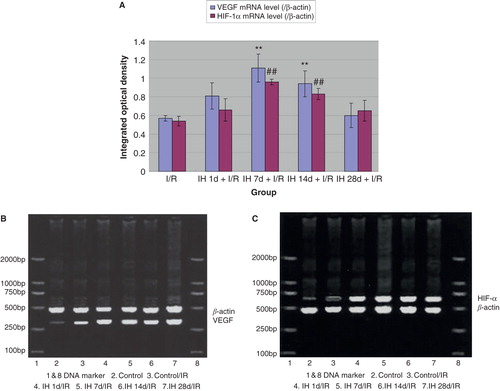
Figure 5. A: Effects of IH on I/R-induced expression of VEGF (gray bars) and HIF-1α (black bars) protein. **P < 0.01 versus I/R controls, ## P < 0.01 versus I/R controls. B: Effects of IH on I/R-induced expression of VEGF and HIF-1α protein.
Effects of IH on apoptosis and Bcl-2 protein level
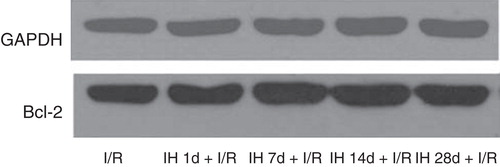
Next, we determined the extent of apoptosis by TUNEL staining and the expression of the anti-apoptotic protein Bcl-2 in rat myocardium. Interestingly, IH treatment did not affect the proportion of TUNEL-positive cells at 1, 7, and 14 days, but increased approximately 4-fold at 28 days after IH (). In contrast, Bcl-2 expression increased at all these time points ( and ). The surgically induced I/R model caused a pronounced increase both in the proportion of apoptotic cells and Bcl-2 expression ( and ). Superimposed IH (1, 7, 14, and 28 days) was effective in decreasing the extent of apoptosis in myocytes (). However, the beneficial effect of IH on Bcl-2 expression was observed only after 7 days of IH ( and ).
Figure 6. Effects of IH on protein levels of Bcl-2 in rat myocardial tissue.
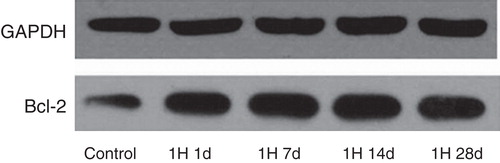
Table II. Effects of IH on apoptosis and Bcl-2 protein levels in rat myocardial tissue.
Table III. Effects of IH on ischemia/reperfusion-induced apoptosis and Bcl-2 protein level in rat myocardial tissue.
Figure 7. Effects of IH on I/R-induced expression of Bcl-2 protein in rat myocardial tissue.
Effects of IH on MDA and SOD concentrations
IH treatment for 1, 7, 14, and 28 days of animals not subject to I/R decreased the concentration of MDA in myocardial tissue, whereas the SOD concentration increased (). I/R injury as such effected increased MDA but decreased SOD concentrations ( and ). As in the group with control rats (without I/R injury), IH treatment effectively decreased the concentrations of MDA but increased those of SOD (). The difference in SOD concentration after 1 day of IH, however, did not attain statistical significance.
Table IV. Effects of IH on MDA and SOD concentrations in rat myocardial tissue.
Table V. Effects of IH on ischemia/reperfusion (I/R)-induced changes of MDA and SOD concentrations in rat myocardial tissue.
Discussion
In the present study, we showed that the treatment of short-term IH in rats reduced the myocardial infarction size. We further found that the HIF-1α and VEGF mRNA and protein expressions were elevated by IH treatment, suggesting that these two important hypoxia-induced factors may contribute to the cardioprotective effects of IH. In addition, both the largest decrease in apoptotic cell number (TUNEL) and the enhanced expression of the anti-apoptotic protein Bcl-2 were observed in the IH 7d + I/R group, further supporting the notion that IH protects the heart from I/R-induced myocardial injury through inhibiting apoptosis at least partly via HIF-1α and VEGF-associated pathways.
The most important finding of our study is that we demonstrated that short-term IH is cardioprotective. The effect reached a peak after 7 days of IH. This intriguing result indicates that the IH with cycles of less than 60 s in duration was capable of protecting the heart from ischemic injury. It has been well established that chronic IH during frequent nocturnal events as occurs in sleep apnea results in various pathologies (Citation16,32). Generally, chronic IH in OSA has been thought to be harmful for overall heart function, being associated with myocyte hypertrophy (Citation33) and apoptosis both in vitro (Citation16) and in vivo (Citation15). Thus, our study is the first report relating to short-term IH, with cycles less than 60 s in duration, demonstrating that this paradigm is, in fact, cardioprotective.
In our study, RVSP and RV/LV+S were both elevated at 28 days after IH treatment, suggesting that long-term IH may not be protective and may eventually be hazardous to the heart. This result was further supported by the TUNEL assay, which demonstrated that the number of apoptotic cells in the IH 28 d group increased markedly compared with the control group. Indeed, similar results were reported in a previous publication. Joyeux-Faure et al. showed that IH for 35 consecutive days increased the sensitivity of hearts to infarction (Citation33). Also, recently, Chen et al. demonstrated that IH for 6 weeks led to increased mean arterial pressure and left ventricle end-diastolic pressure, reduced cardiac output, and cardiomyocyte injury (Citation16). Thus, our results are consistent with these previous reports and suggest that, to conserve the beneficial effects of IH training, the time of IH should be restricted to a short term, whereas long-term IH may induce harmful effects.
The mechanisms underlying the marked organ-protective effects of IH remain to be fully elucidated, although a number of pathways have been suggested. Up-regulation of PKC-δ was reported to be involved in the cardioprotective mechanisms of IH (Citation34-36). Nitric oxide (NO) production has also been associated with vasorelaxation and capillary perfusion in IH-induced cardioprotection (Citation12,37,38). Moreover, IH cardioprotective effects involve up-regulation of heat shock proteins (Hsp) (specifically Hsp 23 and Hsp 70) (Citation39), β1-adrenergic receptor activation (Citation13), function of ATP-dependent potassium channels (Citation40), and PKC-δ activation. We focused on HIF-1α and VEGF in our study as they are critical regulatory proteins in hypoxia-induced adaptation (Citation41). However, the role of these hypoxia-response factors in IH cardioprotective effects is largely unknown. In our study, a convergence between IH cardioprotective effects and expression of HIF-1α and VEGF was observed. The involvement of HIF-1α in IH was first reported in studies of the nervous system. Rapino et al. demonstrated that IH could prevent hypoxia-induced neuronal cell death by up-regulating HIF-1α levels, combined with a low level of IkBα phosphorylation (Citation42). Subsequently, Yang et al. reported that IH activated an HIF-1α-dependent signaling cascade leading to hypoxia/ischemia tolerance against renal I/R injury (Citation43). Our study is the first report regarding the possible involvement of HIF-1α in the cardioprotective effects of IH. HIF-1α activation confers protection against I/R injury, by triggering an increase in expression of genes involved in glycolysis, glucose metabolism, mitochondrial function, cell survival, apoptosis, and resistance to oxidative stress (Citation44). In fact, we observed that IH decreased MDA, and increased SOD concentrations in rat myocardial tissue, which reached a peak at 14 days after IH, suggesting that these changes might be located downstream of alterations in HIF-1α expression.
VEGF is another important factor triggering cellular protection and metabolic alterations in response to oxygen deprivation (Citation45,46). This is particularly evident in angiogenesis and neovascularization after myocardial infarction (Citation47). Currently, there is no information regarding the role of VEGF in IH cardioprotection. In this study, we found a tendency for VEGF expression to change in a similar manner to that of HIF-1α expression. This suggests that VEGF, at least partly, contributes to the cardioprotective effects of IH. It should, however, be noted that in our study the most effective reduction of the extent of myocardial infarction occurred at 7 days after IH treatment, while this beneficial cardioprotective effect gradually declined at later time points. Consistent with this, the up-regulation of HIF-1α and VEGF also reached a peak at 7 days after IH, but then became progressively weaker despite increasing the overall duration of IH treatment. All together, these observations suggest that HIF-1α and VEGF participate in IH-mediated cardiac protection.
In conclusion, we have demonstrated that IH reduces the extent of myocardial infarction. The modulating effect of IH on apoptosis, oxidative stress, HIF-1α, and VEGF may contribute to the protection of IH in this model. Thus, our results may shed some light on the potential importance of HIF-1α and VEGF in IH, and provide new insights into endogenous defense mechanisms of IH.
Acknowledgements
This manuscript has been edited and proof-read by Medjaden Bioscience Limited.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Kuwahira I, Heisler N, Piiper J, Gonzalez NC. Effect of chronic hypoxia on hemodynamics, organ blood flow and O2 supply in rats. Respir Physiol. 1993;92:227–38.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36.
- Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–96.
- Vasquez-Vivar J, Whitsett J, Derrick M, Ji X, Yu L, Tan S. Tetrahydrobiopterin in the prevention of hypertonia in hypoxic fetal brain. Ann Neurol. 2009;66:323–31.
- Granfeldt A, Lefer DJ, Vinten-Johansen J. Protective ischaemia in patients: preconditioning and postconditioning. Cardiovasc Res. 2009;83:234–46.
- Powell SR, Divald A. The ubiquitin-proteasome system in myocardial ischaemia and preconditioning. Cardiovasc Res. 2010;85:303–11.
- Weidemann A, Bernhardt WM, Klanke B, Daniel C, Buchholz B, Campean V, HIF activation protects from acute kidney injury. J Am Soc Nephrol. 2008;19:486–94.
- Furrer K, Deoliveira ML, Graf R, Clavien PA. Improving outcome in patients undergoing liver surgery. Liver Int. 2007;27:26–39.
- Neubauer JA. Invited review: Physiological and pathophysiological responses to intermittent hypoxia. J Appl Physiol. 2001;90:1593–9.
- Lin AM, Chen CF, Ho LT. Neuroprotective effect of intermittent hypoxia on iron-induced oxidative injury in rat brain. Exp Neurol. 2002;176:328–35.
- Jung ME, Simpkins JW, Wilson AM, Downey HF, Mallet RT. Intermittent hypoxia conditioning prevents behavioral deficit and brain oxidative stress in ethanol-withdrawn rats. J Appl Physiol. 2008;105:510–17.
- Manukhina EB, Downey HF, Mallet RT. Role of nitric oxide in cardiovascular adaptation to intermittent hypoxia. Exp Biol Med (Maywood). 2006;231:343–65.
- Mallet RT, Ryou MG, Williams AG Jr, Howard L, Downey HF. Beta1-adrenergic receptor antagonism abrogates cardioprotective effects of intermittent hypoxia. Basic Res Cardiol. 2006;101:436–46.
- Whitehouse JS, Riggle KM, Purpi DP, Mayer AN, Pritchard KA Jr, Oldham KT, The protective role of intestinal alkaline phosphatase in necrotizing enterocolitis. J Surg Res. 2010;163:79–85.
- Belaidi E, Joyeux-Faure M, Ribuot C, Launois SH, Levy P, Godin-Ribuot D. Major role for hypoxia inducible factor-1 and the endothelin system in promoting myocardial infarction and hypertension in an animal model of obstructive sleep apnea. J Am Coll Cardiol. 2009;53:1309–17.
- Chen L, Zhang J, Gan TX, Chen-Izu Y, Hasday JD, Karmazyn M, Left ventricular dysfunction and associated cellular injury in rats exposed to chronic intermittent hypoxia. J Appl Physiol. 2008;104:218–23.
- Serebrovskaya TV, Manukhina EB, Smith ML, Downey HF, Mallet RT. Intermittent hypoxia: cause of or therapy for systemic hypertension? Exp Biol Med (Maywood). 2008;233:627–50.
- Fagan KA. Selected contribution: pulmonary hypertension in mice following intermittent hypoxia. J Appl Physiol. 2001;90:2502–7.
- Greenberg HE, Sica A, Batson D, Scharf SM. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol. 1999;86:298–305.
- Fishbein MC, Maclean D, Maroko PR. Experimental myocardial infarction in the rat: qualitative and quantitative changes during pathologic evolution. Am J Pathol. 1978;90:57–70.
- Taniyama Y, Morishita R, Aoki M, Nakagami H, Yamamoto K, Yamazaki K, Therapeutic angiogenesis induced by human hepatocyte growth factor gene in rat and rabbit hindlimb ischemia models: preclinical study for treatment of peripheral arterial disease. Gene Ther. 2001;8:181–9.
- Taniyama Y, Morishita R, Aoki M, Hiraoka K, Yamasaki K, Hashiya N, Angiogenesis and antifibrotic action by hepatocyte growth factor in cardiomyopathy. Hypertension. 2002;40:47–53.
- Koppelmann T, Pollak Y, Mogilner J, Bejar J, Coran AG, Sukhotnik I. Dietary L-arginine supplementation reduces Methotrexate-induced intestinal mucosal injury in rat. BMC Gastroenterol. 2012;12:41.
- Jang S, Park JW, Cha HR, Jung SY, Lee JE, Jung SS, Silver nanoparticles modify VEGF signaling pathway and mucus hypersecretion in allergic airway inflammation. Int J Nanomedicine. 2012;7:1329–43.
- Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC, Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011;69:360–74.
- Kimura A, Hsu M, Seldin M, Verkman AS, Scharfman HE, Binder DK. Protective role of aquaporin-4 water channels after contusion spinal cord injury. Ann Neurol. 2010;67:794–801.
- Yu S, Patchev AV, Wu Y, Lu J, Holsboer F, Zhang JZ, Depletion of the neural precursor cell pool by glucocorticoids. Ann Neurol. 2010;67:21–30.
- Xia Z, Gu J, Ansley DM, Xia F, Yu J. Antioxidant therapy with Salvia miltiorrhiza decreases plasma endothelin-1 and thromboxane B2 after cardiopulmonary bypass in patients with congenital heart disease. J Thorac Cardiovasc Surg. 2003;126:1404–10.
- Kurrelmeyer KM, Michael LH, Baumgarten G, Taffet GE, Peschon JJ, Sivasubramanian N, Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci USA. 2000;97:5456–61.
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76.
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88:1474–80.
- Zhang W, Si LY. Obstructive sleep apnea syndrome (OSAS) and hypertension: pathogenic mechanisms and possible therapeutic approaches. Ups J Med Sci. 2012;117:370–82.
- Joyeux-Faure M, Stanke-Labesque F, Lefebvre B, Beguin P, Godin-Ribuot D, Ribuot C, Chronic intermittent hypoxia increases infarction in the isolated rat heart. J Appl Physiol. 2005;98:1691–6.
- Kolar F, Jezkova J, Balkova P, Breh J, Neckar J, Novak F, Role of oxidative stress in PKC-delta upregulation and cardioprotection induced by chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2007;292:H224–30.
- Allahdadi KJ, Duling LC, Walker BR, Kanagy NL. Eucapnic intermittent hypoxia augments endothelin-1 vasoconstriction in rats: role of PKCdelta. Am J Physiol Heart Circ Physiol. 2008;294:H920–7.
- Ding HL, Zhu HF, Dong JW, Zhu WZ, Zhou ZN. Intermittent hypoxia protects the rat heart against ischemia/reperfusion injury by activating protein kinase C. Life Sci. 2004;75:2587–603.
- Bertuglia S. Intermittent hypoxia modulates nitric oxide-dependent vasodilation and capillary perfusion during ischemia-reperfusion-induced damage. Am J Physiol Heart Circ Physiol. 2008;294:H1914–22.
- Ivanina AV, Eilers S, Kurochkin IO, Chung JS, Techa S, Piontkivska H, Effects of cadmium exposure and intermittent anoxia on nitric oxide metabolism in eastern oysters, Crassostrea virginica. J Exp Biol. 2010;213:433–44.
- Azad P, Zhou D, Russo E, Haddad GG. Distinct mechanisms underlying tolerance to intermittent and constant hypoxia in Drosophila melanogaster. PLoS One. 2009;4:e5371.
- Zhu HF, Dong JW, Zhu WZ, Ding HL, Zhou ZN. ATP-dependent potassium channels involved in the cardiac protection induced by intermittent hypoxia against ischemia/reperfusion injury. Life Sci. 2003;73:1275–87.
- Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008;15:686–90.
- Rapino C, Bianchi G, Di Giulio C, Centurione L, Cacchio M, Antonucci A, HIF-1alpha cytoplasmic accumulation is associated with cell death in old rat cerebral cortex exposed to intermittent hypoxia. Aging Cell. 2005;4:177–85.
- Yang CC, Lin LC, Wu MS, Chien CT, Lai MK. Repetitive hypoxic preconditioning attenuates renal ischemia/reperfusion induced oxidative injury via upregulating HIF-1 alpha-dependent bcl-2 signaling. Transplantation. 2009;88:1251–60.
- Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006;70:1469–80.
- Chen Y, Hao Q, Kim H, Su H, Letarte M, Karumanchi SA, Soluble endoglin modulates aberrant cerebral vascular remodeling. Ann Neurol. 2009;66:19–27.
- Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67.
- Tammela T, Enholm B, Alitalo K, Paavonen K. The biology of vascular endothelial growth factors. Cardiovasc Res. 2005;65:550–63.