Abstract
The pathophysiology of thromboembolism in atrial fibrillation (AF) is a multifactorial and complex process. Abnormalities of haemostasis, fibrinolysis, endothelium, and platelets have all been described in AF. This prothrombotic state observed in AF appears to be additive to the presence of clinical and echocardiography risk factors for thromboembolism. Nonetheless, the precise mechanistic pathway(s) leading to the prothrombotic state in AF remain to be elucidated. Of note, there are limited data on the influence of genetic polymorphisms in thromboembolic risk associated with AF. On the other hand, the response to coumarin derivatives depends on several factors, such as sex, age, diet, or interacting drugs.
Optimal anticoagulation control is usually hampered by significant interindividual variability in dose requirements for a given target level of anticoagulation. There is increasing evidence that interindividual sensitivity and side-effects to coumarinics may be largely determined genetically. Thus, genetic polymorphisms could explain the individual risk of developing an adverse drug reaction (bleeding) or drug inefficacy (thrombosis) with oral anticoagulation.
In this article, we provide an overview of the limited data about the possible influence of genetic polymorphisms on thromboembolic risk in AF, as well as the genetic influences on anticoagulant drug responsiveness.
Key messages
There is a lack of data in the literature to support the use of SNPs in risk stratification of stroke.
Currently genetic studies are not useful for the stratification of embolic risk in patients with atrial fibrillation, and thus its practical utility is limited.
Pharmacogenetics emerges as a relevant discipline that would help to find the most appropriate treatment to each patient, thus increasing the efficiency and potentially reducing side-effects of treatment.
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia which is associated with increased all-cause mortality, especially from stroke and thromboembolism (Citation1). The risk of thromboembolism is not homogeneous and is associated with several clinical and echocardiographic risk factors (Citation2). Several prominent risk stratification schemes, such as CHADS2 or Framingham risk score, have been developed to help distinguish those patients with AF who are at high risk from those with a risk sufficiently low that anticoagulation might not be beneficial, when considering the associated bleeding risks (Citation3,Citation4). Although many of the clinical variables overlap among the different risk schemes, there are differences in how specific variables are coded, combined, and weighted. These variations may lead to significant differences in whether patients are categorized as being at low or high risk for thromboembolism. Thus, accurate stratification of these patients should ideally target the use of warfarin for patients at highest risk of thromboembolism and reduce the exposure of low-risk patients to the complications of warfarin (Citation5). Abnormalities of haemostasis, fibrinolysis, endothelium, and platelets have all been described in AF, which may increase the risk of thromboembolism in this cardiac arrhythmia. This prothrombotic or hypercoagulable state observed in AF appears to be additive to the presence of clinical and echocardiographic risk factors for thromboembolism. Nonetheless, the precise mechanistic pathway(s) leading to the prothrombotic state in AF remain to be elucidated, although different pathophysiological systems have been hypothesized as possible mechanisms.
Whilst many different underlying mechanisms have been suggested to promote the prothrombotic state in AF (Citation6), the possibility arises that genetic polymorphisms may contribute. However, there are relatively limited data on the influence of genetic background on the stroke and thromboembolic risk associated with AF (Citation7). Only a few studies have linked genetic polymorphisms to the thromboembolic risk secondary to AF, with both positive and negative results (Citation8–11). On the other hand, the response to coumarin derivatives depends on several factors (Citation12), but there is increasing evidence that interindividual sensitivity and side-effects may be genetically determined (Citation13).
In this review article, we provide an overview on the influence of genetic polymorphisms on the prothrombotic state and thromboembolic risk of AF, as well as the variability in patients’ responses to oral anticoagulation. Because current risk schemes have limited overall ability to predict thromboembolism (Citation5), these polymorphisms may provide additional factors to be considered when stratifying the risk of stroke in subjects with AF, which would help target subjects most at risk of thromboembolic complications. Importantly, the knowledge of genetic background could potentially improve the safety and effectiveness of oral anticoagulant drugs.
Search strategy and selection criteria
Published data for this review were identified by searches of MEDLINE and reference lists from relevant articles. Searches were concentrated on the following key words: atrial fibrillation, hypercoagulable state, prothrombotic state, thrombogenesis, endothelial dysfunction, fibrinolytic function, inflammation, D-dimer, P-selectin, inflammatory markers, matrix remodelling, angiogenesis, vascular endothelial growth factor, tissue factor, genetic polymorphisms, thromboembolic risk, stroke, platelet polymorphisms, factor V Leiden, prothrombin G20210A, alpha-fibrinogen T312A, factor XIII Val34Leu, factor VII-323 decanucleotide D/I, beta-fibrinogen 455 G/A, C807T polymorphism, HPA, pharmacogenetic, coumarin derivatives, vitamin K antagonists, CYP2C9 variability, and VKORC1 variability. Representative studies for various reviews about hypercoagulable state and atrial fibrillation were also included. In order to avoid any confusion we here will refer to genes according to HGNC (HUGO [Human Genome Organization] Gene Nomenclature Committee) gene symbol and polymorphisms following HGVS (Human Genome Variation Society) names and rs references.
The hypercoagulable state in atrial fibrillation: a brief overview
Structural abnormalities (for example, dilated atria or poorly contracting dilated left ventricles), heart valve disease (for example, mitral stenosis), and congestive heart failure, which are clinical features commonly associated with stroke and thromboembolism in AF, contribute to the first two components of Virchow's Triad (that is, abnormalities of blood flow and vessels) (Citation14,Citation15).
Abnormalities of haemostasis, coagulation, platelets, and the endothelium are also present in AF, thus fulfilling the third component (‘abnormal blood constituents’) of Virchow's Triad (Citation16–18). These abnormalities of haemostasis and coagulation in AF have been associated with left atrial thrombosis and spontaneous echo contrast (Citation17). There are some data that the hypercoagulable state observed in AF may be additive to the presence of other risk factors for thromboembolism (Citation19–21).
Previous studies have mainly focused on various endothelial damage or dysfunction markers, prothrombotic factors, platelet activation markers, and fibrinolytic function (Citation17,Citation22–25). Indeed, haemostatic abnormalities have been found in many cross-sectional studies of patients with AF (Citation23,Citation26,Citation27). For example, Kumagai et al. first reported increased D-dimer concentrations, a marker for intravascular thrombogenesis, in AF irrespective of any underlying structural heart disease (Citation27). Von Willebrand factor (vWf), an index of endothelial damage/dysfunction, increases in chronic AF and has been related to risk factors, stroke risk strata, and prognosis (Citation28). The presence of platelet activation is recognized in chronic AF but may relate to associated vascular disease or risk factors rather than AF per se (Citation29). Finally, some studies have specifically investigated fibrinolytic function in AF (Citation25,Citation30–32). In other studies, significant correlations were found between tissue-type plasminogen activator (t-PA) antigen level and established endothelial markers (Citation33,Citation34).
There are several mechanism(s) underlying the prothrombotic state in AF. Abnormalities in inflammatory markers (Citation35,Citation36), structural remodelling of atria (matrix metalloproteinase system) (Citation37), platelet activation (Citation38), or abnormalities in growth factors and tissue factor (TF) levels (Citation39–42) have all been studied as systems implicated in the prothrombotic or hypercoagulable state of AF. However, the activation mechanisms of the coagulation and fibrinolytic systems, and therefore the prothrombotic state, are controlled to a substantial degree by genetic factors. Genes influencing activation of haemostasis are likely to be an important component of the overall thrombotic tendency (Citation43). Fewer studies have explored the role of genetic polymorphisms in the hypercoagulable state and the contribution to the thromboembolic risk seen in patients with AF ().
Table I. Studies examining genetic polymorphisms and thromboembolic risk in atrial fibrillation.
Haemostatic polymorphisms
Fibrinogen polymorphisms
FGA T331A polymorphism (rs6050). The conversion of fibrinogen to fibrin gives rise to the formation of fibrin protofibrils, and stabilization of the forming fibrin clot requires cross-linking of adjacent γ- and α-chains by factor (F) XIIIa (Citation44). The γ-γ cross-links contribute to 35% and α-α cross-links to 65% of clot stability (Citation44), highlighting the relative importance of α-fibrinogen cross-linking in determining overall clot stability. This common change of the α-fibrinogen gene supposes a threonine to alanine amino acid substitution at position 331 (T331A) (Citation45). This polymorphism lies close to the FXIIIa cross-linking site at position 328 (Citation44). Thus, T331A could influence clot strength and elasticity by interfering with these FXIIIa-dependent cross-linking processes.
Carter et al. (Citation11) showed that this polymorphism was associated with an increased mortality in patients with stroke and AF compared to those in sinus rhythm. They therefore suggest that possession of A331 may give rise to an increased susceptibility for embolization of thrombus, possibly due to defective FXIII-dependent cross-linking.
FGB g.4577G> A polymorphism (rs1800790). The g.4577G > A polymorphism is located in the promoter region of the β-fibrinogen gene. Carriers of the less frequent allele A (≈0.20 in the general population) have higher fibrinogen levels (≈20%–30%) than do carriers of the G allele (Citation46).
Why is this important? Hyperfibrinogenemia is one important risk factor for cardiovascular disease and stroke (Citation47–52). Fibrinogen influences platelet aggregation as well as blood viscosity. As demonstrated clinically, this polymorphism is associated with coronary atherosclerotic disease (Citation53,Citation54) as well as myocardial infarction (Citation55,Citation56). Bozdemir et al. studied the effect of β-fibrinogen polymorphism on the development of left atrial thrombus or spontaneous echo contrast (Citation57). They found that this polymorphism was associated with both pathologies, suggesting that β-fibrinogen g.4577G > A polymorphism may be a probable marker for the prediction of thromboembolism risk in patients with AF.
F13A1 V34L polymorphism (rs5985). Coagulation FXIII is a tetrameric structure consisting of 2A (active) and 2B subunits. Calcium and thrombin activate FXIII in the final phase of the coagulation process. FXIII plays an essential role in the coagulation process, as activated FXIII catalyses the formation of γ-glutamyl-lysine bonds between fibrin and α2-plasmin inhibitor, increasing the resistance of fibrin to degradation (Citation58). A common G to T polymorphism in exon 2 of the F13A1 gene causes a valine to leucine change at position 34 (V34L), three amino acids upstream to the thrombin cleavage site (Citation59). However, there are controversial data on the role of this polymorphism in cardiovascular disease (Citation60–62). Indeed, the L34 variant displays an increased rate of FXIII activation by thrombin (Citation63), which results in an increased FXIII-specific transglutaminase activity (Citation64) in vivo, with an increased and faster rate of fibrin stabilization (Citation65,Citation66). In accordance, thrombi formed in subjects carrying the L34 variant would take a much longer time for removal from the vascular system.
Marín et al. (Citation67) reported that F13A1 V34L polymorphism was independently associated with the prothrombotic and inflammatory state in AF patients, as evidenced by its association with raised TF and interleukin-6 (IL-6) levels, but not with platelet activation. In a multivariate analysis, IL-6 values were independently associated with the presence of F13A1 polymorphism, sex, and previous thromboembolism (the two last features being clinical risk factors for thromboembolism) (Citation68), whereas TF levels were related to IL-6 concentrations. These findings confirm the observations on TF expression, as induced by local inflammation, in the endothelium of left atrial appendage may be involved in the pathogenesis of clot formation in AF patients (Citation40). The nexus between the faster and more efficient activation of F13A1 L34 and increased IL-6 levels might be the expression of FXIII on the surface of monocytes (Citation44). This would activate a pro-inflammatory response by the monocyte, according to recent studies that have identified a relationship between transglutaminase activity and pro-inflammatory res-ponse in other tissues (Citation69). However, we were unable to find a significant association in 96 patients with cardioembolic stroke secondary to AF and this polymorphism (Citation70). Further studies are needed to clarify the role of this polymorphism in thromboembolic risk associated with AF.
F2 g.25404A > G polymorphism (rs3136517). The prothrombin variant g.25404A > G, a genetic variation in the 3´-untranslated region of the F2 gene, is associated with a moderate increase of prothrombin and thrombin levels, thus increasing the risk of thrombotic events (Citation71). It has a frequency of 1.7% (95% confidence interval 1.3–2.2) in populations of European descent (Citation72). The role of the F2 gene variant in arterial thromboembolism is still debated (Citation73–78).
There are controversial data on the association of the prothrombin g.25404A > G variant and the occurrence of systemic thromboembolism in AF patients (Citation7–9,Citation70,Citation79). Hatzinikolaou-Kotsakou et al. found that this polymorphism may participate in the prothrombotic state of AF patients (Citation79), whilst Pengo et al. observed the association between the presence of this genetic variant and the occurrence of previous systemic embolism (Citation8). However, Poli et al. did not confirm these results in a larger cohort of patients but in multivariate analysis showed an independent association of this genetic variant with AF, suggesting a possible relationship between the presence of prothrombin gene variant and AF per se (Citation9). Also, Roldán et al. did not find a significant association with embolic risk in AF patients (Citation70).
F5 R506Q polymorphism (rs6025). The c.1601A > G change in F5 gene is responsible for factor V Leiden (FVL) mutation, the most common genetic risk factor for venous thromboembolism, and promotes formation of fibrin-rich clots through resistance to activated protein C (Citation80,Citation81). FVL occurs in 4%–6% of individuals in most European populations (Citation82) and in 20%–30% of patients with venous thrombosis (Citation83).
In AF, Go et al. (Citation10) analysed a small proportion of patients participating in the Anticoagulation and Risk Factors in Atrial Fibrillation (ATRIA) study but failed to find an increased risk of ischaemic stroke in carriers of FVL polymorphism. The possible selection biases, such as non-adjustment for classical and consistent embolic risk factors, could have limited the power of the study to clarify the influence of this polymorphism on embolism in AF. In the Stroke Prevention in Atrial Fibrillation III Study (SPAF III), FVL was not associated with increased thromboembolic risk, although there was an independent association with raised levels of prothrombin fragment 1+2 (F1+2), a marker of thrombin generation (Citation84). In a case-control study of 72 referred case patients with a previous history of systemic thromboembolism and 142 age-matched controls, FVL was also not found independently associated with thromboembolism after controlling for hypertension, heart failure, diabetes, and coronary disease (Citation8). Additional groups have not found any significant association between FVL and embolic risk (Citation9,Citation70). Surprisingly, Hatzinikolaou-Kotsakou et al. found a statistical difference in the prevalence of FVL with only 55 patients and 17 controls (Citation79), suggesting that this genetic alteration could coexist in AF and may be a contributor to the development of hypercoagulability in AF patients. However, multivariate analysis was not performed, and the relation between FVL and risk of thromboembolism was not explored.
Thus, there were conflicting results about the influence of FVL and thromboembolic risk in AF. Most of these studies did not find association between the mutation and thromboembolic risk, and small studies could simply reflect type II errors due to lack of power. Further studies are clearly needed.
F7 g.4727_4728ins10 polymorphism (rs5742910). Factor VII (FVII) levels (both coagulant activity and antigen) have a wide interindividual variation in plasma, and both genetic and environmental factors are responsible for such variation (Citation85). The F7 gene has five polymorphic loci that may account for up to 30% of the variance in plasmatic FVII levels; one of them is the g.4727_4728ins10 polymorphism, resulting from a common 10-bp insertion in the F7 promoter region, which diminishes promoter activity by 33% (Citation86), causing a reduction of both FVII antigen and coagulation activity (Citation87,Citation88). Elevated levels of FVII coagulation activity might provide a mechanism for precipitating thrombotic events and, conversely, reduced levels of FVII coagulation activity would diminish the thrombotic process (Citation89).
Roldán et al. (Citation70) tested the possible role of F7 polymorphism on AF thromboembolic risk. The authors studied a consecutive cohort of AF patients who had been admitted to hospital with a thromboembolic ischaemic stroke secondary to AF and compared them with AF patients without stroke: they found a lower prevalence of the g.4727_4728ins10 polymorphism amongst patients without stroke compared to patients with stroke, even after adjusting for clinical risk factors for thromboembolism. This relationship was linked to low F1+2 levels, suggesting that the Ins allele, associated with reduced plasma FVII levels, may confer a lower ischaemic stroke risk in AF. Thus, this allele could influence the stroke risk in AF via modulation of the prothrombotic state associated with AF (Citation70).
Polymorphisms associated to platelet functionality
Platelets are crucial in primary haemostasis, and their adhesion to damaged vessel wall is mainly mediated by the collagen receptor glycoprotein (GP) Ia/IIa, or the primary vWf receptor GPIb/IX (Citation90). Collagen, a major component of the subendothelium (Citation91), is a strong platelet agonist capable of inducing adhesion (Citation92) and activation of platelets (Citation93). The membrane GPIa/IIa, known as integrin α2β1, functions as a general receptor for all collagens exposed in the subendothelial matrix (Citation92). Although this process is essential in limiting blood loss in cases of vascular injuries, in certain circumstances this might lead to an increased risk of arterial thrombosis and, thus, contribute to the pathogenesis of myocardial infarction and ischaemic stroke.
The integrin α2 gene (ITGA2) is located on chromosome 5q11.2. A silent change in the coding region at position g.67214C > T (rs1126643) has been identified. This single nucleotide polymorphism on the ITGA2 gene has been shown to correlate with the platelet GPIa/IIa density (Citation94,Citation95). Then, subjects with the T allele have an increased potential of platelet adhesion and a tendency to arterial thrombosis, increasing the risk of cardiovascular disease.
The integrin αIIbβ3 is the most abundant receptor on the platelet membrane surface involved in platelet adhesion and aggregation (Citation96–98) and is characterized by several inheritable dimorphisms. The two most common and clinically important β3 alleles encode L59 (PlA1 or HPA-1a) and P59 (PlA2 or HPA-1b) (rs5918), with gene frequencies of 0.85 and 0.15, respectively, in the Caucasian population. In 1996 Weiss et al. first reported a prothrombotic role for this polymorphism in myocardial infarction (Citation99). Subsequent studies have given different and, in some cases, contradictory results (Citation100–103).
These platelet polymorphisms were studied by Roldán et al. (Citation70) in 119 patients with non-rheumatic AF and 96 patients with cardioembolic stroke secondary to AF. Of these, only the polymorphism of the ITGA2 gene was associated with an increased risk of cardioembolic stroke independently of prothrombotic state of AF, suggesting that changes in GPIa surface receptor could be associated with an increasing risk of suffering a cardioembolic stroke, independently of other prothrombotic state associated with AF.
Other polymorphisms
The annexin family is integrated by proteins that share structural similarities and have the functional property of binding to phospholipids in the presence of Ca+2 ions.
Annexin 5 (A5) is the most abundant member of this group of proteins, found in many tissues, including blood (Citation104). A5 has various antithrombotic properties, including the prevention of prothrombinase and tenase complexes formation as well as inhibition of platelet aggregation and thrombus formation, both in vitro and in vivo (Citation105,Citation106). The ANXA5 gene contains 12 exons that code for a single polypeptide chain of 319 amino acids (Citation107). The C> T transition at c.-1 of the ANXA5 gene has been described (rs11575945), affecting the Kozak sequence at the nucleotide preceding the initiation ATG codon (Citation108). The c.-1 C> T polymorphism has been shown to play a protective role against myocardial infarction in young patients, and this may be due to an increased translation efficiency resulting in increased levels of A5 (Citation109). Only Roldán et al. have studied its role in the thromboembolic risk in patients with AF where no significant influence was found (Citation70).
There are other polymorphisms that could be associated with the risk of stroke: for example, a common polymorphism in methylenetetrahydrofolate reductase gene (MTHFR), A222V (rs1801133) that promotes hyperhomocysteinemia (Citation110), genetic variants at the plasminogen activator inhibitor type 1 locus (PAI-1) (g. 4332_4333insG; rs1799889) which could impair clot lysis (Citation111), or inflammatory polymorphisms such as g.4880C> G (rs1800795) at IL-6 gene promoter, which could influence the development of postoperative AF (Citation112). However, there is a lack of studies on the association of stroke risk in AF with these mutations. We have only indirect information from substudies, which did not find any significant association between these genotypes and cardioembolic stroke (Citation113–116).
Pharmacogenetics of oral anticoagulant drugs
Pharmacogenetics has emerged as a field that tries to identify specific gene variants that are able to explain the variability in patient response to a given drug (Citation117). Most genetic changes involved in pharmacogenetics are common polymorphisms. Polymorphisms affecting disposition, metabolism, transporters, or targets of the drug could modify the individual response to therapy and probably its side-effects (Citation118). This interindividual heterogeneity is less complex than that observed in the risk to develop cardiovascular diseases, and the role of a single and functional polymorphism affecting any element involved in the treatment could be strong. The pharmacogenetic knowledge will not diminish the patient population in whom a drug is effective but will allow prediction of individual response rather than prolonged and expensive prescribing by trial and error (Citation119).
Coumarin derivatives compete with vitamin K for epoxide reductase, thereby decreasing the amount of reduced vitamin K required for the carboxylation of clotting factors II, VII, IX, and X and protein C, S, and Z (Citation120) (). Vitamin K antagonists (VKAs) are in general characterized by a marked inter- and intra-patient variability. For decades, it has been well known that the maintenance dose of VKAs is influenced by different acquired factors, such as race, dietary vitamin K intake, co-morbid conditions, acute illness, or co-medications (Citation12). Recent advances in pharmacogenetics have suggested that pharmacogenetic-based VKAs therapy could improve the safety and effectiveness of these drugs in the future ().
Figure 1. Vitamin K cycle. The vitamin K cycle regenerates the reduced form of vitamin K from its epoxide form through the vitamin K epoxide reductase (VKOR). Reduced vitamin K is necessary for post-translational gamma-carboxylation of glutamic acid residues of the vitamin K-dependent coagulation factors II, VII, IX, and X and protein C, S, and Z. Gamma-carboxylation is catalysed by a_gamma-glutamyl carboxylase and requires the reduced form of vitamin K, molecular oxygen, and carbon dioxide. Vitamin K antagonists (VKAs) produce their anticoagulant effect interfering with VKOR activity. Adapted from: Francisco Marín et al. J Am Coll Cardiol. 2009;54: 1041–57 (with permission) (Citation117).
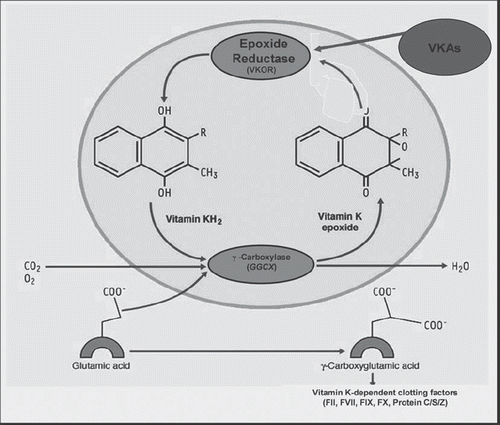
Table II. Studies examining pharmacogenetics of oral anticoagulants.
Genetic polymorphisms affecting VKAs metabolism
The pharmacological effect of coumarins depends on hepatic metabolism, catalysed largely by a member of the cytochrome P450 (CYP) system, CYP2C9, which converts these drugs to inactive metabolites excreted in urine (Citation121). Coumarins are administered in a racemic mixture of R- and S-enantiomers. The R-enantiomer is less active, but clearance of the S-enantiomer is more rapid as it is efficiently metabolized by cytochrome 2C9. CYP2C9*1 is the wild-type allele and is considered the reference sequence. The CYP2C9*2 and CYP2C9*3 alleles encode the R144C (rs1799853) and I359L (rs1057910) variants and are present at allele frequencies of 12% and 8%, respectively, in Caucasians (Citation122). Patients carrying either CYP2C9*2 or CYP2C9*3 polymorphisms showed impaired metabolism of the S-warfarin (the more active enantiomer) increasing the risk of over-anticoagulation (Citation13). Interestingly, the decreased clearance caused by CYP2C9*2 and *3 alleles has a significant clinical impact (Citation123). Thus, the reduction in average warfarin maintenance dose requirement has been demonstrated (Citation124). Similarly, the relationship with bleeding complications, with the time to achieve the maintenance dose, and with over-anticoagulation has been also observed (Citation125–128). Consistent data exist with other coumarin derivatives (Citation129).
Very recently, a new genetic variant in cytochrome 4F2 gene (CYP4F2), V433M (rs2108622), has been reported to be associated with both warfarin and acenocoumarol anticoagulant dose. Individuals carrying the polymorphic M variant have decreased function of the enzyme, increasing the requirements of the anticoagulant drugs (Citation130,Citation131). Interestingly, a recently genome-wide association study (GWAS) of acenocoumarol confirms that, besides age, gender, body mass index, and target international normalized ratio (INR), polymorphism within each of the VKORC1, CYP2C9, CYP4F2, and CYP2C18 genes could explain about 50% of acenocoumarol dosage variation (Citation132).
Genetic polymorphisms affecting VKAs target
The enzyme responsible for the regeneration of vitamin K from vitamin K epoxide is vitamin K epoxide reductase subunit 1, encoded by VKORC1 gene. Common polymorphisms (SNPs) in regulatory regions of the VKORC1 gene correlate strongly with VKAs response. Rieder et al. (Citation133) identified five haplotypes in the VKORC1 gene that are associated with warfarin dose requirements. The molecular mechanism of the warfarin dose response appears to be regulated at the transcriptional level (Citation133). Recently, it has been demonstrated that the SNP g.3588G >A (rs9923231) at promoter region and the tightly linked rs9934438 at intron 1 of VKORC1 gene accurately predict warfarin dose and are suitable biomarkers for warfarin dosing across ethnic populations (Citation134). Moreover, the effect of these polymorphisms may be exacerbated by combination with other polymorphisms, such as c.*4A> G change of CALU gene (rs1043550), an inhibitor of vitamin K-dependent carboxylation (Citation135). Conversely, rare mutations have been also described that reduce VKORC1 activity or result in warfarin resistance (Citation136).
Pharmacogenetics and clinical response to VKAs
As described previously, CYP2C9 genotype predicts ≈10% of the interpatient variability in warfarin dose. Similarly, VKORC1 genotype predicts 25% of such variability. The importance of these strong genetic effects was recognized by the recent relabelling of warfarin by the Food and Drug Administration (FDA) (Citation137). Recently, Takeuchi et al. have reported the first GWAS whose sample size (1,053 Swedish subjects) is sufficiently powered to detect genome-wide significance (P < 1.5 × 10(−7)) for polymorphisms VKORC1, CYP2C9, and CYP4F2 that modestly alter therapeutic warfarin dose (Citation138). The performance of commercial platforms for rapid genotyping of polymorphisms affecting warfarin dose and modelling stable dose requirements based on clinical, physiologic, environmental, and genetic factors has shown promise as a strategic approach to predict individualized stable warfarin dose requirements (Citation139–141). However, it is important to note that patient demographics, clinical factors, and (known) genetic variance combined only explain 45%–55% of the total dose variant (Citation142).
A recent study used clinical and genetic data from 4,043 patients to create a dose algorithm where genetic data were added to clinical variables. This pharmacogenetic approach produces dose recommendations that are significantly closer to the required therapeutic dose than if only clinical data or a fixed dose were used, although the greatest benefits were observed in the approximately 50% of the population that requires higher or lower warfarin dose than the mean (Citation143). In a similar way, the largest prospective warfarin-treated cohort showed how CYP2C9 and VKORC1 significantly influenced warfarin dose and predicted individuals who are predisposed to unstable anticoagulation (Citation144). Another study examined the clinical and genetic profiles of patients steadily anticoagulated with extreme acenocoumarol doses and showed how clinical data, age, and body surface area and three SNPs in VKORC1, CYP2C9, and CYP4F2 genes strongly predicted outlier patients treated with acenocoumarol (Citation145). All these studies suggest that initiation of anticoagulation guided by pharmacogenetics variables would improve clinical outcome. Indeed, both VKORC1 and CYP2C9 genotypes were significant independent predictors of therapeutic dose, especially in the first days, as the partial R2 of genotype was 43% (Citation146).
Pharmacogenetics has the potential to increase benefit and reduce harm in people whose drugs responses are not average. The evidence base for pharmacogenetics testing involves knowing the characteristics of these patients. It is clear that new models will be forthcoming and that these models will continue to evolve with data from large GWAS, and with the inclusion of acquired or environmental factors not considered to date. All these results provide further impetus for conducting large-scale trials assessing patient benefit from genotype-based forecasting of warfarin dose.
Limitations
Results of previous studies should be taken with certain cautions. Most of these are limited by their cross-sectional nature, small number of patients, the possible lack of other cardiovascular risk factors, the difficulty to quantify the exact duration and frequency of the paroxysmal atrial fibrillation, survival bias implied in a disease association study, and other potential limitations. Ethnic differences of the patients included in the studies may represent a problem when generalizing findings to the global population. Also, the need for cautious interpretation of GWAS should be stressed. Past experience suggests that for some disorders as many as 20,000 to 30,000 case subjects and a similar number of comparison subjects could be required to obtain highly robust findings for the validity and reproducibility of candidate gene approaches. This raises important questions regarding resource allocation. For example, the next phase of genetic studies will involve a combination of increasingly large GWAS analyses (for common SNP and copy number variant associations) and resequencing studies (for rare variants).
Conclusions
The thromboembolic risk of AF is a multifactorial complex which involves different systems. In this context, genetics may contribute additional and individual information that help physicians to stratify risk of stroke for these patients. Many important polymorphisms have been associated with a hypercoagulable state and embolic risk. However, there is a lack of data in the literature to support the use of SNPs in risk stratification of stroke. Currently genetic studies are not useful for the stratification of embolic risk in patients with AF, and thus the practical utility is limited. New research developments, based on GWAS studies of well defined large cohorts, should help to characterize better the different genetic profiles of embolic risk in these patients.
On the other hand, pharmacogenetics emerges as a relevant discipline that would help to find the most appropriate treatment for each patient, thus increasing the efficiency and potentially reducing side-effects of treatment. All patients are different, and the identification of the genetic basis of such heterogeneity will help to identify high-risk patients and to predict individualized stable warfarin dose requirements. Genetics will help the development of more personalized medicine.
Declaration of interest: Supported in part by a ResearchGrant by “Fundación Española del Corazón para Investigación Básica y Clínicaen Cardiología”. Dr Hernández-Romero holds a postdoctoral position funded by the Instituto de Salud Carlos III.
Related Research Data
References
- Benjamin EJ, Wolf PA, D'Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–52.
- Fuster V, Ryden LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, . American College of Cardiology/American Heart Association Task Force on Practice Guidelines; European Society of Cardiology Committee for Practice Guidelines; European Heart Rhythm Association; Heart Rhythm Society. ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006; 114:e257–354.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285:2864–70.
- Wang TJ, Massaro JM, Levy D, Vasan RS, Wolf PA, D'Agostino RB, . A risk score for predicting stroke or death in individuals with new-onset atrial fibrillation in the community: the Framingham Heart Study. JAMA. 2003;290:1049–56.
- Fang MC, Go AS, Chang Y, Borowsky L, Pomernacki NK, Singer DE; for the ATRIA Study Group. Comparison of risk stratification schemes to predict thromboembolism in people with nonvalvular atrial fibrillation. J Am Coll Cardiol. 2008;51:810–5.
- Lip GYH. The prothrombotic state in atrial fibrillation: The atrium, the endothelium… and tissue factor? Thromb Res. 2003;111:133–5.
- Marín F, Roldán V, Tello A, Lip GY. Genetic polymorphisms and thromboembolic risk in atrial fibrillation. J Thromb Thrombolysis. 2004;18:151–2.
- Pengo V, Filippi B, Biasiolo A, Pegoraro C, Noventa F, Iliceto S. Association of the G20210A mutation in the factor II gene with systemic embolism in nonvalvular atrial fibrillation. Am J Cardiol. 2002;90:545–7.
- Poli D, Antonucci E, Cecchi E, Betti I, Valdré L, Mugnaini C, . Thrombophilic mutations in high-risk atrial fibrillation patients: high prevalence of prothrombin gene G20210A polymorphism and lack of correlation with thromboembolism. Thromb Haemost. 2003;90:1158–62.
- Go AS, Reed GL, Hylek EM, Phillips KA, Liu L, Henault LE, . Factor V Leiden and risk of ischemic stroke in nonvalvular atrial fibrillation: the anticoagulation and risk factors in atrial fibrillation (ATRIA) study. J Thromb Thrombolysis. 2003;15:41–6.
- Carter AM, Catto AJ, Grant PJ. Association of the α-fibrinogen Thr312Ala polymorphism with poststroke mortality in subjects with atrial fibrillation. Circulation. 1999;99:2423–6.
- James AH, Britt RP, Raskino CL, Thompson SG. Factors affecting the maintenance dose of warfarin. J Clin Pathol. 1992;45:704–6.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics. 2003;13:247–52.
- Chiang CW, Lo SK, Kuo CT, Cheng NJ, Hsu TS. Noninvasive predictors of systemic embolism in mitral stenosis. An echocardiographic and clinical study of 500 patients. Chest. 1994;106:396–9.
- Cioffi G, Pozzoli M, Forni G, Franchini M, Opasich C, Cobelli F, Tavazzi L. Systemic thromboembolism in chronic heart failure. A prospective study in 406 patients. Eur Heart J. 1996;17:1381–9.
- Virchow R. Phlogose und thrombose im Geraβsystem. Virchow R. Gesammelte Abhandlungen zur wissenchaftlichen Medizin. Frankfurt: Von Meidinger Sohn: 1856. 458–636.
- Heppell RM, Berkin KE, McLenachan JM, Davies JA. Haemostatic and haemodynamic abnormalities associated with left atrial thrombosis in non-rheumatic atrial fibrillation. Heart. 1997;77:407–11.
- Lip GYH. Hypercoagulability and haemodinamic abnormalities in atrial fibrillation. Heart. 1997;77:395–6.
- Lip GYH, Gibbs CR. Does heart failure confer a hypercoaguble state? Virchow's triad revisited. J Am Coll Cardiol. 1999;33:1424–6.
- Secchi LA, Zingaro L, Catena C, Casaccio D, De Marchi S. Relationship of fibrinogen levels and hemostatic abnormalities with organ damage in hypertension. Hypertension. 2000;36:978–85.
- Marín F, Roldán V, Monmeneu JV, Bodí V, Fernández C, García de Burgos F, . Prothrombotic state and elevated levels of plasminogen activator inhibitor-1 in mitral stenosis with and without atrial fibrillation. Am J Cardiol. 1999; 84:862–4.
- Lip GYH, Lowe GDO, Rumley A, Dunn FG. Increased markers of thrombogenesis in chronic atrial fibrillation: effects of warfarin treatment. Br Heart J. 1995;73: 527–33.
- Lip GYH, Lip PL, Zafiris J, Watson RDS, Lowe GDO. Fibrin D-dimer and beta-thromboglobulin as a markers of thrombogenesis and platelet activation in atrial fibrillation. Circulation. 1996;94:425–31.
- Feng D, D'Agostino RB, Silbershatz H, Lipinska I, Massaro J, Levy D, . Haemostatic state and atrial fibrillation (The Framingham Offspring Study). Am J Cardiol. 2001;87: 168–71.
- Furui H, Taniguchi N, Yamauchi K, Sotobata I, Saito H, Inagaki H. Effects of treadmill exercise on platelet function, blood coagulability and fibrinolytic activity in patients with atrial fibrillation. Jpn Heart J. 1987;28:177–84.
- Asakura H, Hifumi S, Jokaji H, Saito M, Kumabashiri I, Uotani C, . Prothrombin fragment F1 + 2 and thrombin-antithrombin III complex are useful markers of the hypercoagulable state in atrial fibrillation. Blood Coagul Fibrinolysis. 1992;3:469–73.
- Kumagai K, Fukunami M, Ohmori M, Kitabatake A, Kamada T, Hoki N. Increased intracardiovascular clotting in patients with chronic atrial fibrillation. J Am Coll Cardiol. 1990;16:377–80.
- Blann AD, Lip GYH. The endothelium in atherothrombotic disease: assessment of function, mechanisms and clinical implications. Blood Coagul Fibrinolysis. 1998;9: 297–306.
- Blann AD, Lip GYH. Is soluble P-selectin a new marker of platelet activation? Atherosclerosis. 1997;128:135–8.
- Feinberg WM, Macy E, Cornell ES, Nightingale SD, Pearce LA, Tracy RP, . Plasmin-α2-antiplasmin complex in patients with atrial fibrillation. Thromb Haemost. 1999; 82:100–3.
- Marín F, Roldán V, Marco P, Martínez JG, Toral A, García de Burgos F, . Improvement of fibrinolytical function in chronic rheumatic atrial fibrillation after anticoagulation. Rev Esp Cardiol. 1999;52:25–30.
- Roldán V, Marín F, Marco P, Martínez JG, Calatayud R, Sogorb F. Hypofibrinolysis in atrial fibrillation. Am Heart J. 1998;136:956–60.
- Blann AD, Dobrotova M, Kubisz P, McCollum CN. Von Willebrand factor, soluble P-selectin, tissue plasminogen activator and plasminogen activator inhibitor in atherosclerosis. Thromb Haemost. 1995;74:626–30.
- Takahashi H, Ito S, Hanano M, Wada K, Niwano H, Seki Y, . Circulating thrombomodulin as a novel endothelial cell marker: comparison of its behaviour with von Willebrand factor and tissue-type plasminogen activator. Am J Hematol. 1992;4:32–9.
- Thambidorai SK, Parakh K, Martin DO, Shah TK, Wazni O, . Relation of C-reactive protein correlates with risk of thromboembolism in patients with atrial fibrillation. Am J Cardiol. 2004;94:805–7.
- Maclean PS, Tait RC, Rumley A, Mcmahon AD, Lowe GD. Anticoagulation with warfarin downregulates inflammation. J Thromb Haemost. 2003;1:1838–9.
- Marin F, Roldan V, Climent V, Garcia A, Marco P, Lip GYH. Is thrombogenesis in atrial fibrillation related to matrix metalloproteinase-1 and its inhibitor TIMP-1? Stroke. 2003; 34:1181–6.
- Kamath S, Blann AD, Lip GYH. Platelets and atrial fibrillation. Eur Heart J. 2001;22:2233–42.
- Chung NA, Belgore F, Li-Saw-Hee FL, Conway DS, Blann AD, Lip GYH. Is the hypercoagulable state in atrial fibrillation mediated by vascular endothelial growth factor? Stroke. 2002;33:2187–91.
- Nakamura Y, Nakamura K, Fukushima-Kusano K, Ohta K, Matsubara H, Hamuro T, . Tissue factor expression in atrial endothelia associated with non-valvular atrial fibrillation: possible involvement in intracardiac thrombogenesis. Thromb Res. 2003;111:137–42.
- Goldsmith I, Kumar P, Carter P, Blann AD, Patel RL, Lip GYH. Atrial endocardial changes in mitral valve disease: a scanning electron microscopy study. Am Heart J. 2000; 140:777–84.
- Fukuchi M, Watanabe J, Kumagai K, Katori Y, Baba S, Fukuda K, . Increased von Willebrand factor in the endocardium as a local predisposing factor for thrombogenesis in overloaded human atrial appendage. J Am Coll Cardiol. 2001;37:1436–42.
- Ariëns RA, de Lange M, Snieder H, Boothby M, Spector TD, Grant PJ. Activation markers of coagulation and fibrinolysis in twins: heritability of the prethrombotic state. Lancet. 2002;359:667–71.
- Muszbek L, Adany R, Mikkola H. Novel aspects of blood coagulation factor XIII. I: structure, distribution, activation, and function. Crit Rev Clin Lab Sci. 1996;33: 357–421.
- Baumann RE, Henschen AH. Human fibrinogen polymorphic site analysis by restriction endonuclease digestion and allele-specific polymerase chain reaction amplification: identification of polymorphisms at positions Aa312 and Bb448. Blood. 1993;82:2117–24.
- Thomas A, Green F, Kelleher C, Wilkes HC, Brennan PJ, Meade TW, . Variation in the promotor region of the ß fibrinogen gene is associated with plasma fibrinogen levels in smokers and non-smokers. Thromb Haemost. 1991;65: 487–90.
- Heinrich J, Assmann G. Fibrinogen and cardiovascular risk. J Cardiovasc Risk. 1995;2:197–205.
- DiMinno G, Mancini M. Measuring plasma fibrinogen to predict stroke and myocardial infarction. Arteriosclerosis. 1990;10:1–7.
- Lip GY. Fibrinogen and cardiovascular disorders. QJM. 1995;88:155–65.
- Wilhemsen L, Svardsudd K, Korsan-Bengtsen K, Larsson B, Welin L, Tibblin G. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984;311:501–5.
- Kannel WB, Wolf PA, Castelli WP, D'Agostino RB. Fibrinogen and the risk of cardiovascular disease: the Framingham study. JAMA. 1987;258:1183–6.
- Eber B, Schumacher M. Fibrinogen: its role in the hemostatic regulation in atherosclerosis. Semin Thromb Hemost. 1993;19:104–7.
- Green F, Hamsten A, Blomback M, Humphries S. The role of beta-fibrinogen genotype in determining plasma fibrinogen levels in young survivors of myocardial infarction and healthy controls from Sweden. Thromb Haemost. 1993; 70:915–20.
- Yu Q, Safavi F, Roberts R, Marian AJ. A variant of the beta fibrinogen is a genetic risk factor for coronary artery disease and myocardial infarction. J Invest Med. 1996;44:154–9.
- Behague I, Poirier O, Nicaud V, Evans A, Arveiler D, Luc G, . Beta fibrinogen gene polymorphisms are associated with plasma fibrinogen and coronary artery disease and myocardial infarction: the ECTIM Study. Circulation. 1996;93:440–9.
- Scarabin PY, Bara L, Ricard S, Poirier O, Cambou JP, Arveiler D, . Genetic variation at the beta-fibrinogen locus in relation to plasma fibrinogen concentrations and risk of myocardial infarction: the ECTIM Study. Arterioscler Thromb. 1993;13:886–91.
- Bozdemir V, Kirimli O, Akdeniz B, Aslan A, Kala V, Ozel E, . Beta-fibrinogen 455 G/A gene polymorphism is associated with the left atrial thrombus and severe spontaneous echo contrast in atrial fibrillation. Eur Heart J. 2008;29(abst suppl):259.
- Muszbek L, Yee VC, Hevessy Z. Blood coagulation factor XIII: structure and function. Thromb Res. 1999;94: 271–305.
- Mikkola H, Syrjala M, Rasi V, Vahtera E, Hamalainen E, Peltonen L, . Deficiency in the A-subunit of coagulation factor XIII: two novel point mutations demonstrate different effects on transcript levels. Blood. 1994; 84:517–25.
- Kohler HP, Stickland MH, Ossei-Gernig N, Carter A, Mikkola H, Grant PJ. Association of a common polymorphism in the factor XIII gene with myocardial infarction. Thromb Haemost. 1998;79:8–13.
- Aleksic N, Ahn C, Wang YW, Juneja H, Folsom AR, Boerwinkle E, . Factor XIIIA Val34Leu polymorphism does not predict risk of coronary heart disease. Arterioscler Thromb Vasc Biol. 2002;22:348–52.
- Corral J, González-Conejero R, Iniesta JA, Rivera J, Martínez C, Vicente V. The FXIII Val34Leu polymorphism in venous and arterial thromboembolism. Haematologica. 2000;85: 293–7.
- Ariëns RAS, Lai TS, Weisel JW, Greenberg CS, Grant PJ. Role of factor XIII in fibrin clot formation and effects of genetic polymorphism. Blood. 2002;100:743–54.
- Kangsadalampai S, Board PG. The Val34Leu polymorphism in the A subunit coagulation factor XIII contributes to the large normal range in activity and demonstrates that the activation peptide plays a role in catalytic activity. Blood. 1998;92:2766–70.
- Ariëns RAS, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000;96:988–95.
- Balogh I, Szöke G, Kárpáti L, Wartiovaarar U, Katona E, Komáromi I, . Val34Leu polymorphism of plasma FXIII: biochemistry and epidemiology in familial thrombophilia. Blood. 2000;96:2479–86.
- Marín F, Corral J, Roldán V, González-Conejero R, del Rey ML, Sogorb F, . Factor XIII Val34Leu polymorphism modulates the prothrombotic and inflammatory state associated with atrial fibrillation. J Mol Cell Cardiol. 2004;37:699–704.
- Hart RG, Halperin JL. Atrial fibrillation and thromboembolism: a decade of progress in stroke prevention. Ann Intern Med. 1999;131:688–95.
- Kim SY, Jeitner TM, Steinert PM. Transglutaminases in disease. Neurochem Int. 2002;40:85–103.
- Roldán V, Marín F, González-Conejero R, García-Honrubia A, Martí S, Alfaro A, . Factor VII-323 decanucleotide D/I polymorphism in atrial fibrillation: Implications for the prothrombotic state and stroke risk. Ann Med. 2008;40:553–9.
- Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3´-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88:3698–703.
- Rosendaal FR, Doggen CJ, Zivelin A, Arruda VR, Aiach M, Siscovick DS, . Geographic distribution of the 20210A G to A prothrombin variant. Thromb Haemost. 1998;79: 706–8.
- Redondo M, Watzke HH, Stucki B, Sulzer I, Biasiutti FD, Binder BR, . Coagulation factors II, V, VII, and X, prothrombin gene 20210G◊A transition, and factor V Leiden in coronary artery disease: high factor V clotting activity is an independent risk factor for myocardial infarction. Arterioscler Thromb Vasc Biol. 1999;19:1020–5.
- Eikelboom JW, Baker RI, Parsons R, Taylor RR, van Bockxmeer FM. No association between the 20210G/A prothrombin gene mutation: premature coronary artery disease. Thromb Haemost. 1998;80:878–80.
- Russo G, Girelli D, Olivieri O, Guarini P, Manzato F, Pizzolo F, . G20210A prothrombin gene polymorphism and prothrombin activity subjects with or without angiographically documented coronary artery disease. Circulation. 2001;103:2436–40.
- Gardemann A, Arsic T, Katz N, Tillmanns H, Hehrlein FW, Haberbosch W. The factor II G20210A and factor V G1691A gene transition and coronary heart disease. Thromb Haemost. 1999;81:208–13.
- Rosendaal FR, Siscovick DS, Schwartz SM, Psaty BM, Raghunathan TE, Vos HL. A common prothrombin variant (20210 G to A) increases the risk of myocardial infarction in young women. Blood. 1997;90:1747–50.
- De Stefano V, Chiusolo P, Paciaroni K, Casorelli I, Rossi E, Molinari M, . Prothrombin G20210A mutant genotype is a risk factor for cerebrovascular ischemic disease in young patients. Blood. 1998;91:3562–5.
- Hatzinikolau-Kotsakou E, Kartasis Z, Tziakas D, Hotidis A, Stakos D, Tsatalas K, . Atrial fibrillation and hypercoagulability: dependent on clinical factors or/and on genetic alterations? J Thromb Thrombolys. 2003;16:155–61.
- Middeldorp S, Meinardi JR, Koopman MM, van Pampus EC, Hamulyák K, van Der Meer J, . A prospective study of asymptomatic carriers of the factor V Leiden mutation to determine the incidence of venous thromboembolism. Ann Intern Med. 2001;135:322–7.
- Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Eng J Med. 1995;332:912–7.
- Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet. 1995;346:1133–4.
- Koster T, Rosendaal FR, de Ronde H, Briet E, Vandenbroucke JP, Bertina RM. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet. 1993;342:1503–6.
- Feinberg WM, Pearce LA, Hart RG, Cushman M, Cornell ES, Lip GY, . Markers of thrombin and platelet activity in patients with atrial fibrillation: correlation with stroke among 1531 participants in the stroke prevention in atrial fibrillation III study. Stroke. 1999;30:2547–53.
- Bernardi F, Marchetti G, Pinotti M, Arcieri P, Baroncini C, Papacchini M, . Factor VII polymorphisms contribute about one third of the factor VII level variation in plasma. Arterioscl Thromb Vasc Biol. 1996;16:72–6.
- Pollak ES, Hung HL, Godin W, Overton GC, High KA. Functional characterization of the human factor VII 5′-flanking region. J Biol Chem. 1996;19:1738–47.
- Sacchi E, Tagliabue L, Scoglio R, Baroncini C, Coppola R, Bernardi F, . Plasma factor VII levels are influenced by a polymorphism in the promoter region of the FVII gene. Blood Coagul Fibrinolysis. 1996;72:114–7.
- Bernardi F, Arcieri P, Bertina RM, Chiarotti F, Corral J, Pinotti M, . Contribution of factor VII genotype to activated FVII levels. Differences in genotype frequencies between northern and southern European populations. Arterioscl Thromb Vasc Biol. 1997;17:2548–53.
- Corral J, Iniesta JA, González-Conejero R, Villalón M, Vicente V. Polymorphism of clotting factors modify the risk for primary intracraneal haemorrhage. Blood. 2001;97: 2979–82.
- Clemetson KJ, Clemetson JM. Platelet collagen receptors. Thromb Haemost. 2001;86:189–97.
- Baungartner HR. Platelet interaction with collagen fibrils in flowing blood: I. Reaction of human platelets with alphachymotrypsin-digested subendothelium. Thromb Haemost. 1977;37:1–16.
- Sixma JJ, van Zanten H, Huizinga EG, van der Plas RM, Verkley M, Wu YP. Platelet adhesion to collagen: an update. Thromb Haemost. 1997;78:434–8.
- Alberio L, Dale GL. Flow cytometric analysis of platelet activation by different collagen types present in the vessel wall. Br J Haematol. 1998;102:1212–8.
- Kunicki TJ, Kritzik M, Annis DS, Nugent DJ. Hereditary variation in platelet integrin alpha 2 beta 1 density is associated with two silent polymorphisms in the alpha 2 gene coding sequence. Blood. 1997;89:1939–43.
- Corral J, Gonzalez-Conejero R, Rivera J, Ortuno F, Aparicio P, Vicente V. Role of the 807 C/T polymorphism of the alpha2 gene in platelet GP Ia collagen receptor expression and function-effect in thromboembolic diseases. Thromb Haemost. 1999;81:951–6.
- Calvete JJ. Clues for understanding the structure and function of a prototypic human integrin: the platelet glycoprotein IIb/IIIa complex. Thromb Haemost. 1994;72:1–15.
- Shattil SJ. Function and regulation of the β3 integrins in hemostasis and vascular biology. Thromb Haemost. 1995; 74:149–55.
- Lefkovits J, Plow EF, Topol EJ. Platelet glycoprotein IIb/IIIa receptors in cardiovascular medicine. N Engl J Med. 1995;332:1553–9.
- Weiss EJ, Bray PF, Tayback M, Schulman SP, Kickler TS, Becker LC, . A polymorphism of a platelet glycoprotein receptor as an inherited risk factor for coronary thrombosis. N Engl J Med. 1996;334:1090–4.
- Zotz RB, Winkelmann BR, Nauck M, Giers G, Maruhn-Debowski B, Marz W, . Polymorphism of platelet membrane glycoprotein IIIa: human platelet antigen 1b (HPA-1b/PlA2) is an inherited risk factor for premature myocardial infarction in coronary artery disease. Thromb Haemost. 1998;79:731–5.
- Zotz RB, Winkelmann BR, Muller C, Boehm BO, Marz W, Scharf RE. Association of polymorphisms of platelet membrane integrins alpha IIb(beta)3 (HPA-1b/Pl) and alpha2(beta)1 (alpha807TT) with premature myocardial infarction. J Thromb Haemost. 2005;3:1522–9.
- Streifler JY, Rosenberg N, Chetrit A, Eskaraev R, Sela A, Dardik R, . Cerebrovascular events in patients with significant stenosis of the carotid artery are associated with hyperhomocysteinemia and platelet antigen-1(Leu33Pro) polymorphism. Stroke. 2001;32:2753–8.
- Cole VJ, Staton JM, Eikelboom JW, Hankey GJ, Yi Q, Shen Y, . Collagen platelet receptor polymorphisms integrin alpha2beta1 C807T and GPVI Q317L and risk of ischemic stroke. J Thromb Haemost. 2003;1:963–70.
- Kaetzel MA, Hazarika P, Dedman JR. Differential tissue expression of three 35-kDa annexin calcium-dependent phospholipid binding proteins. J Biol Chem. 1989;264: 14463–70.
- Van Heerde WL, de Groot PG, Reutelingsperger C. The complexity of the phospholipid binding protein annexin V. Thromb Haemost. 1995;73:172–9.
- Thiagarajan P, Benedict CR. Inhibition of arterial thrombosis by recombinant annexin V in a rabbit carotid artery injury model. Circulation. 1997;96:2339–47.
- Cookson BT, Engelhardt S, Smith C, Bamford HA, Prochazka M, Tait JF. Organization of the human annexin V (ANX5) gene. Genomics. 1994;20:463–7.
- van Heerde W, Jumilly AL, Lux P, Meyer K. Mutations in the annexin V gene, a new risk factor for venous thrombosis? [abstract no. 1691]. Thromb Haemost. 1999;S:537.
- Gonzalez-Conejero R, Corral J, Roldán V, Martinez C, Marin F, Rivera J, . A common polymorphism in the annexin V Kozak sequence (1C/T) increases efficiency and plasma levels of annexin V, and decreases the risk of myocardial infarction in young patients. Blood. 2002;100:2081–6.
- Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Mathews RG, . A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–3.
- Eriksson P, Kallin B, van't Hooft FM, Båvenholm P, Hamsten A. Allele-specific increase in basal transcription of the plasminogen-activator inhibitor 1 gene is associated with myocardial infarction. Proc Natl Acad Sci U S A. 1995;92: 1851–5.
- Gaudino M, Andreotti F, Zamparelli R, Di Castelnuovo A, Nasso G, Burzotta F, . The -174G/C interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation. 2003;108 Suppl 1:II195–9.
- Goracy I, Cyryłowski L, Kaczmarczyk M, Fabian A, Koziarska D, Goracy J, . C677T polymorphism of the methylenetetrahydrofolate reductase gene and the risk of ischemic stroke in Polish subjects. J Appl Genet. 2009; 50:63–7.
- Kawamoto R, Kohara K, Oka Y, Tomita H, Tabara Y, Miki T. An association of 5,10-methylenetetrahydrofolate reductase (MTHFR) gene polymorphism and ischemic stroke. J Stroke Cerebrovasc Dis. 2005;14:67–74.
- Adamski MG, Turaj W, Slowik A, Wloch-Kopec D, Wolkow P, Szczudlik A. A-G-4G haplotype of PAI-1 gene polymorphisms −844 G/A, HindIII G/C, and −675 4G/5G is associated with increased risk of ischemic stroke caused by small vessel disease. Acta Neurol Scand. 2009;120:94–100.
- Jood K, Ladenvall P, Tjärnlund-Wolf A, Ladenvall C, Andersson M, Nilsson S, . Fibrinolytic gene polymorphism and ischemic stroke. Stroke. 2005;36:2077–81.
- Marín F, González-Conejero R, Capranzano P, Bass TA, Roldán V, Angiolillo DJ. Pharmacogenetics in cardiovascular antithrombotic therapy. J Am Coll Cardiol. 2009; 54:1041–57.
- Roses AD. Pharmacogenetics and future drug development and delivery. Lancet. 2000;355:1358–61.
- Roses AD. Pharmacogenetics and the practice of medicine. Nature. 2000;405:857–65.
- Hirsh J, Fuster V, Ansell J, Halperin JL; American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation. 2003;107:1692–711.
- Rettie AE, Korzekwa KR, Kunze KL, Lawrence RF, Eddy AC, Aoyama T, . Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem Res Toxicol. 1992;5:54–9.
- Yasar U, Eliasson E, Dahl ML, Johansson I, Ingelman-Sundberg M, Sjöqvist F. Validation of methods for CYP2C9 genotyping: frequencies of mutant alleles in a Swedish population. Biochem Biophys Res Commun. 1999;254: 628–31.
- Linder MW, Looney S, Adams JE 3rd, Johnson N, Antonino-Green D, Lacefield N, . Warfarin dose adjustments based on CYP2C9 genetic polymorphisms. J Thromb Thrombolysis. 2002;14:227–32.
- Taube J, Halsall D, Baglin T. Influence of cytochrome P-450 CYP2C9 polymorphisms on warfarin sensitivity and risk of over-anticoagulation in patients on long-term treatment. Blood. 2000;96:1816–9.
- Aithal GP, Day CP, Kesteven PJ, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding Complications. Lancet. 1999;353:717–9.
- Higashi MK, Veenstra DL, Kondo LM, Wittkowsky AK, Srinouanprachanh SL, Farin FM, . Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA. 2002;287:1690–8.
- Margaglione M, Colaizzo D, D'Andrea G, Brancaccio V, Ciampa A, Grandone E, . Genetic modulation of oral anticoagulation with warfarin. Thromb Haemost. 2000;84: 775–8.
- Peyvandi F, Spreafico M, Siboni SM, Moia M, Mannucci PM. CYP2C9 genotypes and dose requirements during the induction phase of oral anticoagulant therapy. Clin Pharmacol Ther. 2004;75:198–203.
- Visser LE, van Vliet M, van Schaik RH, Kasbergen AA, De Smet PA, Vulto AG, . The risk of overanticoagulation in patients with cytochrome P450 CYP2C9*2 or CYP2C9*3 alleles on acenocoumarol or phenprocoumon. Pharmacogenetics. 2004;14:27–33.
- Caldwell MD, Awad T, Johnson JA, Gage BF, Falkowski M, Gardina P, . CYP4F2 genetic variant alters required warfarin dose. Blood. 2008;111:4106–12.
- Pérez-Andreu V, Roldán V, Antón AI, García-Barberá N, Corral J, Vicente V, . Pharmacogenetic relevance of CYP4F2 V433M polymorphism on acenocoumarol therapy. Blood. 2009;113:4977–9.
- Teichert M, Eijgelsheim M, Rivadeneira F, Uitterlinden AG, van Schaik RH, Hofman A, . A genome-wide association study of acenocoumarol maintenance dosage. Hum Mol Genet. 2009;18:3758–68.
- Rieder MJ, Reiner AP, Gage BF, Nickerson DA, Eby CS, McLeod HL, . Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med. 2005;352:2285–93.
- Wang D, Chen H, Momary KM, Cavallari LH, Johnson JA, Sadée W. Regulatory polymorphism in vitamin K epoxide reductase complex subunit 1 (VKORC1) affects gene expression and warfarin dose requirement. Blood. 2008;112: 1013–21.
- González-Conejero R, Corral J, Roldán V, Ferrer F, Sánchez-Serrano I, Sánchez-Blanco JJ, . The genetic interaction between VKORC1 c1173t and calumenin a29809g modulates the anticoagulant response of acenocoumarol. J Thromb Haemost. 2007;5:1701–6.
- Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hörtnagel K, Pelz HJ, . Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–41.
- Gage BF, Lesko LJ. Pharmacogenetics of warfarin: regulatory, scientific, and clinical issues. J Thromb Thrombolysis. 2008;25:45–51.
- Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, Soranzo N, . A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009;5:e1000433.
- King CR, Porche-Sorbet RM, Gage BF, Ridker PM, Renaud Y, Phillips MS, . Performance of commercial platforms for rapid genotyping of polymorphisms affecting warfarin dose. Am J Clin Pathol. 2008;129:876–83.
- Schwarz UI, Ritchie MD, Bradford Y, Li C, Dudek SM, Frye-Anderson A, . Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med. 2008;358:999–1008.
- Gage BF, Eby C, Johnson JA, Deych E, Rieder MJ, Ridker PM, . Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther. 2008;84:326–31.
- Cooper GM, Johnson JA, Langaee TY, Feng H, Stanaway IB, Schwarz UI, . A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008;112:1022–7.
- International Warfarin Pharmacogenetic Consortium; Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, Lee MT, . Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med. 2009;360: 753–64.
- Wadelius M, Chen LY, Lindh JD, Eriksson N, Ghori MJ, Bumpstead S, . The largest prospective warfarin-treated cohort supports genetic forecasting. Blood. 2009;113: 784–92.
- Pérez-Andreu V, Roldán V, López-Fernández MF, Antón AI, Alberca I, Corral J, . Pharmacogenetics of acenocoumarol in patients with extreme dose requirements. J Thromb Haemost. 2010;8:1012–7.
- Ferder NS, Eby CS, Deych E, Harris JK, Ridker PM, Milligan PE, . Ability of VKORC1 and CYP2C9 to predict therapeutic warfarin dose during the initial weeks of therapy. J Thromb Haemost. 2010;8:95–100.