
Abstract
The platelet-derived growth factor (PDGF) family of mitogens exerts vital functions during embryonal development, e.g. in the central nervous system, where PDGF drives the proliferation of oligodendrocyte precursors. PDGF and PDGF receptors are co-expressed in human glioblastoma (GBM). Whether an aberrant activation of the PDGF receptor pathway is a driving force in glioma development has remained an open question. In experimental animals, overexpression of PDGF has convincingly been shown to induce tumors, both in wild-type animals (marmoset, rat, mouse) and in mice with targeted deletions of suppressor genes, e.g. Tp53 or Ink4A. Targeting the PDGF receptor in tumor-bearing mice leads to growth inhibition and reversion of the transformed phenotype. Findings of PDGF receptor amplification or mutations in human GBM are strong indicators of a causative role of the PDGF receptor pathway. However, clinical trials using PDGF receptor antagonists have been disappointing. In conclusion, a PDGF receptor profile may be a biomarker for a subgroup of GBM originating from a PDGF receptor-responsive cell. Although compelling experimental and clinical evidence supports the notion that the PDGF receptor pathway is a driver in GBM, formal proof is still missing.
Introduction
The widely cited articles by Hanahan and Weinberg on the hallmarks of cancer (Citation1) have highlighted the importance of self-sufficiency in growth factor receptor signaling, mediated by aberrant ligand-induced activation, constitutively activated and mutated receptors, or aberrant activation of downstream signals. The idea that cancer cells are growth-stimulated by autocrine growth factors has been around for quite a while, even before the term ‘autocrine’ was used to describe the phenomenon (Citation2). For instance, Howard Temin referred to it in his pioneering work on the mechanism of RNA tumor virus-induced cell transformation (Citation3,4). The field was tremendously boosted by the discovery that the transforming v-sis gene of simian sarcoma virus (SSV) is a retroviral version of the cellular platelet-derived growth factor (PDGF) B-chain gene (Citation5,6) and that the transforming activity of the gene is mediated by autocrine PDGF-BB (Citation7).
In my own search for suitable sources for novel growth factors, I was encouraged by the early studies by Temin and others to use serum-free conditioned culture media from human established cancer cell lines. The human osteosarcoma cell line U-2 OS, established by Jan Pontén (Citation8), turned out positive as it was found to produce growth-promoting factor(s) which could be partially purified (Citation9). Further purification and structural and functional analyses revealed a striking resemblance to PDGF (Citation10). Subsequently, the osteosarcoma cell-derived growth factor was shown to be identical to PDGF-AA (Citation11). Using the recently developed PDGF receptor-binding assay, I found that one of our clonal human glioblastoma (GBM) cell lines, U-343 MGa Cl2, produced large amounts of PDGF receptor-competing activity. Through subsequent studies it became evident that the cells produce significant amounts of PDGF (Citation12,13), mainly PDGF-AA (Citation14). This finding was the starting-point for a new research avenue, PDGF in human and experimental brain tumors (reviewed in (Citation15,16)).
PDGF and PDGF receptors
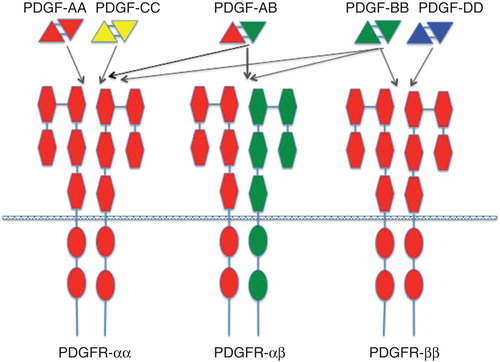
The platelet-derived growth factor (PDGF) family consists of covalently linked hetero- or homodimers of A-, B-, C-, and D-chains (PDGF-AA, -AB, -BB, -CC, and -DD) (for comprehensive reviews on PDGF and PDGF receptors, see (Citation17,18)). The ligands bind to and activate heterodimeric α and β tyrosine kinase receptors with ligand specificities outlined in . Several intracellular signaling pathways are engaged by PDGF receptor activation among which the Ras-MAPK, PI3K, and PLCγ pathways are most studied. Receptor activation culminates in cell cycle initiation, DNA synthesis, and mitosis. PDGF is not only involved in cell cycle regulation but also cell migration and chemotaxis (Citation19-21). PDGF and PDGF receptors fulfill important functions in development, e.g. in kidney, lung, intestine, and brain tissues, mainly through paracrine receptor activation (Citation17).
Figure 1. Binding specificities of PDGF ligands and receptors. The PDGF isoforms are made up as homo- or heterodimers of antiparallel subunits covalently linked by two S-S bonds. The ligands bind to and dimerize PDGF α and β receptors with specificities depicted in the figure. The receptors have extracellular domains with five immunoglobulin domains. Each receptor has an intracellular, split tyrosine kinase domain.
PDGF in glial development
A ground-breaking study by Richardson et al. (Citation22) showed that cultured oligodendrocyte precursor cells (OPCs) from the developing rat optic nerve are growth-stimulated by PDGF, present in astrocyte-conditioned medium. A subsequent study showed that the OPCs express PDGFRα (Citation23). These findings have been confirmed by in vivo experiments in mice, where PDGF-AA has been shown to drive the proliferation of OPCs (Citation24-26). OPCs are likely to be stimulated in vivo by paracrine PDGF released from astrocytes and neurons (Citation27). No such autocrine stimulation by normal OPCs during development has been demonstrated. Interestingly, PDGF-AA seems to be a rate-determining factor for OPC proliferation (Citation28), but not for myelination; it is only in the total absence of PDGF that myelination becomes negatively affected (Citation26). Continuous infusion of PDGF into the subventricular zone of the mouse brain induces precursor cell proliferation and hyperplastic, glioma-like lesions. These lesions are reversible and cannot be defined as full-blown malignancies (Citation29). In conclusion, in normal development PDGF drives the expansion of undifferentiated OPCs, and an excess of PDGF leads to an increase in OPC number beyond the physiological need. Apparently, there is no negative feedback mechanism operating in OPCs to control their PDGF-induced proliferation. Rather OPC proliferation seems to be controlled at the level of PDGF available for receptor activation.
PDGF-induced gliomas in animal models
Experiments performed by Friedrich Deinhardt and collaborators showed that SSV is tumorigenic in marmoset monkeys (Citation30,31). A complete record of Deinhardt’s experiments was unfortunately never published, but we were lucky to obtain the original paraffin-embedded material from Deinhardt’s experiments and could publish the histology of brain tumors induced by intracerebral SSV injections into newborn animals (Citation32). Interestingly, the virus caused lesions indistinguishable from human glioblastoma with all its hallmarks: cellular pleomorphism, necroses, and microvascular proliferations. The morphology was mixed, with oligodendroglioma-like, astrocytic, and anaplastic areas present. We now know that the transforming gene v-sis of SSV is a retroviral version of the PDGFB gene and that the transforming activity of v-sis is exerted by an autocrine PDGF receptor activation. Thus, an autocrine and uncontrolled PDGF receptor activation of target cells in primate brain in a retroviral context leads to the development of brain tumors displaying all the hallmarks of GBM. In this respect, SSV infection in marmosets has a more dramatic effect than continuous infusion in mouse brain (Citation29).
A number of studies have shown that intracerebral delivery of PDGF-encoding retroviral vectors in rodents leads to the development of brain tumors at high frequency. A recombinant Moloney murine leukemia virus encoding PDGFB (MoMuLV/PDGFB) injected together with replication-competent helper virus (MoMuLV) was found to induce glioma-like lesions in wild-type mice (Citation33). The tumors mostly resembled human glioblastoma with extensive necrotic areas. Some tumors displayed characteristics of primitive neuroectodermal tumors, and some were phenocopies of human low-grade oligodendrogliomas. Only a minority of the cells, identified as infiltrating astrocytes, were glial fibrillary acidic protein (GFAP)-positive, whereas a high proportion were nestin and PDGFRα-positive. These characteristics suggested that the tumors were derived from a PDGFRα-positive neural progenitor transformed by a persistent autocrine activation of the PDGF receptor signaling pathway. Eric Holland and collaborators have made important discoveries and significantly contributed to our understanding of PDGF-induced brain tumors in the mouse. A useful mouse model was developed, where PDGF is encoded by a recombinant avian RCAS virus and delivered intracerebrally in transgenic mice with a targeted expression of the virus receptor tv-a under the control of GFAP (GTv-a mice) or nestin (NTv-a mice) promoters (Citation34). RCAS-PDGFB generated brain tumors in both types of transgenic mice: oligodendrogliomas in NTv-a mice, and oligodendrogliomas or oligoastrocytomas in GTv-a mice. Tumor latency was shortened, and malignancy was enhanced in mice with homozygous deletion of the Ink4a-Arf locus, which is a common aberration in human GBM. Using the 2’,3’-cyclic nucleotide 3’-phosphodiesterase (CNPase) promoter to drive the expression of tv-a in transgenic mice (CTv-a mice), Lene Uhrbom’s research group was able to target PDGF expression to cells of the oligodendrocyte lineage. In the absence of other genetic aberrations, RCAS-PDGFB generated low-grade oligodendrogliomas (Citation35), whereas high-grade oligodendrogliomas as well as astrocytomas were induced in Cdkn2a null (p16Ink4a-/-, p19Arf-/-) mice (Citation36). Collectively, these data show that a single oncogene, i.e. Pdgfb, can cause tumors with different cellular origin (GFAP-positive astrocytes, nestin-positive neural stem cells, and CNPase-positive oligodendrocyte progenitors). The impact of these findings on the identification of the cell of origin of human glioblastoma has been discussed (Citation37). PDGFB has also been shown to induce GBM-like tumors when expressed as a transgene under the control of the Gfap promoter in Tp53 null, but not wild-type, mice (Citation38).
High expression of PDGFB, generated by deleting the translation-inhibiting sequence in the 5´ end of PDGFB cDNA inserted into the RCAS vector, was shown to give rise to high-grade tumors in wild-type mice, whereas the full-length sequence mostly induced low-grade oligodendrogliomas (Citation39). Tumor grade is thus dose-dependent. Low expression of PDGF causes low-grade tumors, and high expression yields high-grade tumors. In this context it is interesting to note that the v-sis gene lacks this particular sequence (Citation40), which may explain why SSV causes high-grade tumors in marmosets. The finding that an oncogene coding for a structurally and functionally normal growth factor can cause high-grade tumors is difficult to understand given the widely accepted concept that malignant tumors are caused by multiple genetic events and display a diversity of phenotypic aberrations (Citation1). This question was addressed decades ago (Citation41) but has not yet been fully answered. There are three main alternatives.
Mitogenic stimulation and unrestricted cell proliferation induced by excessive amounts of PDGF is accompanied by secondary genetic aberrations causing tumor progression. This idea was adopted in a recent review (Citation42), where replicative stress was supposed to be the driving force behind the mutagenic events. Direct evidence for this hypothesis, e.g. whole-genome sequence data for PDGF-induced tumors, is, however, lacking.
PDGF synthesis driven by retroviral vectors synergizes with insertional mutagenesis of complementary oncogenes or suppressor genes. Retroviral long terminal repeats are strong promoters and enhancers, which could activate cellular genes at a distance from the insertion sites. Further, integrated proviral DNA could theoretically interrupt genes and give rise to truncation or inactivation of the gene product. In order to probe this idea and search for genes that synergize with PDGF in gliomagenesis, Uhrbom et al. (Citation33) constructed a recombinant MoMuLV coding for a full-length PDGFB cDNA. The recombinant virus was combined with a replication-competent helper virus to increase the frequency of proviral insertions in order to render the genetic screen more effective. The experiment generated 108 brain tumors from which 647 insertion sites were obtained with as many as 66 common insertion sites (Citation43), reviewed in (Citation44). A number of the tagged genes are known to be involved in oncogenesis, e.g. Trp53, Eef1a1, Gli, Fos, and Ccnd, providing proof of concept for the genetic screen. The vast majority of tagged genes, however, had not previously been implicated in oncogenesis or PDGF signaling. One of these, Sox5, was subsequently found to be a suppressor of PDGF-induced brain tumors and cause cellular senescence (Citation45). Five insertions in the Nfix locus were found, the functional implication of which is still unknown and worth pursuing. NFIX has been shown to be a critical factor in development and to induce quiescence in neural stem cells (Citation46,47). Insertion into the Nfix locus may interfere with the differentiation program and thereby synergize with PDGF in tumor development. Whether insertional mutagenesis also operates in the RCAS-PDGF-induced brain tumors is an open question. The oncogenic potential of retroviruses was dramatically illustrated by the clinical trials on gene therapy in X-linked severe combined immunodeficiency (XSCID) patients a few years ago. Three out of 11 patients developed T cell tumors, all of which caused by proviral insertions in the LMO2 locus (Citation48,49).
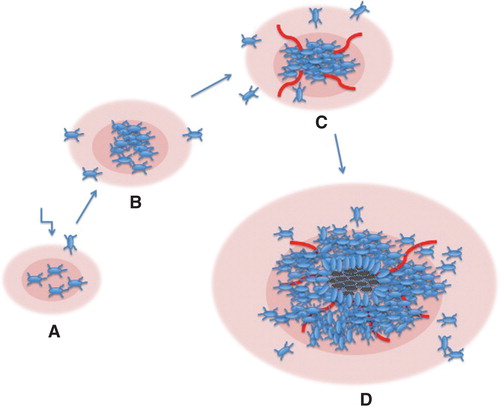
The generation of high-grade gliomas by PDGF overexpression is caused by an autocrine activation of transformed cells as well as recruitment of non-transformed cells, which are attracted to the tumor site by paracrine activation. In fact, PDGF virus-induced gliomas in the rat contain a large number of untransformed cells, which apparently have migrated and populated the tumor (Citation50). According to this model, PDGF is sufficient to drive tumor formation in the absence of other genetic aberrations. This model may seem unlikely and less attractive than those described above. The model presumes that an excessive, PDGF-induced, cell proliferation is not controlled by any feedback regulation. Interestingly, this may indeed be the case. In PDGF-transgenic mice, the number of PDGF-responsive oligodendrocyte precursors was proportional to the supply of PDGF and seemingly unsaturable (Citation51). In the case of a local production of autocrine/paracrine PDGF, the pool of stimulated cells may increase progressively to the point where hypoxia drives angiogenesis and where necroses may occur in severely hypoxic areas. Such a process may then yield tumors, which closely resemble glioblastoma multiforme (see for a schematic representation of this model).
Figure 2. Schematic model for the development of high-grade gliomas in experimental animals following infection with PDGF-encoding retroviruses. A. Cells are infected, produce PDGF, and start to proliferate in an uncontrolled fashion. B. The PDGF-transformed population expands, and neighboring cells are attracted by paracrine stimulation. C. Cell density has increased, and local hypoxia elicits an angiogenic response. D. The expanding population has grown beyond the supply of nutrients, and necroses appear. Pink areas denote PDGF released by the tumor cells.
PDGF and PDGF receptors in human glioma
Our observation that human glioma cells in culture produce PDGF became an incentive for studies of the expression of PDGF and PDGF receptors in human glioma. A rather coherent picture has emerged from these studies, pointing to a causative role for PDGF in gliomagenesis. PDGFA and PDGFα receptors are co-expressed in glioblastoma (Citation52) and oligodendroglioma (Citation53). The PDGFRα gene is amplified, mutated, or rearranged in a fraction of glioblastoma tumors (Citation54-60). These findings strongly suggest that genetic aberrations and overactivity of the PDGFα receptor signaling pathway are important events in the development of a subset of glioblastomas. This view is strengthened by transcriptome analysis of glioblastoma, which has indicated that the proneural subgroup of GBM is characterized by aberrations in the PDGF/PDGFR pathway (Citation60). Indeed, overactivity of the PDGFα receptor pathway may be an initiating event in human glioblastoma as a result of an increased PDGFA gene dosage generated by chromosomal non-disjunction by the occurrence of multiple copies of chromosome 7, which harbors the PDGF A-chain gene (Citation61).
Driver or biomarker?
In the discussion on the causative role of PDGF and PDGFR in the development of human brain tumors, we may be guided by an analogous discussion over a century ago regarding micro-organisms as etiologic agents in human disease. This subject was brought up by Henle and Koch and resulted in what became known as Koch’s postulates (reviewed in (Citation62)). Although Koch’s postulates cannot be directly translated into the etiology or pathogenesis of a non-viral cancer, such as glioblastoma multiforme, they can be adjusted as follows:
PDGF receptor signaling must be aberrantly activated in brain tumor cells. As has been summarized above, PDGF and PDGFR are expressed at high levels in most GBM. Since the cell of origin of human GBM is yet unknown, we cannot make any direct comparison between the levels of activation in the normal cell(s) of origin versus GBM cells. However, the PDGFRα gene is amplified in a subset of GBM, which strongly indicates that it confers a selective growth advantage and thus has a mechanistic role in the pathogenesis of the tumor. The recurrent findings of receptor mutations and rearrangements, albeit rare, are strong indicators of a mechanistic role, although we cannot formally rule out the possibility that these changes are passenger mutations and not drivers.
Aberrant activation of PDGF receptors in the cell of origin must cause GBM in vivo. As described above, studies in marmosets, rats, and mice have provided convincing evidence that overexpression of PDGF, mainly PDGFB but also PDGFA (Citation61), causes GBM in experimental animals. It needs to be said, however, that these findings in no way prove that similar mechanisms operate in human GBM.
Blocking the PDGFR signal transduction pathway in GBM should retard the growth of glioma cells in vivo. There are a few examples where PDGF antagonists have been shown to retard cell growth in GBM cultures (Citation63,64). However, clinical trials on the use of PDGF receptor kinase inhibitors have been disappointing (Citation65,66). These results are in striking contrast to studies on PDGFB-induced gliomas in mice, where low-molecular-weight PDGF receptor kinase inhibitors are efficacious in reverting the transformed phenotype and cause tumor growth retardation in vivo (Citation67). It is also notable that imatinib, a low-molecular-weight PDGFR inhibitor, is an effective drug in the treatment of other PDGF-driven tumors such as gastrointestinal stroma tumors with PDGFRα mutations and dermatofibrosarcoma tuberans with an activating PDGFB translocation (reviewed in (Citation68,69)). In the absence of in-depth pharmacokinetic and pharmacodynamic studies in GBM, we cannot draw firm conclusions regarding the potential of PDGF receptor antagonists in the treatment of GBM. However, the possibility remains that aberrant activation of the PDGFR signaling pathway is an initial event in the proneural and mesenchymal subgroups of GBM (Citation61) but is rendered redundant during tumor progression. This would point to a hit-and-run mechanism in contrast to the clinically more favorable situation of oncogene addiction (Citation70,71) where oncogene inactivation leads to the death of tumor cells but spares normal cells. A third possibility could be that PDGFR activation is a driver in a subset GBM in all stages but intervention of the pathway needs to be combined with an interference with other aberrations, e.g. in the PTEN, TP53, INK4A/ARF pathways.
In conclusion, a PDGF receptor profile, such as has been demonstrated in the proneural subgroup of GBM, is likely to be a marker of the cell of origin. Such tumors are probably derived from PDGFRα-positive cells. There is also considerable evidence that a constitutively active PDGFR pathway is a driver in a subset of GBM, although formal proof is still lacking.
Targeting the PDGFR, or other signaling pathways, in GBM has so far not been clinically successful. Maybe an unprejudiced search for GBM antagonists by high-throughput screening will be more fruitful than the ‘intelligent design’ of targeted drugs. A promising substance has recently been identified (Citation72). There is currently a vibrant activity in basic, translational, and clinical research on GBM as well as in cancer research in general. It is my personal belief that these research activities will bear fruit and translate into a better treatment of GBM, which until now has remained a tumor with a dismal prognosis.
Acknowledgements
My own work cited in the text has been generously supported by grants from the Swedish Cancer Society, the Swedish Research Council, and the Swedish Childhood Cancer Foundation.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
Additional information
Notes on contributors

Bengt Westermark
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
- Sporn MB, Todaro GJ. Autocrine secretion and malignant transformation of cells. N Engl J Med. 1980;303:878–80.
- Temin HM. Studies on carcinogenesis by avian sarcoma viruses. 3. The differential effect of serum and polyanions on multiplication of uninfected and converted cells. J Natl Cancer Inst. 1966;37:167–75.
- Temin HM. Control of multiplication of uninfected rat cells and rat cells converted by murine sarcoma virus. J Cell Physiol. 1970;75:107–19.
- Doolittle RF, Hunkapiller MW, Hood LE, Devare SG, Robbins KC, Aaronson SA, et al. Simian sarcoma virus onc gene, v-sis, is derived from the gene (or genes) encoding a platelet-derived growth factor. Science. 1983;221:275–7.
- Waterfield MD, Scrace GT, Whittle N, Stroobant P, Johnsson A, Wasteson A, et al. Platelet-derived growth factor is structurally related to the putative transforming protein p28sis of simian sarcoma virus. Nature. 1983;304:35–9.
- Johnsson A, Betsholtz C, Heldin CH, Westermark B. Antibodies against platelet-derived growth factor inhibit acute transformation by simian sarcoma virus. Nature. 1985;317:438–40.
- Ponten J, Saksela E. Two established in vitro cell lines from human mesenchymal tumours. Int J Cancer. 1967;2:434–47.
- Westermark B, Wasteson A. The response of cultured human normal glial cells to growth factors. Adv Metab Disord. 1975;8:85–100.
- Heldin CH, Westermark B, Wasteson A. Chemical and biological properties of a growth factor from human-cultured osteosarcoma cells: resemblance with platelet-derived growth factor. J Cell Physiol. 1980;105:235–46.
- Heldin CH, Johnsson A, Wennergren S, Wernstedt C, Betsholtz C, Westermark B. A human osteosarcoma cell line secretes a growth factor structurally related to a homodimer of PDGF A-chains. Nature. 1986;319:511–14.
- Nister M, Heldin CH, Wasteson A, Westermark B. A platelet-derived growth factor analog produced by a human clonal glioma cell line. Ann N Y Acad Sci. 1982;397:25–33.
- Nister M, Heldin CH, Wasteson A, Westermark B. A glioma-derived analog to platelet-derived growth factor: demonstration of receptor competing activity and immunological crossreactivity. Proc Natl Acad Sci USA. 1984;81:926–30.
- Hammacher A, Nister M, Westermark B, Heldin CH. A human glioma cell line secretes three structurally and functionally different dimeric forms of platelet-derived growth factor. Eur J Biochem. 1988;176:179–86.
- Nazarenko I, Hede SM, He X, Hedren A, Thompson J, Lindstrom MS, et al. PDGF and PDGF receptors in glioma. Ups J Med Sci. 2012;117:99–112.
- Westermark B. Glioblastoma–a moving target. Ups J Med Sci. 2012;117:251–6.
- Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312.
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–316.
- Deuel TF, Senior RM, Huang JS, Griffin GL. Chemotaxis of monocytes and neutrophils to platelet-derived growth factor. J Clin Invest. 1982;69:1046–9.
- Grotendorst GR, Chang T, Seppa HE, Kleinman HK, Martin GR. Platelet-derived growth factor is a chemoattractant for vascular smooth muscle cells. J Cell Physiol. 1982;113:261–6.
- Westermark B, Siegbahn A, Heldin CH, Claesson-Welsh L. B-type receptor for platelet-derived growth factor mediates a chemotactic response by means of ligand-induced activation of the receptor protein-tyrosine kinase. Proc Natl Acad Sci USA. 1990;87:128–32.
- Richardson WD, Pringle N, Mosley MJ, Westermark B, Dubois-Dalcq M. A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell. 1988;53:309–19.
- Pringle N, Collarini EJ, Mosley MJ, Heldin CH, Westermark B, Richardson WD. PDGF A chain homodimers drive proliferation of bipotential (O-2A) glial progenitor cells in the developing rat optic nerve. EMBO J. 1989;8:1049–56.
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, et al. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–82.
- Fruttiger M, Calver AR, Kruger WH, Mudhar HS, Michalovich D, Takakura N, et al. PDGF mediates a neuron-astrocyte interaction in the developing retina. Neuron. 1996;17:1117–31.
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, et al. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 1999;126:457–67.
- Fruttiger M, Calver AR, Richardson WD. Platelet-derived growth factor is constitutively secreted from neuronal cell bodies but not from axons. Curr Biol. 2000;10:1283–6.
- Woodruff RH, Fruttiger M, Richardson WD, Franklin RJ. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci. 2004;25:252–62.
- Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, et al. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–99.
- Deinhardt F. Biology of primate retroviruses. In Klein G, editor. Viral oncology. New York: Raven Press; 1980. p 357–98.
- Wolfe LG, Deinhardt F, Theilen GH, Rabin H, Kawakami T, Bustad LK. Induction of tumors in marmoset monkeys by simian sarcoma virus, type 1 (Lagothrix): a preliminary report. J Natl Cancer Inst. 1971;47:1115–20.
- Nister M, Westermark B. Mechanisms of altered growth control. Growth factors. In Bigner DD, McLendon RE, Bruner JM, editors. Russel and Rubinstein’s Pathology of tumors of the nervous system. 6th ed. London: Arnold; 1998. p 83–116.
- Uhrbom L, Hesselager G, Nister M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–9.
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–25.
- Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009;28:2266–75.
- Lindberg N, Jiang Y, Xie J, Bolouri H, Kastemar M, Olofsson T, et al. Oncogenic 1 signaling is dominant to cell of origin and dictates astrocytic or2 oligodendroglial tumor development from oligodendrocyte precursor cells. J Cell Sci. 2014; In press.
- Jiang Y, Uhrbom L. On the origin of glioma. Ups J Med Sci. 2012;117:113–21.
- Hede SM, Hansson I, Afink GB, Eriksson A, Nazarenko I, Andrae J, et al. GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia. 2009;57:1143–53.
- Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004;64:4783–9.
- Devare SG, Reddy EP, Robbins KC, Andersen PR, Tronick SR, Aaronson SA. Nucleotide sequence of the transforming gene of simian sarcoma virus. Proc Natl Acad Sci USA. 1982;79:3179–82.
- Westermark B, Johnsson A, Betsholtz C, Heldin CH. Biological properties of simian sarcoma virus and its oncogene product. In Hofschneider PH, Munk K, editors. Viruses in human tumors. Contributions to oncology. Vol. 24 Karger, Basel, München, Paris, London, New York, New Delhi, Singapore, Tokyo, Sydney; 1987. p 51–61.
- Lindberg N, Holland EC. PDGF in gliomas: more than just a growth factor? Ups J Med Sci. 2012;117:92–8.
- Johansson FK, Brodd J, Eklof C, Ferletta M, Hesselager G, Tiger CF, et al. Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci USA. 2004;101:11334–7.
- Johansson Swartling F. Identifying candidate genes involved in brain tumor formation. Ups J Med Sci. 2008;113:1–38.
- Tchougounova E, Jiang Y, Brasater D, Lindberg N, Kastemar M, Asplund A, et al. Sox5 can suppress platelet-derived growth factor B-induced glioma development in Ink4a-deficient mice through induction of acute cellular senescence. Oncogene. 2009;28:1537–48.
- Heng YH, McLeay RC, Harvey TJ, Smith AG, Barry G, Cato K, et al. NFIX regulates neural progenitor cell differentiation during hippocampal morphogenesis. Cereb Cortex. 2014;24:261–79.
- Martynoga B, Mateo JL, Zhou B, Andersen J, Achimastou A, Urban N, et al. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev. 2013;27:1769–86.
- Dave UP, Jenkins NA, Copeland NG. Gene therapy insertional mutagenesis insights. Science. 2004;303:333.
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–19.
- Assanah MC, Bruce JN, Suzuki SO, Chen A, Goldman JE, Canoll P. PDGF stimulates the massive expansion of glial progenitors in the neonatal forebrain. Glia. 2009;57:1835–47.
- van Heyningen P, Calver AR, Richardson WD. Control of progenitor cell number by mitogen supply and demand. Curr Biol. 2001;11:232–41.
- Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52:3213–19.
- Di Rocco F, Carroll RS, Zhang J, Black PM. Platelet-derived growth factor and its receptor expression in human oligodendrogliomas. Neurosurgery. 1998;42:341–6.
- Fleming TP, Saxena A, Clark WC, Robertson JT, Oldfield EH, Aaronson SA, et al. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992;52:4550–3.
- Kumabe T, Sohma Y, Kayama T, Yoshimoto T, Yamamoto T. Overexpression and amplification of alpha-PDGF receptor gene lacking exons coding for a portion of the extracellular region in a malignant glioma. Tohoku J Exp Med. 1992;168:265–9.
- Kumabe T, Sohma Y, Kayama T, Yoshimoto T, Yamamoto T. Amplification of alpha-platelet-derived growth factor receptor gene lacking an exon coding for a portion of the extracellular region in a primary brain tumor of glial origin. Oncogene. 1992;7:627–33.
- Martinho O, Longatto-Filho A, Lambros MB, Martins A, Pinheiro C, Silva A, et al. Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. Br J Cancer. 2009;101:973–82.
- Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M, et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010;24:2205–18.
- Rand V, Huang J, Stockwell T, Ferriera S, Buzko O, Levy S, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci USA. 2005;102:14344–9.
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300.
- Evans AS. Causation and disease: the Henle-Koch postulates revisited. Yale J Biol Med. 1976;49:175–95.
- Hagerstrand D, He X, Bradic Lindh M, Hoefs S, Hesselager G, Ostman A, et al. Identification of a SOX2-dependent subset of tumor- and sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro Oncol. 2011;13:1178–91.
- Vassbotn FS, Ostman A, Langeland N, Holmsen H, Westermark B, Heldin CH, et al. Activated platelet-derived growth factor autocrine pathway drives the transformed phenotype of a human glioblastoma cell line. J Cell Physiol. 1994;158:381–9.
- Dresemann G, Weller M, Rosenthal MA, Wedding U, Wagner W, Engel E, et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol. 2010;96:393–402.
- Reardon DA, Dresemann G, Taillibert S, Campone M, van den Bent M, Clement P, et al. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br J Cancer. 2009;101:1995–2004.
- Uhrbom L, Nerio E, Holland EC. Dissecting tumor maintenance requirements using bioluminescence imaging of cell proliferation in a mouse glioma model. Nat Med. 2004;10:1257–60.
- Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–43.
- Heldin CH. Autocrine PDGF stimulation in malignancies. Ups J Med Sci. 2012;117:83–91.
- Jonkers J, Berns A. Oncogene addiction: sometimes a temporary slavery. Cancer Cell. 2004;6:535–8.
- McCormick F. Cancer therapy based on oncogene addiction. J Surg Oncol. 2011;103:464–7.
- Kitambi SS, Toledo EM, Usoskin D, Wee S, Harisankar A, Svensson R, et al. Vulnerability of glioblastoma cells to catastrophic vacuolization and death induced by a small molecule. Cell. 2014;157:313–28.