
Abstract
Background. Sodium palmitate causes apoptosis of β-cells, and the anti-apoptotic protein Bcl-2 has been shown to counteract this event. However, the exact mechanisms that underlie palmitate-induced pancreatic β-cell apoptosis and through which pathway Bcl-2 executes the protective effect are still unclear.
Methods. A stable Bcl-2-overexpressing RINm5F cell clone (BMG) and its negative control (B45) were exposed to palmitate for up to 8 h, and cell viability, mitochondrial membrane potential (Δψm), reactive oxygen species (ROS) generation, endoplasmic reticulum (ER) stress, and NF-κB activation were studied in time course experiments.
Results. Palmitate exposure for 8 h resulted in increased cell death rates, and this event was partially counteracted by Bcl-2. Bcl-2 overexpression promoted in parallel also a delayed induction of GADD153/CHOP and a weaker phosphorylation of BimEL in palmitate-exposed cells. At earlier time points (2–4 h) palmitate exposure resulted in increased generation of ROS, a decrease in mitochondrial membrane potential (Δψm), and a modest increase in the phosphorylation of eIF2α and IRE1α. BMG cells produced similar amounts of ROS and displayed the same eIF2α and IRE1α phosphorylation rates as B45 cells. However, the palmitate-induced dissipation of Δψm was partially counteracted by Bcl-2. In addition, basal NF-κB activity was increased in BMG cells.
Conclusions. Our results indicate that Bcl-2 counteracts palmitate-induced β-cell death by maintaining mitochondrial membrane integrity and augmenting NF-κB activity, but not by affecting ROS production and ER stress.
Introduction
Type 2 diabetes is often associated with obesity and increased plasma free fatty acid (FFA) levels. When pancreatic insulin-producing cells are exposed to FFA for prolonged time periods, β-cell apoptosis can be observed (Citation1). The exact mechanisms that underlie palmitate-induced pancreatic β-cell apoptosis are still unclear. It may be that palmitate induces several different events, including endoplasmic reticulum (ER) stress (Citation2), mitochondrial perturbations (Citation3), generation of reactive oxygen species (Citation3), ceramide, and NO (Citation4), altered Ca2+ homeostasis (Citation5), and others that together or individually promote β-cell death.
The anti-apoptotic protein Bcl-2 has been found to be localized in the nucleus, mitochondria, and ER and may play an important role in the regulation of cell death induced by palmitate (Citation6). In non-β-cells, palmitate-induced apoptosis was associated with dissipation of the mitochondrial membrane potential (Δψm), two events that were counteracted by Bcl-2 overexpression (Citation6). FFAs may, at least in part, decrease Δψm by acting as uncouplers of mitochondrial respiration. The uncoupling effect of FFAs is associated with not only a decreased Δψm, but also increased respiration, enhanced reactive oxygen species (ROS) generation, lower ATP levels, and mitochondrial swelling (Citation3).
Besides acting at the mitochondrial level, Bcl-2 may also affect ER stress-induced apoptosis since ER-targeted Bcl-2 expression protected against tunicamycin-induced ER stress in renal tubular cells (Citation7), possibly via modulation of the permeability of the ER membrane to Ca2+ (Citation8). The ER of the pancreatic β-cell is highly active due to the engagement in synthesis and release of insulin. When ER homeostasis is lost, β-cells will initiate the unfolded protein response (UPR) to balance the protein synthesis demand against the ER protein-folding capacity. PKR-like ER kinase (PERK), inositol requiring ER-to-nucleus signal kinase 1 (IRE1), and activating transcription factor 6 (ATF6) are three ER membrane-located ER stress transducers activated by palmitate (Citation2). Active PERK phosphorylates the eukaryotic translation initiation factor (eIF2α), thereby generally attenuating protein translation and decreasing the functional demand on the ER. Meanwhile, phosphorylation of eIF2α induces activating transcription factor 4 (ATF4) expression. ATF4 is a transcription factor that regulates expression of C/EBP homologous protein (CHOP), also named growth arrest and DNA damage-inducible protein 153 (GADD153). Active IRE1 splices X-box binding protein 1 (XBP1) mRNA. Spliced XBP1 (XBP1s) activates many UPR genes required for protein folding, maturation, and degradation. Active ATF6 augments the expression of ER chaperones, such as BiP/GRP78 and GRP94, to increase the protein folding capacity. If the UPR fails to alleviate the ER stress, cells undergo apoptosis, characterized by induction of CHOP and c-Jun N-terminal kinase (JNK), activation of caspase 12, down-regulation of Bcl-2, activation of Bax/Bak, Bim, NOXA, PUMA, and p53 (Citation9).
In view of the general uncertainty as to how FFAs kill β-cells, and that Bcl-2 intervenes in specific steps in FFA-induced cell death, we have delineated the early time course of palmitate-induced mitochondrial and ER stress events, and how Bcl-2 interferes in these pathways, with the aim to understand better the primary palmitate-induced signaling events leading to β-cell death.
Materials and methods
Materials
The chemicals were obtained from the following sources: RPMI 1640, fetal bovine serum, L-glutamine, benzylpenicillin, streptomycin, Hanks’ balanced salt solution, trypsin, carbonyl cyanide p-(trifluoromethoxy)phenyl-hydrazone (FCCP), bisbenzimide (Hoechst 33342), propidium iodide (PI), 5, 5′, 6, 6′-tetrachloro-1, 1′, 3, 3′-tetraethylbenzimidazol carbocyanine iodide (JC-1), sodium palmitate, dichlorofluorescein-diacetate (DCFH-DA), alkaline phosphatase (from bovine intestinal mucosa), and albumin bovine serum (fatty acid and endotoxin free) from Sigma-Aldrich, St. Louis, MO, USA; Bcl-2 and GADD153 (F-168) antibodies from Santa Cruz Biotechnology, Dallas, TX, USA; phospho-IRE1α from Affinity BioReagents, Golden, CO, USA; phospho-eIF2α, cleaved caspase 3, and Bim from Cell Signaling, Danvers, MA, USA; and a horseradish-peroxidase-labeled donkey anti-rabbit or anti-mouse antibody from GE Healthcare, Uppsala, Sweden. All other chemicals of analytical grade were obtained from E. Merck, Darmstadt, Germany.
Cell culture
RINm5F cells overexpressing Bcl-2 were generated previously (Citation10) by stable liposome-mediated transfection with an episomally maintained multicopy bovine papilloma virus (BPV)-derived BMG neo vector (Citation11) containing the mouse bcl-2 cDNA (Citation12) inserted downstream of a metallothionein promoter. Stable clones with increased Bcl-2 expression (BMG cells) were isolated, and RINm5F cells transfected with an empty BPV-derived neo-containing vector were used as control (B45 cells). The cell clones were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM L-glutamine and benzylpenicillin (100 U/mL), streptomycin (0.1 mg/mL) at 37°C in humidified air with 5% CO2.
FFA preparation and cell treatment
Palmitate was dissolved in 50% ethanol, heated to 45°C, and used at a 1:100 dilution. During FFA exposure, cells were cultured in the RPMI 1640 medium supplemented with 0.5 mM palmitate, 0.5% BSA or 1% BSA (FFA:BSA molar ratio of 6.6:1 and 3.3:1, respectively). The control condition contained the same amount of vehicle. All cultures were supplemented with 1% FBS.
Determination of cell viability
After exposure to 0.5 mM palmitate (0.5% BSA or 1% BSA), or 1 μg/mL carbonyl cyanide p-(trifluoromethoxy)phenyl-hydrazone (FCCP) (solubilized in 50% ethanol and used in a 1:100 dilution) for 8 h, BMG and B45 cells were vital stained with 20 μg/mL bisbenzimide and 10 μg/mL propidium iodide for 15 min at 37°C. Free-floating cells and cell attached to the culture dish were then washed separately with PBS, pooled and examined by inverted fluorescence microscopy (excitation at 365 nm for HO and at 546 nm for PI). In each experimental group 3000–5000 cells were examined, red fluorescence (propidium iodide) and blue fluorescence (bisbenzimide) were quantified using Adobe Photoshop software, and the ratio red over blue was calculated as a relative measure of cell viability.
Determination of the mitochondrial membrane potential (Δψm) by JC-1 fluorescence
The mitochondrial membrane potential was semiquantitatively determined using the fluorescent probe JC-1. BMG and B45 cells were incubated for 1, 2, 3, 4, 5, 6, 7, 8 h at 37°C with 0.5 mM palmitate (0.5% BSA). FCCP (1 μg/mL) was also used in the incubation as a positive control of mitochondrial uncoupling. During the last 30 min of incubation, each group of the cells was incubated in the same medium as described before with the additional supplementation of 4 μM JC-1 (solubilized in DMSO, diluted 1:100). After careful washings of the cells, the fluorescence was determined using the Kodak Image Station 4000 MM with excitation wavelength at 465 nm and emission wavelength at 530 nm (green, JC-1 monomers at depolarized membrane potential) and 585 nm (red, J-aggregates at hyperpolarized membrane potential), respectively. The fluorescence ratio of red over green was used to monitor changes in mitochondrial membrane potential (Citation13).
Determination of ROS by DCFH-DA fluorescence
The production of ROS was estimated using the oxidation-sensitive fluorescent probe DCFH-DA. BMG and B45 cells were cultured for 1, 2, 3 h at 37°C with 0.5 mM palmitate (0.5% BSA). At the end of incubation, 10 μM DCFH-DA was applied to each group of cells for 20 min, followed by flow cytometry analysis with BD FACSCalibur (BD Biosciences, San Jose, CA, USA). Fluorescence emission from DCF (green) was detected at a wavelength of 530 nm.
Immunoblot analysis
After incubation with palmitate for 1–8 h, BMG or B45 cells were washed in cold PBS and lysed on ice in SDS-sample buffer (2% SDS, 0.15 M Tris, pH 6.8, 10% glycerol, 5% β-mercaptoethanol, bromophenol blue, and 2 mM phenylmethylsulfonyl fluoride). The samples were boiled for 5 min and separated on 10% or 12% SDS-PAGE gels. Proteins were transferred to Hybond-P membranes (GE Healthcare). The membranes were pre-blocked in 2.5% BSA for 1 h and then incubated with Bcl-2 (1:200), cleaved caspase 3 (1:1000), GADD153 (1:200), phospho-IRE1α (1:1000), phospho-eIF2α (1:1000), methyl-Lys221-p65, Bim (1:1000) antibodies overnight at 4°C. After secondary antibody incubation, the bound antibodies were visualized with the Kodak Image Station 4000 MM using ECL plus (GE Healthcare). The band intensities were calculated using Kodak Molecular Imaging Software 4.5.1 SE. Total protein loading and transfer onto the membranes was visualized by amido black staining.
Statistical analysis
Data are presented as means ± SEM. Statistical significance between two experimental conditions was analyzed using paired Student’s t test. Statistical significance: *p < 0.05, #p < 0.01.
Results
Overexpression of Bcl-2 in RINm5F cells
To confirm the possibility that overexpression of Bcl-2 might increase resistance to palmitate-induced β-cells death and to investigate through which mechanism Bcl-2 overexpression might execute its protective effect, a bcl-2-transfected insulin-producing rat pancreatic RINm5F cell line BMG was used in subsequent experiments (Citation10). BMG cells came from the stable clones of RINm5F cells overexpressing Bcl-2 protein 3–4-fold, as assessed by Western blot analysis (). B45 cells, which were transfected with an empty BPV-derived neo-containing vector and expressed low levels of Bcl-2, were used as control.
Figure 1. Expression of Bcl-2 in neo (B45) and bcl-2 (BMG)-transfected RINm5F cell clones and effects of palmitate and FCCP on B45 and BMG-transfected cell viability. A: Expression of Bcl-2 in BMG and B45 cell clones. B: Effects of palmitate and FCCP on B45 and BMG-transfected cell viability. RIN cell clones were incubated with 0.5 mM palmitate (0.5% BSA or 1% BSA + 1% FBS) or 1 μg/mL FCCP for 8 h. Results are means ± SEM for five separate experiments. * denotes p < 0.05 using paired Student’s t test when comparing versus corresponding control. C: One representative immunoblot showing Bcl-2 expression during the 8-h incubation with 0.5 mM palmitate (0.5% BSA). D: One representative immunoblot showing cleaved caspase 3 levels from five experiments. E: Mean optical density measurements of the immunoblots of cleaved caspase 3. The results are expressed as percentages of the control (B45 cells; Time zero) and shown as means ± SEM for five separate experiments. * denotes p < 0.05 using paired Student’s t test.
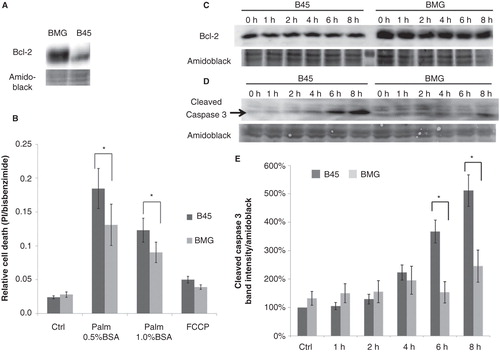
Palmitate-induced cell death was partially counteracted by Bcl-2 overexpression
To investigate whether Bcl-2 protects against saturated FFA-induced cell death, B45 and BMG cells were incubated with 0.5 mM palmitate complexed with 0.5% BSA or 1% BSA (FFA:BSA: molar ratio of 6.6:1 and 3.3:1, respectively) for 8 h. Relative measurements of cell death rate given by bisbenzimide and propidium iodide staining showed that 0.5 mM palmitate complexed with both 0.5% BSA or 1% BSA caused increased cell death. Palmitate:BSA at the ratio of 3.3:1 induced less cell death than the ratio of 6.6:1, which might be due to the higher toxicity of unbound free fatty acid. Bcl-2 overexpression promoted a partial protection against both 0.5 mM palmitate (0.5% BSA) (p = 0.025) and 0.5 mM palmitate (1% BSA) treatments (p = 0.029) (). Bcl-2 overexpression tended to protect against the uncoupler FCCP, but this did not reach statistical significance. The overexpression of Bcl-2 was maintained through the 8-h incubation with 0.5 mM palmitate complexed with 0.5% BSA (). The Bcl-2 overexpression-induced partial protection against 0.5 mM palmitate (0.5% BSA) was further confirmed by analysis of cleaved caspase 3 activation ().
Palmitate-induced GADD153/CHOP induction was delayed by Bcl-2 overexpression
As an important event of palmitate-induced β-cell death, levels of the transcription factor GADD153/CHOP were analyzed at an interval of 2 h during the 8 h of palmitate exposure. The induction of GADD153/CHOP protein levels, which occurred after 6 h of palmitate exposure, was markedly delayed or counteracted by Bcl-2 ().
Figure 2. Effects of palmitate on GADD153 (CHOP) expression in B45 and BMG cells. RIN cell clones were incubated with 0.5 mM palmitate (0.5% BSA) for 8 h. A: Mean optical density measurements of the immunoblots of CHOP. Protein values were normalized to amido black staining of total protein. The results are expressed as percentages of the control (B45 cells; Time zero) and shown as means ± SEM. * denotes p < 0.05 using paired Student’s t test when comparing protein induction in BMG cells with corresponding B45 cells. B: One representative immunoblot of CHOP expression from five experiments is shown.
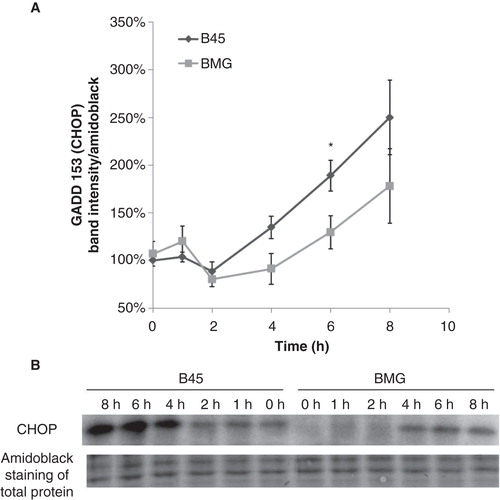
Palmitate induced BimEL phosphorylation, which was attenuated by Bcl-2 overexpression
Palmitate has been shown to induce the proapoptotic BH3-only protein Bcl-2-like 11 (Bim) up-regulation (Citation14), and Bim activity may be controlled by several posttranscriptional phosphorylation events at multiple serine and threonine residues (Citation15). For instance, BimEL, the longest splice form of Bim, can be phosphorylated by c-Jun N-terminal kinase (JNK) or MAPK-p38 at serine 65 to increase its proapoptotic activity (Citation16,17). Using an anti-Bim antibody, we observed two bands of BimEL (). The upper band is probably the phosphorylated form of BimEL as verified by phosphatase treatment (). When the ratio of BimEL upper (phosphorylated) to lower (non-phosphorylated) bands was calculated, a significant decrease of BimEL phosphorylation was observed in BMG cells at 4 and 8 h of palmitate incubation ().
Figure 3. Effects of palmitate on ratio of BimEL upper to lower band intensity in B45 and BMG cells. Cell clones were incubated with cytokines (IL-1β 20 ng/mL and IFN-γ 20 ng/mL) for 1 h or 0.5 mM palmitate (0.5% BSA) for 2–8 h. After the treatments, the cells were lysed and analyzed by immunoblotting with anti-Bim antibody. BimEL is the longest splice form (23 kDa) of Bim. Two bands of BimEL were observed, and the upper band is probably the phosphorylated form of BimEL as verified by phosphatase treatment. A: Ratio of BimEL upper (phosphorylated) to lower (non-phosphorylated) bands. The results are expressed as percentages of the control (B45 cells; Time zero) and shown as means ± SEM. * denotes p < 0.05 and # denotes p < 0.01 when comparing BMG cells with corresponding B45 cells. B: One representative immunoblot of BimEL phosphorylation from four experiments is shown. C: One representative immunoblot of phosphatase treatment.
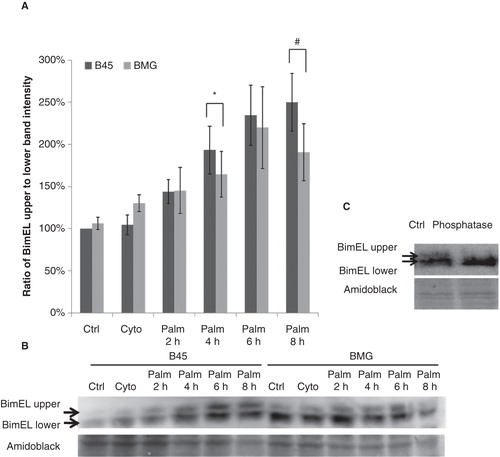
Palmitate induced a gradual decrease of mitochondrial membrane potential (Δψm), which was counteracted by Bcl-2
Partial mitochondrial uncoupling is induced by palmitate in pancreatic islets (Citation3), and Bcl-2 overexpression can stabilize the Δψm in cytokine-treated RINm5F cells (Citation18). We therefore investigated whether Bcl-2 overexpression affected palmitate-induced alterations in JC-1 fluorescence. A 60 min exposure to palmitate promoted a small increase in Δψm in both B45 and BMG cells (), possibly due to an increase in mitochondrial fatty acid oxidation. However, at 3–8 h, the Δψm was instead gradually decreased in response to palmitate. Bcl-2 overexpression counteracted the palmitate-induced loss of Δψm at 3 and 8 h of palmitate exposure.
Figure 4. A: Effects of palmitate on JC-1 fluorescence in B45 and BMG cells. BMG and B45 cells were cultured for 1–8 h with 0.5 mM palmitate, 0.5% BSA. Data are expressed as the 585/530 nm fluorescence ratio normalized to the control of each time point. Results are means ± SEM for three separate experiments. # denotes p < 0.01 using paired Student’s t test when comparing uncoupling rate of BMG cells with corresponding B45 cells. The basal JC-1 fluorescence ratio at time zero is B45: 100 ± 5% and BMG: 86 ± 22%. B: Effects of palmitate on DCF fluorescence in B45 and BMG cells. BMG and B45 cells were cultured for 1, 2, or 3 h with 0.5 mM palmitate (0.5% BSA). Data are expressed as 530 nm fluorescence normalized to the control of each time point. Results are means ± SEM for three separate experiments. The basal signal of DCF fluorescence at time zero is B45: 100 ± 8%, and BMG: 97 ± 8%.
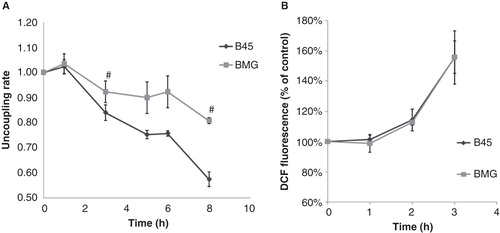
Bcl-2 overexpression failed to counteract palmitate-induced ROS generation
The magnitude of the mitochondrial membrane potential is known to influence rates of mitochondrial ROS production (Citation19). We therefore analyzed ROS levels in response to palmitate and Bcl-2 overexpression. Using the hydrogen peroxide and the hydroxyl radical sensor DCFH-DA, we observed increased ROS generation in response to a 2 h palmitate exposure and that this effect was even stronger at 3 h in both B45 and BMG cells (). Bcl-2 overexpression failed to counteract palmitate-induced ROS generation at all time points.
Effects of Bcl-2 overexpression on palmitate-induced ER stress signaling
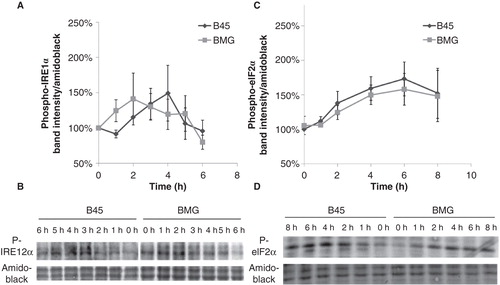
The Bcl-2 protein is known to modulate ER stress responses (Citation20). We therefore investigated the initiation of palmitate-induced ER stress in B45 and BMG cells. The activation of two ER stress sensors eIF2α and IRE1α was analyzed by immunoblotting with phospho-specific antibodies at an interval of 1 or 2 h during the palmitate exposure. We observed that the phosphorylation of IRE1α was only weakly increased by palmitate and that this event was not affected by Bcl-2 overexpression (). The phosphorylation of eIF2α was also not significantly affected by Bcl-2 overexpression ().
Figure 5. Effects of palmitate on phospho-IRE1α and phospho-eIF2α expression in B45 and BMG cells. RIN cell clones were incubated with 0.5 mM palmitate (0.5% BSA) for 6 or 8 h. Western blots were quantified, and protein values were normalized to amido black staining of total protein. Mean optical density measurements of the immunoblots for phospho-IRE1α (A) and phospho-eIF2α (C). The results are expressed as percentages of the control (B45 cells; Time zero) and shown as means ± SEM. B: One representative immunoblot of phospho-IRE1α expression from five experiments is shown. D: One representative immunoblot of phospho-eIF2α expression from six experiments is shown.
Effects of palmitate on Lys221-p65 methylation in B45 and BMG cells
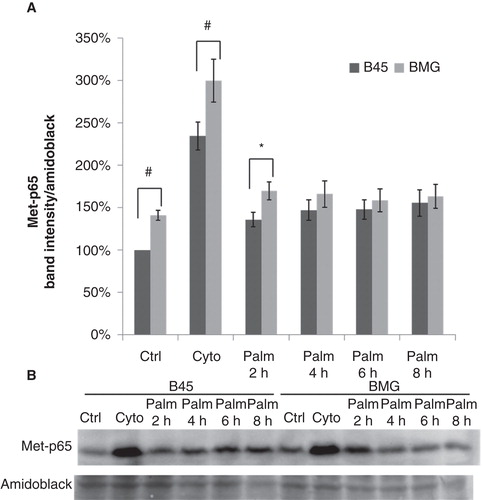
Recently, palmitate has been shown to induce NF-κB activation in human islets (Citation21) and a mouse β-cell line (Citation22). The activation of most p65-dependent genes has been reported to rely on lysine methylation of the p65 subunit (Citation23,24). Therefore we performed a time course study of Lys221-p65 methylation to monitor palmitate-induced NF-κB activation in B45 and BMG cells. Cytokine incubation, IL-1β (20 ng/mL) and IFN-γ (20 ng/mL) for 1 h, was used here as a positive control for NF-κB activation. Palmitate promoted in B45 cells, but not in BMG cells, a modest but significant increase in p65 methylation at 2, 4, 6, and 8 h (p < 0.05 using paired Student’s t test when comparing to time zero). A significant increase in BMG cell p65 methylation was observed at basal conditions and after 2 h of palmitate exposure when compared to B45 cells, which indicated an enhanced NF-κB activation through Bcl-2 overexpression. The augmented NF-κB pathway in BMG cells was still capable of being further activated as we observed higher methylation rates of p65 when exposed to cytokines for 1 h ().
Figure 6. Effects of palmitate on Lys221-p65 methylation in B45 and BMG cells. Cell clones were incubated with cytokines (IL-1β 20 ng/mL and IFN-γ 20 ng/mL) for 1 h or 0.5 mM palmitate (0.5% BSA) for 2–8 h. After the treatments, the cells were lysed and analyzed by immunoblotting with anti-Methyl-Lys221-p65 antibody (Citation23). Immunoblots were quantified, and protein values were normalized to amido black staining of total protein. A: Cytokines and palmitate induced methylation of p65. The results are expressed as percentages of the control (B45 cells; Time zero) and shown as means ± SEM. * denotes p < 0.05 and # denotes p < 0.01 using paired Student’s t test when comparing BMG cells with corresponding B45 cells. B: One representative immunoblot of Lys221-p65 methylation from seven experiments is shown.
Discussion
In this study we investigated the impact of Bcl-2 on early mitochondrial and ER events induced by sodium palmitate in insulin-producing RINm5F cells. Because β-cell levels of Bcl-2 decrease in response to prolonged exposure periods of high palmitate concentrations (Citation4) and because factors that protect against lipotoxicity, i.e. GLP-1 and hepatocyte growth factor, increase Bcl-2 expression (Citation25,26), it is likely that the anti-apoptotic Bcl-2 protein mediates a protective action against FFA-induced β-cell dysfunction and death. Indeed, we observed partial resistance against palmitate-induced cell death in cells overexpressing Bcl-2. However, the mechanisms by which Bcl-2 promotes this beneficial effect are not well characterized. Bcl-2 is known primarily to counteract proapoptotic mitochondrial events leading to a loss of Δψm, mitochondrial permeability transition (MPT), and release of cytochrome c. Moreover, Bcl-2 is also found at the ER where it reduces ER stress-mediated apoptosis, possibly via counteraction of proapoptotic Bax/Bak and modulation of ER calcium homeostasis (Citation20). To address these possibilities, we investigated early palmitate-induced alterations in mitochondrial membrane potential and observed that Bcl-2 overexpression mediated protection against the gradual decrease in Δψm starting at 3 h of palmitate exposure. Palmitate-induced loss of Δψm was first reported by us in 1999 (Citation3), and, more recently, it has been observed that palmitate, depending on dose and time of exposure, promotes both limited MPT opening, associated with impaired insulin secretion, and full MPT opening, associated with apoptosis (Citation27). Since Bcl-2 overexpression protected both at an early (3 h) and at a late time point (8 h) against palmitate-induced MPT, it is possible that Bcl-2 acts via inhibition of both limited and full MPT pore opening.
Sodium palmitate promotes β-cell production of ROS (Citation3), and antioxidants protect, at least in part, against FFA-induced β-cell dysfunction in vitro and in vivo (Citation28). In the present investigation ROS production was stimulated already at 2 h after addition of palmitate, and Bcl-2-overexpressing cells produced similar amounts of ROS as control cells. The source of palmitate-induced oxygen free radicals is not known but may be of mitochondrial (Citation29) and/or plasma membrane (NADPH-oxidase) (Citation30) origin. In either case, our results suggest that palmitate-induced ROS production occurs prior and independently of the gradual decrease in mitochondrial Δψm, indicating that the loss of Δψm may have resulted from the increased ROS production. Interestingly, it has been observed that Bcl-2 overexpression protects against AMPK-induced β-cell apoptosis without affecting production of oxygen radicals (Citation31). Furthermore, ROS are recognized activators of cytochrome c release and MPT (Citation32). Thus, it is possible that palmitate-induced ROS production causes opening of mitochondrial pores, and that this latter event is counteracted by Bcl-2.
We also report that Bcl-2 overexpression did not affect early palmitate-induced eIF2α and IRE1α phosphorylation events. This argues against a protective role of Bcl-2 at the β-cell ER membrane and instead for the notion that ER stress occurs as a consequence of augmented ROS production, or some other upstream event. The exact role of palmitate-induced ER stress in β-cell death has presently not been investigated, but the finding that BMG cells were partially protected against palmitate-induced cell death, but not against early ER stress events, diminishes the putative importance of the ER stress pathway in β-cell apoptosis.
GADD153/CHOP is thought to play a direct role in palmitate-induced β-cell apoptosis as siRNA-mediated knockdown of GADD153/CHOP has been observed to lower apoptosis rates (Citation9). We observed here that Bcl-2 overexpression delayed or counteracted palmitate-induced GADD153/CHOP induction at 6–8 h. This is a somewhat surprising finding because increased eIF2α phosphorylation is generally associated with subsequent expression of GADD153/CHOP (Citation9). However, it may also be that the palmitate-induced expression of GADD153/CHOP is to a large extent controlled by non-ER stress-related pathways. For example, it has long been known that ceramide, Fas, and JNK all promote GADD153/CHOP induction (Citation33,34). Furthermore, it has been reported that the anti-apoptotic transcription factor NF-κB acts as a repressor of the GADD153 gene (Citation35). Thus, as we observed enhanced NF-κB signaling in BMG cells, both at basal conditions and when treated with palmitate for 2 h, it is possible that Bcl-2 attenuates GADD153/CHOP induction, at least in part, by maintaining a higher activity of NF-κB, and not by modulating ER stress events.
Bim has also been shown to be involved in palmitate-induced β-cell apoptosis (Citation14), and we observed increased Bim phosphorylation in response to palmitate at 4–8 h. It appears that JNK phosphorylates Bim at amino acid ser65 and that this event is associated with increased apoptosis (Citation36). As overexpression of Bcl-2 has been shown to suppress JNK activation (Citation37), it is possible that palmitate-induced BimEL phosphorylation is mediated, at least in part, by JNK activation. In similarity with GADD153/CHOP as discussed above, Bcl-2 may suppress Bim activation in β-cells by maintaining mitochondrial integrity and/or up-regulating NF-κB activity.
In summary, we propose a hypothetical model in which palmitate induces the following chain of events: 1) increased ROS production (2 h); 2) decreased mitochondrial membrane potential and increased ER stress (3–4 h); 3) augmented expression of GADD153/CHOP and increased Bim phosphorylation (4–8 h); and 4) cell death (8 h). In addition, our results are compatible with the view that Bcl-2 protects β-cells against palmitate by maintaining mitochondrial membrane integrity, and not by affecting early ROS production. Thus, mitochondria may play a pivotal role in the control of β-cell survival and function in type 2 diabetes.
Acknowledgements
This work was supported in part by the Swedish Diabetes Association, the family Ernfors Fund, and the Novo-Nordisk Foundation.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes. 2001;50:1771–7.
- Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, Wakeham MC, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci. 2008;121:2308–18.
- Carlsson C, Borg LA, Welsh N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology. 1999;140:3422–8.
- Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51:1437–42.
- Gwiazda KS, Yang TL, Lin Y, Johnson JD. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab. 2009;296:E690–701.
- de Pablo MA, Susin SA, Jacotot E, Larochette N, Costantini P, Ravagnan L, et al. Palmitate induces apoptosis via a direct effect on mitochondria. Apoptosis. 1999;4:81–7.
- Bhatt K, Feng L, Pabla N, Liu K, Smith S, Dong Z. Effects of targeted Bcl-2 expression in mitochondria or endoplasmic reticulum on renal tubular cell apoptosis. Am J Physiol Renal Physiol. 2008;294:F499–507.
- Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci USA. 1994;91:6569–73.
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61.
- Saldeen J. Cytokines induce both necrosis and apoptosis via a common Bcl-2-inhibitable pathway in rat insulin-producing cells. Endocrinology. 2000;141:2003–10.
- Karasuyama H, Melchers F. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4 or 5, using modified cDNA expression vectors. Eur J Immunol. 1988;18:97–104.
- Deng G, Podack ER. Suppression of apoptosis in a cytotoxic T-cell line by interleukin 2-mediated gene transcription and deregulated expression of the protooncogene bcl-2. Proc Natl Acad Sci USA. 1993;90:2189–93.
- Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 1991;88:3671–5.
- Akazawa Y, Cazanave S, Mott JL, Elmi N, Bronk SF, Kohno S, et al. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J Hepatol. 2010;52:586–93.
- Hubner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. 2008;30:415–25.
- Becker EB, Howell J, Kodama Y, Barker PA, Bonni A. Characterization of the c-Jun N-terminal kinase-BimEL signaling pathway in neuronal apoptosis. J Neurosci. 2004;24:8762–70.
- Cai B, Chang SH, Becker EB, Bonni A, Xia Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J Biol Chem. 2006;281:25215–22.
- Barbu A, Welsh N, Saldeen J. Cytokine-induced apoptosis and necrosis are preceded by disruption of the mitochondrial membrane potential (Deltapsi(m)) in pancreatic RINm5F cells: prevention by Bcl-2. Mol Cell Endocrinol. 2002;190:75–82.
- Newsholme P, Haber EP, Hirabara SM, Rebelato EL, Procopio J, Morgan D, et al. Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol. 2007;583:9–24.
- Hetz CA. ER stress signaling and the BCL-2 family of proteins: from adaptation to irreversible cellular damage. Antioxid Redox Signal. 2007;9:2345–55.
- Igoillo-Esteve M, Marselli L, Cunha DA, Ladrière L, Ortis F, Grieco FA, et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia. 2010;53:1395–405.
- Choi HJ, Hwang S, Lee SH, Lee YR, Shin J, Park KS, et al. Genome-wide identification of palmitate-regulated immediate early genes and target genes in pancreatic beta-cells reveals a central role of NF-kappaB. Mol Biol Rep. 2012;39:6781–9.
- Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M, et al. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci USA. 2010;107:46–51.
- Mokhtari D, Li T, Lu T, Welsh N. Effects of imatinib mesylate (Gleevec) on human islet NF-kappaB activation and chemokine production in vitro. PLoS One. 2011;6:e24831.
- Buteau J, El-Assaad W, Rhodes CJ, Rosenberg L, Joly E, Prentki M. Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia. 2004;47:806–15.
- Santangelo C, Matarrese P, Masella R, Di Carlo MC, Di Lillo A, Scazzocchio B, et al. Hepatocyte growth factor protects rat RINm5F cell line against free fatty acid-induced apoptosis by counteracting oxidative stress. J Mol Endocrinol. 2007;38:147–58.
- Koshkin V, Dai FF, Robson-Doucette CA, Chan CB, Wheeler MB. Limited mitochondrial permeabilization is an early manifestation of palmitate-induced lipotoxicity in pancreatic beta-cells. J Biol Chem. 2008;283:7936–48.
- Oprescu AI, Bikopoulos G, Naassan A, Allister EM, Tang C, Park E, et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes. 2007;56:2927–37.
- Maestre I, Jordan J, Calvo S, Reig JA, Ceña V, Soria B, et al. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology. 2003;144:335–45.
- Michalska M, Wolf G, Walther R, Newsholme P. Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic beta-cell dysfunction. Biosci Rep. 2010;30:445–53.
- Cai Y, Martens GA, Hinke SA, Heimberg H, Pipeleers D, Van de Casteele M. Increased oxygen radical formation and mitochondrial dysfunction mediate beta cell apoptosis under conditions of AMP-activated protein kinase stimulation. Free Radic Biol Med. 2007;42:64–78.
- Costantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem. 1996;271:6746–51.
- Kultz D, Madhany S, Burg MB. Hyperosmolality causes growth arrest of murine kidney cells. Induction of GADD45 and GADD153 by osmosensing via stress-activated protein kinase 2. J Biol Chem. 1998;273:13645–51.
- Brenner B, Koppenhoefer U, Weinstock C, Linderkamp O, Lang F, Gulbins E. Fas- or ceramide-induced apoptosis is mediated by a Rac1-regulated activation of Jun N-terminal kinase/p38 kinases and GADD153. J Biol Chem. 1997;272:22173–81.
- Schapansky J, Olson K, Van Der Ploeg R, Glazner G. NF-kappaB activated by ER calcium release inhibits Abeta-mediated expression of CHOP protein: enhancement by AD-linked mutant presenilin 1. Exp Neurol. 2007;208:169–76.
- Santin I, Moore F, Colli ML, Gurzov EN, Marselli L, Marchetti P, et al. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic beta-cell apoptosis via regulation of the BH3-only protein Bim. Diabetes. 2011;60:3279–88.
- Koo MS, Kwo YG, Park JH, Choi WJ, Billiar TR, Kim YM. Signaling and function of caspase and c-jun N-terminal kinase in cisplatin-induced apoptosis. Mol Cells. 2002;13:194–201.