Abstract
We here report a case of diabetic ketoacidosis at onset of type 1 diabetes after a prolonged period of starvation due to anorexia nervosa. A 53-year-old female with a history of anorexia nervosa was admitted to the psychiatric clinic due to psychotic behaviour and inability to take care of herself. Twenty-four hours after admission she was transferred to the clinic of internal medicine due to altered mental status, and laboratory screening revealed a pH of 6.895 and blood glucose concentration of 40 mmol/L. Due to the unusual combination of prolonged starvation and diabetic ketoacidosis we implemented some modifications of existing treatment guidelines and some special considerations regarding nutrition in order to prevent a re-feeding syndrome.
Introduction
Diabetic ketoacidosis (DKA) is a feared and potentially lethal complication in diabetic patients, with a 5% mortality rate. DKA is the cause of half of all deaths in individuals with type 1 diabetes (T1D) under the age of 24 (Citation1) and is characterized by hyperglycaemia, acidosis, electrolyte changes, and dehydration due to hypoinsulinaemia (Citation2), with the diagnostic criteria: ketonaemia >3 mmol/L; blood glucose >11 mmol/L, and venous bicarbonate <15 mmol/L and/or a venous pH <7.3 (Citation3). Most cases of DKA occur due to an infection, trauma, pancreatitis, or poor medication compliance in patients with known diabetes, but T1D can also present as DKA (Citation1). Another condition that causes an altered metabolism and metabolic acidosis is severe starvation where ketone bodies are produced as in DKA (Citation4). However, starvation leads to hypoglycaemia, and the loss of fluid is therefore not as severe as in DKA. Patients with anorexia nervosa (AN) display several metabolic and endocrine changes including impaired glucose tolerance caused by long-standing starvation (Citation5,6) and in its most severe form leads to severe electrolyte disturbances and even multi-organ failure (Citation6). The combination of long-term starvation due to AN and DKA therefore represents a challenge with high demands on intensive care monitoring. We here report on the case of a 53-year-old woman with long-standing AN who presented at the emergency room after a prolonged period of starvation with onset of T1D and severe ketoacidosis. The patient has given a written consent to the publication of this case report.
Case report
A 53-year-old woman with a history of AN since adolescence was admitted to the psychiatric clinic at the Uppsala University Hospital due to psychotic behaviour and inability to take care of herself. There were reports of paranoid schizophrenia and personality disorders, but her contacts with the psychiatric clinic were sparse, and she used no medications. According to her relatives she had isolated herself the last two weeks, and over the last two months she had barely been eating at all. Twenty-four hours after admission to the psychiatric clinic she was admitted to the clinic of internal medicine and presented at the emergency room (ER) in a cachectic state with hypothermia (32.6°C). Her mental status was altered (reaction level scale 2), and she barely responded to questions, being close to stupor. There were clinical signs of severe dehydration and muscle atrophy. Her breathing was shallow with 30 bpm, whereas blood pressure and pulse rate were normal, 110/60 mmHg and 77 bpm (see for a summary of the initial physical examination). She denied alcohol and any substance abuse. An initial arterial blood gas analysis displayed pH 6.895, pCO2 0.93 kPa, pO2 22 kPa, and P-glucose 40.6 mmol/L. There were no signs of infection, and electrolytes were normal (see for a summary of laboratory screening). She was immediately admitted to the intensive care unit (ICU), where rehydration was initiated with warm fluid combined with re-warming with heated blankets. Bicarbonate (100 mL) was administered i.v. in order to reverse acidosis; pH increased to 7.1, and blood glucose decreased to 35 mmol/L. Six hours after ICU admission insulin infusion was started with initially 0.5 IU/h (0.0128 IU/kg) combined with 5 mmol potassium/h. The patient's pH was normalized 15 hours after admission to the ICU, and the blood glucose concentration was kept around 15 mmol until 36 hours after admission. Variations of pH, glucose concentration, and arterial pCO2 and pO2 during the first 24 hours have been displayed in . Laboratory screening at the ICU revealed a P-C-peptide concentration of 0.14 nmol/L (ref. 0.4–1.5 nmol/L), a HbA1c of 151 mmol/mol (ref. <46 mmol/mol), and presence of both glutamic acid decarboxylase (GAD) (74 IE/mL, ref. <5) and insulinoma antigen 2 (IA2) antibodies (69 kE/L, ref. <8).
Table I. Summary of vital signs from the initial physical examination at the emergency room.
Table II. Summary of the initial laboratory screening.
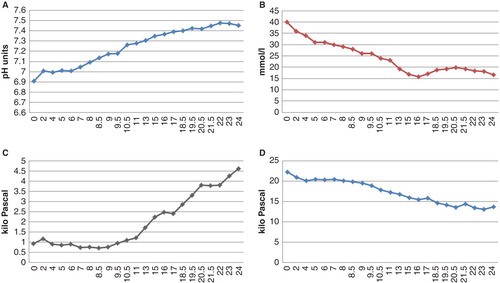
Figure 1. Plot of pH, plasma glucose concentration, and arterial gas values during the first 24 hours of care at the ICU. Bicarbonate was administered i.v. during the first hour, and the insulin infusion was initiated six hours after admission to the ICU. A: pH value, normalized (7.35) after 15 hours. B: Glucose concentration (mmol/L) continuously decreased up until 15 hours after admission when glucose infusion was initiated. C: Arterial CO2 displayed extremely low values, <1 kPa. After 11 hours there was an increase which correlated to an increased pH level. CO2 was not normalized until 30 hours after admission. D: Arterial O2 continuously decreased and reached normal levels (12.6 kPa) after 25 hours.
Due to the risk of re-feeding syndrome, the intake of calories was restricted to 500 kcal over the first 24 hours and then increased when the patient was able to drink nutritional drinks. The risk of re-feeding syndrome is especially high in patients with a BMI <16, recent weight loss, and electrolyte abnormalities (Citation7). In order to prevent re-feeding syndrome, especially carbohydrates should be restricted whereas proteins and fats seem to be less of a threat (Citation8). However, in this case administration of i.v. glucose (120 kcal) in the first 24 hours could not be avoided, since plasma glucose concentrations had to be maintained around 15 mmol/L during the insulin infusion in order to prevent the risk of brain oedema. Remaining energy intake was administered per os as nutritional drinks with a balanced protein, fat, and carbohydrate content. A transthoracic echocardiogram was performed in order to exclude ventricular dysfunction and mitralic valve prolapse which can be observed after starvation and can potentially be lethal, especially in combination with re-feeding syndrome (Citation9). An expanded health history revealed that the patient had rapidly lost 10 kilograms of weight over the last two weeks and had polydipsia and polyuria. It was noted that her niece had T1D but there was no family history of diabetes.
As pH and blood glucose concentrations normalized, the patient improved; she was transferred to an internal medicine ward, and after optimization of the insulin regime she was transferred to the psychiatric clinic for continuous psychiatric care. She is currently on treatment with Insulin Glargine and Insulin Aspart, and on her last clinical control her fasting glucose was 16.1 mmol/L (ref. 4.0–6.0), HbA1c 82 mmol/mol (ref. 31–46), and her P-C-peptide had further decreased to 0.093 nmol/L (ref. 0.4–1.5). There have been no additional episodes of DKA, and her weight has increased to 55 kg with a BMI of 18.2 kg/m2 (ref. 20–25). In , there is a summary of laboratory screening six months post-DKA.
Table III. Laboratory screening six months after the DKA.
Discussion
We have not found any reports in the literature of DKA in patients after prolonged starvation. In the acute phase we made some alterations to the treatment guidelines for DKA (Citation1,3) that we believed necessary. First of all bicarbonate was administered in order to reverse acidosis, which is usually not recommended. In fact, the use of bicarbonate in the treatment of DKA is somewhat controversial (Citation10), and there is no evidence of beneficial effects on mortality or morbidity in patients with a pH between 6.9 and 7.1 (Citation11). Bicarbonate treatment can cause an increase of CO2 in the cerebral spinal fluid and thereby worsen cerebral acidosis unless treatment is initiated to reverse the acidosis by other means. In this case the initial pH was 6.895, and in such severe cases of acidosis there is no randomized evidence-based guidelines to rely on (Citation12), although it is recommended to give bicarbonate. The second adjustment we made was the delayed insulin treatment. According to the guidelines insulin treatment should be initiated immediately with a continuous infusion of 0.1 IU/kg/h (Citation1). In this case we chose to administer fluid only during the first hours after admission to the ICU, which decreased P-glucose from 40 to 35 mmol/L. In starvation insulin secretion is significantly reduced, and in acute AN the response to glucose is impaired (Citation5,6). Immediate administration of insulin could therefore have been lethal due to a rapid shift in osmolality and influx of potassium from plasma into cells, thereby risking cardiac and respiratory failure (Citation6,8).
According to the DKA treatment guidelines P-glucose was kept at 15 mmol/L by a balanced glucose infusion. However, in order to prevent a re-feeding syndrome administration of carbohydrates should be avoided or kept to a minimum and nutrients should preferentially be administered as a combination of proteins and carbohydrates (Citation6). It is not possible to fulfil both these recommendations from the guidelines, and therefore we had to choose a common path, adhering to the first.
The brain and renal medulla are both dependent on glucose and ketone bodies for energy metabolism (Citation4), and therefore the formation of ketone bodies is an important mechanism in the absence of glucose. The liver plays a central role in order to maintain energy metabolism. During starvation free fatty acids will be metabolized into ketone bodies. Furthermore, metabolism will shift from storing glucose as glycogen to gluconeogenesis. Insulin has a counteracting mechanism in this scenario, but both in starvation and T1D the decrease of insulin stimulates gluconeogenesis along with stimulatory effects from epinephrine, cortisone, and glucagon. In starvation this will result in hypo- or normoglycaemia, whereas in T1D it leads to hyperglycaemia. In this unique case these two states of metabolic acidosis coincide, and most likely glycogen stores in both liver and muscle were already depleted at the onset of T1D, which then resulted in hyperglycaemia due to gluconeogenesis, increasing formation of ketone bodies, and a severe metabolic acidosis.
In patients with a psychiatric diagnosis there is a risk of delay before diagnosis due to both a patient and doctor delay. In this particular case the patient had been admitted to the psychiatric clinic 24 hours before she was admitted to the clinic of internal medicine. By controlling blood glucose in the pre-hospital setting her hyperglycaemia would have been diagnosed directly and treatment could have been initiated immediately. In several case reports antipsychotics, both conventional and atypical, have been described to cause both hyperglycaemia and DKA (Citation13,14). However, this was not a differential diagnosis since the patient had not been prescribed any antipsychotic drugs. Even though the metabolic control is not perfect, the patient is now doing fine. She has gained weight and has not suffered from any additional DKA.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32:1335–43.
- Sherry NA, Levitsky LL. Management of diabetic ketoacidosis in children and adolescents. Paediatr Drugs. 2008;10:209–15.
- Savage MW, Dhatariya KK, Kilvert A, Rayman G, Rees JA, Courtney CH, Joint British Diabetes Societies guideline for the management of diabetic ketoacidosis. Diabet Med. 2011;28:508–15.
- Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr. Brain metabolism during fasting. J Clin Invest. 1967;46:1589–95.
- Casper RC. Carbohydrate metabolism and its regulatory hormones in anorexia nervosa. Psychiatry Res. 1996;62:85–96.
- Usdan LS, Khaodhiar L, Apovian CM. The endocrinopathies of anorexia nervosa. Endocr Pract. 2008;14:1055–63.
- Smith T, Elia M. Artificial nutrition support in hospital: indications and complications. Clin Med. 2006;6:457–60.
- Apovian CM, McMahon MM, Bistrian BR. Guidelines for refeeding the marasmic patient. Crit Care Med. 1990;18:1030–3.
- Schocken DD, Holloway JD, Powers PS. Weight loss and the heart. Effects of anorexia nervosa and starvation. Arch Intern Med. 1989;149:877–81.
- Viallon A, Zeni F, Lafond P, Venet C, Tardy B, Page Y, Does bicarbonate therapy improve the management of severe diabetic ketoacidosis? Crit Care Med. 1999;27:2690–3.
- Morris LR, Murphy MB, Kitabchi AE. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med. 1986;105:836–40.
- Latif KA, Freire AX, Kitabchi AE, Umpierrez GE, Qureshi N. The use of alkali therapy in severe diabetic ketoacidosis. Diabetes Care. 2002;25:2113–14.
- Avella J, Wetli CV, Wilson JC, Katz M, Hahn T. Fatal olanzapine-induced hyperglycemic ketoacidosis. Am J Forensic Med Pathol. 2004;2:172–5.
- Campanella LM, Lartey R, Shih R. Severe hyperglycemic hyperosmolar nonketotic coma in a nondiabetic patient receiving aripiprazole. Ann Emerg Med. 2009;53:264–6.