Dear Sir,
I read with interest a recent article by Antonodimitrakis and colleagues in your journal presenting a case of acquired hemophagocytic lymphohistiocytosis (HLH) in a 60-year-old woman suffering from diabetes mellitus type 2 (Citation1). The reported patient developed a life-threatening HLH associated with a reactivation of an Epstein–Barr virus (EBV) infection and was successfully treated by means of corticosteroids alone.
Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome of an exaggerated inflammatory reaction triggered by various inherited and/or acquired factors (Citation2). The literature on the topic of HLH in adults is limited; however, acquired (i.e. secondary) forms of HLH (e.g. infection-associated HLH, I-HLH; autoimmune-associated HLH, A-HLH; malignancy-associated HLH, M-HLH) are the most prevalent.
Antonodimitrakis et al. wrote in their article that ‘the patient's age showed that a familial form of HLH was unlikely' (Citation1). Familial forms of HLH (FHL) are autosomal recessive disorders related to different mutations in genes encoding proteins required for lymphocyte cytotoxicity (i.e. PRF1, UNC13D, STX11, STXBP2) (Citation2). Although FHL usually arises in infants and the vast majority of HLH cases in adults are acquired, it should be stressed that, albeit rarely, FHL can occur in adulthood, including older individuals (so-called late-onset FHL) (Citation3–5). Therefore, the possibility of late-onset FHL in adults with HLH cannot be univocally excluded before tests of NK/T cell degranulation and activity, as well as genetic testing, have been performed.
A retrospective study from Japan revealed that the frequency of different forms of HLH in adults varied depending on the age bracket (Citation6). Among HLH patients aged 15–29 years, I-HLH was the most common (68% of cases). It was caused in equal parts by EBV-HLH (34%) and I-HLH other than EBV-HLH (34%). In this age group, the second most common cause of HLH was A-HLH (22%), followed by M-HLH (10%). In the group of patients aged 30–59 years, M-HLH occurred only slightly less frequently than I-HLH (37% and 47%, respectively), followed by A-HLH (9%) and HLH after hematopoietic stem cell transplantation (7%). In the group of patients aged ≥60 years, however, malignancy was the most frequent cause of HLH (68%), followed by I-HLH (26%) and A-HLH (6%) (Citation6).
A recent, retrospective, population-based study from our group showed the annual incidence of M-HLH in adults to be 1:280,000 per year, or 0.36/100,000 individuals per year (Citation7). The results of this study were limited by the small population of the Swedish region of northern Halland but were strengthened by the long observation period of over 14 years.
Although M-HLH in East Asia is most often associated with NK/T cell lymphoproliferative malignancies, M-HLH in Europe tends to develop more frequently in the course of the hematological malignancies (Citation6,7). Importantly, it is worth to remember that M-HLH can occur before or during the treatment of known malignancy, or as the first manifestation of an occult malignancy (Citation7,8). Therefore an episode of HLH with unclear etiology in the older individual should always serve as a warning signal for a yet undiagnosed malignancy, leading to further careful cancer diagnostics and follow-up, as was done in the case reported by Antonodimitrakis and colleagues.
The patient reported by Antonodimitrakis et al. underwent repeated biopsies, which failed to show hemophagocytosis in affected organs (Citation1). The diagnosis of HLH can be confirmed in some cases by cytological and/or histopathological evaluation of bone-marrow, spleen, liver, lymph nodes, and cerebrospinal fluid (Citation8,9). Cytohistological examination reveals accumulation of lymphocytes and histiocytes (macrophages), sometimes with hemophagocytic activity (). Although examination of bone-marrow slides has the highest sensitivity for the detection of HLH, approximately 20% of patients lack features of HLH on their initial bone-marrow examination (Citation9). Hemophagocytic activity is a rather late sign of advanced HLH (Citation7). Therefore, further search for hemophagocytic activity of macrophages is encouraged if hemophagocytosis is not proven at the time of HLH onset (Citation8). If the bone-marrow specimen is not conclusive, serial marrow aspirates over time may be helpful as well as a biopsy obtained from other organs (Citation8).
Figure 1. May–Grünwald–Giemsa stain of bone marrow aspirate smear showing four macrophages displaying massive hemophagocytic activity (magnification ×400).
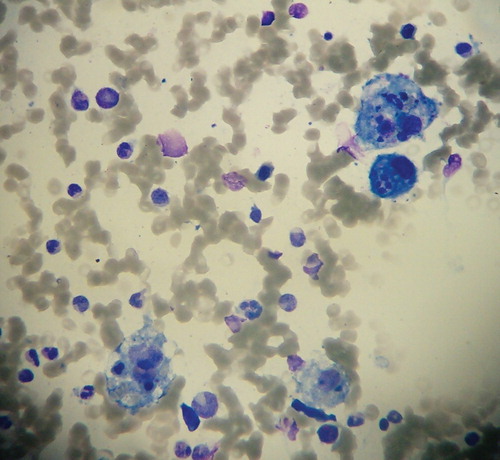
All forms of HLH, even when treated in a timely manner, can be fatal. M-HLH has the worst prognosis compared to other forms of HLH (Citation2,6,7). Ishii et al. found that 5-year survival reaches only 12% in patients with M-HLH associated with T/NK cell non-Hodgkin lymphoma (NHL), 48% in patients with M-HLH associated with B cell NHL, 54% in patients with FHL, and 83%–90% in patients with either I-HLH or A-HLH (Citation6). The therapy of any form of HLH should focus on: 1) suppression of the hyperinflammatory status by destruction of activated CD8+ T lymphocytes and macrophages, and 2) treatment of any existing HLH triggers (Citation2,8). In cases of FHL, an additional aim is the correction of the underlying immune defect by allogeneic stem cell transplantation (allo-SCT) (Citation9).
Treatment of acquired HLH is not standardized so far and remains highly variable across clinical centers. HLH treatment categories include: 1) proapoptotic chemotherapy with etoposide (50–150 mg/m2/dose i.v.), and 2) immunosuppressive drugs, targeting the hyperactivated macrophages (e.g. etoposide, corticosteroids, intravenous immunoglobulin (IVIG)) and T cells (e.g. corticosteroids, cyclosporine A (CyA)) (Citation2,8). Obviously, if possible, treatment of any existing trigger of HLH is mandatory (Citation8).
Antonodimitrakis et al. successfully treated their patient with corticosteroids only (Citation1). It may be a sufficient approach to employ corticosteroids (with or without IVIG) to control hyperinflammation as a frontline therapy in I-HLH and A-HLH (particularly of milder grades) (Citation2). After the improvement of the complete blood count and resolution of coagulopathy, steroids are slowly tapered down to avoid relapses of HLH. Corticosteroid-resistant non-responders may benefit from second-line therapies, such as CyA (Citation2). If there is no response to the aforementioned drugs (corticosteroids, IVIG, CyA), use of the HLH-2004 protocol including etoposide is recommended (Citation2,7,8). Thus, patients with acquired HLH could be started on therapy without etoposide, as long as treatment adjustments are made rapidly in refractory cases. If HLH is driven by EBV infection, monoclonal anti-CD20 antibodies (i.e. rituximab) that deplete B lymphocytes, the predominant type of cells harboring EBV virus, should be used (Citation10). Anecdotal reports have also shown the efficacy of allo-SCT in refractory or recurrent acquired HLH (e.g. EBV-HLH, M-HLH) (Citation11,12).
Since HLH can be encountered by various specialists in the medical field, knowledge of this entity and its diagnostic criteria should be familiar to all physicians. Treatment of HLH is difficult, long-lasting, and often associated with a high morbidity and mortality. The article by Antonodimitrakis and colleagues is a valuable voice in the debate over adult HLH.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
References
- Antonodimitrakis P, Wassberg C, Gerovasileiou S, Back J, Hällgren R, Olsen B. Fulminant hemophagocytic lymphohistiocytosis secondary to a reactivated EBV infection: A case report. Ups J Med Sci. 2013;118:42–5.
- Machaczka M, Sydor W, Rucińska M, Szostek M, Musiał J. Autoimmune-associated hemophagocytic syndrome/macrophage activation syndrome. In: Fang-Ping Huang, editor. Autoimmune disorders—current concepts and advances from bedside to mechanistic insights. Rijeka: InTech—Open Access Publisher; 2011. p 79–104; ISBN 978-953-307-653-9.
- Nagafuji K, Nonami A, Kumano T, Kikushige Y, Yoshimoto G, Takenaka K, et al. Perforin gene mutations in adult-onset hemophagocytic lymphohistiocytosis. Haematologica. 2007;92:978–81.
- Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1. MUNC13-4, and STXBP2 are associated with adult-onset familial hemophagocytic lymphohistiocytosis. Blood. 2011;118:5794–8.
- Sieni E, Cetica V, Piccin A, Gherlinzoni F, Sasso FC, Rabusin M, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649.
- Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86:58–65.
- Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52:613–19.
- Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
- Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunotherapy and bone marrow transplantation. Blood. 2002;100:2367–73.
- Balamuth NJ, Nichols KE, Paessler M, Taechey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007;29:569–73.
- Ohga S, Kudo K, Ishii E, Honjo S, Morimoto A, Osugi Y, et al. Hematopoietic stem cell transplantation for familial hemophagocytic lymphohistiocytosis and Epstein- Barr virus-associated hemophagocytic lymphohistiocytosis in Japan. Pediatr Blood Cancer. 2010;54:299–306.
- Machaczka M, Nahi H, Karbach H, Klimkowska M, Hägglund H. Successful treatment of recurrent malignancy- associated hemophagocytic lymphohistiocytosis with a modified HLH-94 immunochemotherapy and allogeneic stem cell transplantation. Med Oncol. 2012;29:1231–6.