
Abstract
Background. The clinical presentation of Gaucher disease (GD), an inherited lysosomal storage disorder caused by the deficient activity of the lysosomal enzyme glucocerebrosidase, is highly variable, and three clinical types are distinguished based upon the presence of neurologic symptoms. Thrombocytopenia, anemia, hepatosplenomegaly, and bone manifestations are the most typical signs of GD type 1 (GD1).
Case presentation. We present the case of an unsplenectomized man suffering from heterozygous GD1 with mutations of c.1226A>G (N370S) and RecNci I (L444P, A456P, and V460V) in the GBA1 gene, who developed recurrent pulmonary aspergillosis caused by Aspergillus fumigatus and a mycobacterial infection caused by Mycobacterium avium. Despite long-lasting therapy of both aspergillosis (including antifungal drugs and surgery), and the mycobacterial infection (triple therapy with rifampicin, ethambutol, and clarithromycin), recurrent positivity for M. avium and A. fumigatus was detected.
Conclusions. Symptomatic lung involvement and an increased susceptibility to pulmonary infections are uncommon in GD and, if present, are often associated with more severe disease manifestations. To our knowledge, this is the first published report on the association of GD and pulmonary aspergillosis and mycobacterial infection. It illustrates the increased susceptibility of untreated GD patients to opportunistic pulmonary infections and ineffective eradication of these infections despite adequate therapy.
Introduction
Gaucher disease (GD) is an autosomal recessive disorder, resulting from a deficient activity of the lysosomal enzyme glucocerebrosidase (GBA), which causes an accumulation of the sphingolipid glucosylceramide in the cells of the reticulo-endothelial system (Citation1). There are approximately 300 known mutations in the GBA1 gene, located on the long arm of chromosome 1 (1q21) that can cause GD (Citation2). However, it is difficult to simply define a correlation between phenotype and genotype, and GD is well recognized for its striking but unexplained phenotypic diversity.
The clinical presentation of GD is variable. Decreased GBA activity leads to excess storage of glucosylceramide in the cells of the monocyte-macrophage system throughout the body (Citation1). Three clinical types of GD are distinguished based upon the absence (type 1) or presence (types 2 and 3) of neurologic symptoms as well as the dynamics of the development of clinical signs. Thrombocytopenia, anemia, hepatosplenomegaly, and bone manifestations are the most typical signs of GD type 1 (GD1), which is the most prevalent form of GD (Citation1-3). Pulmonary involvement is considered rare in GD1 (Citation4-7).
In the European Union, there are two treatment options available for patients with GD: enzyme replacement therapy (ERT) with macrophage-targeted recombinant glucocerebrosidase (Cerezyme®, Genzyme Corporation, Cambridge, MA, USA, or VPRIV®, Shire HGT, Lexington, MA, USA), and substrate reduction therapy (SRT) with miglustat (N-butyldeoxynojirimycin, Zavesca®, Actelion Pharmaceuticals, Allschwil, Switzerland) (Citation1,3,8-10).
Here, we present the case of an adult man suffering from heterozygous untreated GD1, who developed pulmonary fibrosis complicated by recurrent pulmonary aspergillosis caused by Aspergillus fumigatus and a pulmonary mycobacterial infection caused by Mycobacterium avium. Informed consent has been provided by the patient.
Case presentation
A 65-year-old man, originating from northern Greece but living in Sweden for over 30 years, had been referred to the Hematology Center Karolinska for a consultation concerning suspected Gaucher disease. His past medical history was significant for several co-morbidities (). Because of splenomegaly (18 cm) and mild thrombocytopenia, a bone marrow (BM) examination was performed at the age of 51, disclosing the presence of foamy macrophages classified as Gaucher cells (GCs). However, further investigations aimed at the confirmation of GD diagnosis were not performed at that time.
Table I. Some clinical characteristics of the patient.
At the age of 54 years, he developed malaise and a chronic cough. He was a smoker for 25 years but quit at the age of 50. Radiological examination disclosed mild pulmonary fibrosis and aspergillosis in the right lung. The microbiological examination of secretions from a bronchoalveolar lavage (BAL) showed growth of Aspergillus fumigatus and mycobacteria other than tuberculosis (MOTT). Antifungal therapy with itraconazole (100 mg 2 × 2 p.o.) was administered for 6 weeks, but the MOTT infection was not treated. Of note, several weeks after therapy onset, the patient's clinical course was complicated by pulmonary bleeding (the coagulation work-up was normal apart from moderate thrombocytopenia). The fungal infection did not fully respond to therapy, and 2 years after diagnosis the patient underwent a partial resection of the right lung due to an aspergilloma (). After surgery, his general status improved rapidly.
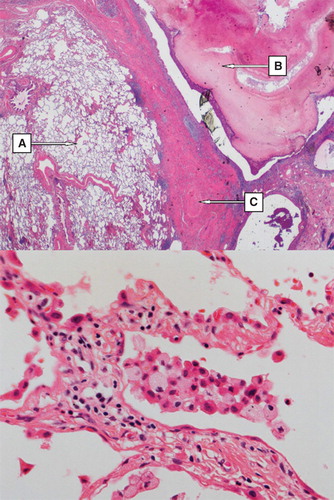
Figure 1. Upper part: overview of the right lung resectate showing (A) emphysema, (B) tumor-forming fungal growth (i.e. mycetoma) in the bronchial lumen, (C) fibrosis of the lung parenchyma (H&E, magnification × 50). Lower part: Right lung resectate showing mild interstitial lymphocytic inflammatory infiltrate; intra-alveolar aggregate of inflammatory cells including foamy macrophages (Gaucher cells) (H&E, magnification × 200).
At age of 59, the patient developed shortness of breath and cough. Echocardiography showed septal left ventricular hypertrophy, normal systolic function of the left ventricle (EF of 50%–60%), signs of diastolic dysfunction, mild insufficiency of both aortic and pulmonary valves, and a slightly elevated pressure in the pulmonary artery (PA) of 42 mmHg. Repeated microbiological examination of the BAL secretions revealed a growth of Aspergillus fumigatus and Aspergillus nidulans, but was negative for MOTT. HIV serology was negative, excluding acquired immunodeficiency syndrome (AIDS). Voriconazole was administered for aspergillosis periodically for 2 years (at the age 61–62 years). Afterwards, the patient's symptoms regressed, and control spirometry showed only moderately decreased results (VC 3.72 L, 82%; FEV1 3.04 L, 84%; FEV1/SVC 82%; FVC 3.72 L, 82%). Chest X-ray did not show any active pathological lesions. However, computed tomography imaging (CT) revealed pronounced fibrosis of lung parenchyma.
At the time of hematologic consultation (at the age of 65), thrombocytopenia and hyperferritinemia were noted. The final diagnosis of GD1 was confirmed by the absence of glucocerebrosidase activity in peripheral blood leukocytes and an increased activity of plasma chitotriosidase. Further direct DNA sequencing of the GBA1 gene revealed heterozygous mutations in the GBA1 gene ().
The patient's status consisting of splenomegaly, thrombocytopenia, and an increased susceptibility to pulmonary infections, which was probably due to lung involvement in the course of GD, was assessed as an indication for commencing treatment of GD. The patient was started on miglustat, which was considered the first line of GD1 treatment at that time due to the worldwide supply shortage of Cerezyme® during 2009–2010 (Citation9). He received the commercially available miglustat capsules at a dose of 100 mg three times a day orally.
Interestingly, the patient experienced an unusually rapid improvement of the whole blood platelet count (PLT), which normalized already after 2 months of miglustat therapy. Unfortunately, the patient developed malaise, diarrhea, poor appetite, weight loss (10 kg, 16% of his baseline body weight), and mild tremor for which miglustat was deemed accountable. During this time, the patient also developed cough, and a microbiological examination of the BAL secretions revealed growth of Aspergillus fumigatus (sensitive to voriconazole) and Mycobacterium avium. Inflammatory activation was observed at that time as presented in .
The patient discontinued miglustat after 4 months of treatment due to the adverse events and the above-mentioned pulmonary infections. Voriconazole was administered, and after 10 days his cough decreased but, after some days, returned, alongside malaise and night sweats. CT of the chest disclosed a new infiltrate in the lower lobe of the right lung, despite antifungal therapy (). Treatment with rifampicin, ethambutol, and clarithromycin for Mycobacterium avium was initiated. After 4 months of therapy, the patient was stronger, his cough and night sweats were clearly reduced, and he had regained 10 kg of body weight.
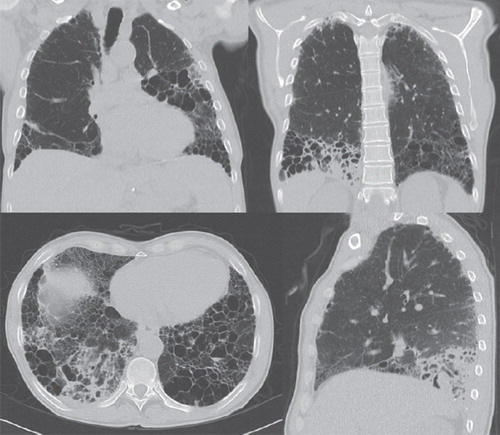
Figure 2. Chest CT scans at the time of aspergillosis relapse and diagnosis of M. avium infection. New infiltrates located dorsally in the right lower lobe and in the left lower lobe.
After the Cerezyme® supply shortage had ceased, the patient was offered to start ERT. Unfortunately, he strongly refused administration of ERT despite our recommendations. Now, almost 2.5 years after the initiation of the triple therapy, and with intermittent periods of negativity, the patient is recurrently positive for Mycobacterium avium in his sputum, despite 8 months of triple therapy administered during the preceding year. During the periods of deterioration in his general condition, he was also positive for Aspergillus fumigatus in sputum.
Discussion
Symptomatic lung involvement is rarely present in GD1 and usually is not the major manifestation of the disease (Citation3-7). Sporadic reports, mostly concerning children, have demonstrated the occurrence of pulmonary hypertension, bilateral interstitial lung disease, hypoxia due to intrapulmonary arterio-venous shunts (most often as a complication of long-standing, severe liver disease, i.e. hepatopulmonary syndrome), or increased susceptibility to pulmonary infections (Citation5-7). Pulmonary involvement may be a direct result of infiltration with GCs or may be secondary, due to extensive hepatic disease or to mechanical compression caused by hepatosplenomegaly. Pulmonary lesions in GD have been described as diffuse, reticulonodular infiltrates or lesions of a miliary pattern on chest X-rays (Citation7). Moreover, pulmonary involvement correlates more often with severe GD manifestations, including the neuronopathic forms of GD (types 2 and 3) (Citation4). In patients with the Norrbottnian type of GD, gibbous malformations due to rib-cage and thorax deformities additionally compromise lung function.
On the other hand, an autopsy study by Lee and Yousem disclosed that as much as 30% of GD patients showed ‘significant’ lung involvement (Citation11). Although significance criteria were not defined by the authors, they recognized three histological patterns of lung abnormalities in GD: 1) interstitial infiltration by GCs invading the peribronchial, perivascular, and septal regions with associated fibrosis; 2) GCs occluding pulmonary capillaries with secondary pulmonary hypertension; and 3) alveolar infiltration by GCs.
Patients with GD1 may have an increased susceptibility to infections due to alterations in their immune system, which could be the result of impaired splenic function or splenectomy, abnormalities in macrophage function, lymphopenia, neutropenia due to marked hypersplenism, abnormalities of humoral immunity (e.g. hypogammaglobulinemia), impaired superoxide production by monocytes, or impaired response to some mitogens (Citation12-15). Impaired host defenses against microbial infections that are benign to healthy individuals can cause significant morbidity in GD patients (Citation16,17). Although the exact mechanisms that lead to increased susceptibility to infections in GD have not yet been elucidated, impaired chemotaxis of granulocytes and defective monocyte function in GD patients have been described (Citation13,15,18). Macrophages loaded with excessive amounts of glucocerebroside may not be able to respond to microbiological pathogens as effectively as normal cells, resulting in an attenuated anti-bacterial response (Citation13,14).
The activity of chitotriosidase, a macrophage enzyme that is believed to play a role in protection against fungal infection, is significantly elevated in untreated GD (Citation3). Berger et al. found that a GD patient with invasive Candida albicans infection displayed decreased, although still high, activity of plasma chitotriosidase (Citation17). The authors speculated that this decrease may have caused a susceptibility to fungal infection.
According to published data, at least in some patients with GD1, administration of ERT has a potential to influence positively the course of pulmonary involvement (Citation19). Since ERT effectively decreases plasma glucocerebroside, which could ameliorate the adverse effects of stored glucocerebroside on proper macrophage activity, ERT should be considered as ancillary therapy in patients with GD associated with recurrent or chronic infections (Citation14,20).
Of note, increased susceptibility to severe opportunistic infections in GD such as invasive aspergillosis and mycobacterial infections has never been reported before. The present case indicates that pulmonary involvement documented as lung fibrosis can result in both an increased susceptibility to opportunistic infections as well as subsequent ineffective eradication of Aspergillus fumigatus and Mycobacterium avium from the lungs of the patients with untreated GD1. An intriguing question arises from the present case about the impact of malfunctioning macrophages and whether ERT could play a role in the restoration of their function. Unfortunately, the patient has refused ERT (or any other form of GD therapy).
Interestingly, numerous large macrophages with a striated cytoplasm resembling Gaucher cells (so-called pseudo-Gaucher cells) have been described in patients with mycobacterial infections, often with M. avium (Citation21). In the light of the present case, it is important to keep in mind in such cases a possibility of undiagnosed Gaucher disease associated with pulmonary infection.
To the best of our knowledge, this paper is the first published report on the association of GD1 with pulmonary aspergillosis and mycobacterial infection. Our case indicates the importance of careful differential diagnostics in patients with recurrent pulmonary aspergillosis as well as mycobacterial infections, since it could be reasonable to exclude GD in subjects in whom macrophages resembling GCs can be identified and/or those who do not respond adequately to antimicrobial therapy.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Zimran A. How I treat Gaucher disease. Blood. 2011;118:1463–71.
- Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29:567–83.
- Machaczka M, Hast R, Dahlman I, Lerner R, Klimkowska M, Engvall M, et al. Substrate reduction therapy with miglustat for type 1 Gaucher disease: a retrospective analysis from a single institution. Ups J Med Sci. 2012;117:28–34.
- Dreborg S, Erikson A, Hagberg B. Gaucher disease – Norrbottnian type. General clinical description. Eur J Pediatr. 1980;133:107–18.
- Myers B. Gaucher's disease of the lungs. Br Med J. 1937;2:8–10.
- Schneider EL, Epstein CJ, Kaback MJ, Brandes D. Severe pulmonary involvement in adult Gaucher's disease. Report of three cases and review of the literature. Am J Med. 1977;63:475–80.
- Kerem E, Elstein D, Abrahamov A, Bar Ziv Y, Hadas-Halpern I, Melzer E, et al. Pulmonary function abnormalities in type I Gaucher disease. Eur Resp J. 1996;9:340–5.
- Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebicek M, et al. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481–5.
- Hollak C, vom Dahl S, Aerts JM, Belmatoug N, Bembi B, Cohen Y, et al. Force majeure: therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher disease. Blood Cells Mol Dis. 2010;44:41–7.
- Machaczka M, Klimkowska M, Hägglund H. Unexpected cure from cutaneous leukocytoclastic vasculitis in a patient treated with N-butyldeoxynojirimycin (miglustat) for Gaucher disease. Adv Med Sci. 2012;57:169–73.
- Lee RE, Yousem SA. The frequency and type of lung involvement in patients with Gaucher's disease. Lab Invest. 1988;58:54A.
- Machaczka M, Lerner R, Klimkowska M, Hägglund H. Treatment of multiple myeloma in patients with Gaucher disease. Am J Hematol. 2009;84:694–6.
- Liel Y, Rudich A, Nagauker-Shriker O, Yermiyahu T, Levy R. Monocyte dysfunction in patients with Gaucher disease: evidence for interference of glucocerebroside with superoxide generation. Blood. 1994;83:2646–53.
- Marodi L, Kaposzta R, Toth J, Laszlo A. Impaired microbicidal capacity of mononuclear phagocytes from patients with type I Gaucher disease: partial correction by enzyme replacement therapy. Blood. 1995;86:4645–9.
- Aker M, Zimran A, Abrahamov A, Horowitz M, Matzner Y. Abnormal neutrophil chemotaxis in Gaucher disease. Br J Haematol. 1993;83:187–91.
- Finkelstein R, Nachum Z, Reissman P, Reiss ND, Besser M, Trajber I, et al. Anaerobic osteomyelitis in patients with Gaucher's disease. Clin Infect Dis. 1992;15:771–3.
- Berger LA, Warwick R, Mehta A. Isolated Candida infection of the pterygoid muscles in a patient with Gaucher's disease. AJR Am J Roentgenol. 2001;176:1332–3.
- Burstein Y, Zakuth V, Rechavi G, Spirer Z. Abnormalities of cellular immunity and natural killer cells in Gaucher's disease. J Clin Lab Immunol. 1987;23:149–51.
- Goitein O, Elstein D, Abrahamov A, Hadas-Halpern I, Melzer E, Kerem E, et al. Lung involvement and enzyme replacement therapy in Gaucher's disease. QJM. 2001;94:407–15.
- Zimran A, Abrahamov A, Aker M, Matzner Y. Correction of neutrophil chemotaxis defect in patients with Gaucher disease by low-dose enzyme replacement therapy. Am J Hematol. 1993;43:69–71.
- Dunn P, Kuo MC, Sun CF. Pseudo-Gaucher cells in mycobacterial infection: a report of two cases. J Clin Pathol. 2005;58:1113–14.