Abstract
Lutein is recovered at high concentration in the human macula lutea. Recent studies suggest that this micronutrient might be implicated in prevention of age-related macular degeneration.
Objective. To identify genes which affect blood and retina lutein concentrations among candidate genes (intestinal sterol transporters and carotenoid oxygenases).
Design. A comparative plus an observational study.
Participants. Twenty-nine healthy subjects for the comparative study and 622 subjects for the observational study.
Intervention and methods. All the participants were genotyped for single nucleotide polymorphisms (SNPs) in the candidate genes. Fasting plasma lutein concentrations were measured in all the participants and after 6 months' supplementation, with either a lutein-rich supplement or a placebo, in the 29 subjects who participated in the comparative study. Macular pigment optical density (MPOD), which is a measure of macula concentration of lutein, was measured before and after the dietary intervention in the 29 subjects. Associations between SNPs and plasma lutein and MPOD were assessed by partial least square (PLS) regression followed by univariate analysis. Observed associations between SNPs and plasma lutein were verified by haplotype-based association analysis in the cohort of 622 subjects.
Main outcome measures. Plasma lutein levels and MPOD.
Results. Six SNPs in four genes (ABCG8, BCMO1, CD36, and NPC1L1) explained 25% and 38% of the plasma and MPOD variance, respectively. Subjects with TT at the BCMO1 rs7501331 locus had lower (P < 0.05) plasma lutein than CT subjects. Subjects with CC at the CD36 rs13230419 locus had lower (P < 0.05) plasma lutein than subjects who carried a T allele. The association between CD36 and plasma lutein was confirmed in the cohort of 622 subjects. Subjects with TT at the BCMO1 rs7501331 locus had a higher (P < 0.05) MPOD, and subjects with GG at rs1761667 CD36 locus had a higher (P < 0.05) MPOD than those with an A allele.
Conclusions. These results suggest that BCMO1 and CD36 are implicated in plasma and retina concentrations of lutein and that genetic variants in these genes can modulate blood and retina concentrations of lutein.
| Abbreviations | ||
| BCDO2 | = | β-carotene dioxygenase 2 |
| BCMO1 | = | β-carotene monoxygenase 1 |
| BMI | = | body mass index |
| HPLC | = | high-pressure liquid chromatography |
| MPOD | = | macular pigment optical density |
| PLS | = | partial least square regression |
| SNP | = | single nucleotide polymorphisms |
Key messages
Age-related macular degeneration is a degenerative eye disease that is due, at least in part, to free radicals, and several studies have suggested that antioxidant micronutrients, such as vitamin C, E, carotenoids, and selenium, may participate in the defense of the retina against free radicals.
The main carotenoids found in the human retina are the xanthophylls, lutein, zeaxanthin, and meso-zeaxanthin.
Our results suggest that genetic variants in BCMO1 and CD36 modulate plasma and retina lutein concentrations.
Introduction
Age-related macular degeneration is an degenerative eye disease that is due, at least in part, to free radicals (Citation1). Several studies have suggested that antioxidant micronutrients, such as vitamin C, E, carotenoids, and selenium, may participate in the defense of the retina against free radicals. The main carotenoids found in the human retina are the xanthophylls, lutein, zeaxanthin, and meso-zeaxanthin (Citation2,Citation3). Macular meso-zeaxanthin apparently originates from the metabolism of lutein (Citation4), while lutein and zeaxanthin originate from the diet. Lutein and zeaxanthin are mainly obtained from fruits and vegetables. It is assumed that carotenoids are extracted from plant cells in the upper part of the gastrointestinal tract (Citation5,Citation6), solubilized in fat lipid droplets (Citation7), and incorporated into mixed micelles (Citation8). Mixed micelles are assumed to carry carotenoids to the intestinal brush border where they are absorbed by the enterocytes (Citation9).
Mechanisms involved in intestinal absorption of carotenoids were initially studied by the Hollander group (Citation10,Citation11). This group concluded that the intestinal absorption of β-carotene, and, by extension, of all the carotenoids, is passive (Citation12). However, this dogma was refuted by recent studies that have shown that absorption of several carotenoids (Citation13–17) involves an enterocyte apical membrane protein, scavenger receptor class B type I (SR-BI), which has been involved in cholesterol uptake. Interestingly, the result obtained in the study dedicated to lutein absorption suggested that other transporters are probably involved (Citation13). We hypothesized that these transporters may be sterol transporters because of the involvement of SR-BI and because they have low substrate specificity. At present, there are several sterol transporters that have been identified: 1) NPC1L1, which is apparently the main protein involved in cholesterol uptake (Citation18) and which has recently been found to be involved in vitamin E uptake (Citation19); 2) SR-BI, which is involved in the uptake of cholesterol (Citation14), carotenoids (Citation13,Citation14,Citation17), and vitamin E (Citation20); 3) ABCG5 and ABCG8, which are involved in phytosterols and cholesterol efflux back into the intestinal lumen (Citation21); and 4) ABCA1, which is mainly located at the basolateral side of the enterocyte and is involved in cholesterol (Citation22) and tocopherol efflux (Citation23,Citation24).
It is assumed that, after absorption, lutein is incorporated into chylomicrons and transported to the liver. A fraction of lutein is then incorporated into very low density lipoproteins (VLDL) and distributed to peripheral tissues by lipoproteins (Citation25–27). A recent study has suggested that SR-BI is involved in xanthophyll uptake by retina cells (Citation28). Since the sterol transporters mentioned above are expressed not only in the intestine but also in various other tissues, we hypothesized that they are good candidates for retina uptake of lutein. Finally, it is assumed that β-carotene monoxygenase 1 (BCMO1) and/or β-carotene dioxygenase 2 (BCDO2), which are involved in cleavage of provitamin A carotenoids in retinal and apo-carotenals, respectively (Citation29), might be involved in the metabolism of lutein and thus in its blood and tissue concentration. This is supported by a recent study showing that a nonsense mutation in BCDO2 was associated with the yellow skin phenotype in sheep, suggesting a broad specificity of this enzyme for carotenoids (Citation30).
The main objective of the present study was to assess whether some sterol transporters and carotene oxygenases are involved in blood and macular concentrations of lutein. To attain our goal, we studied associations between single nucleotide polymorphisms (SNPs) of genes that encode these transporters and the two carotene oxygenases, and blood and macula concentrations of lutein in 29 healthy subjects. Observed associations between genetic variants and plasma lutein were further verified in a cohort of 622 subjects.
Materials and methods
Subject number and characteristics
Since there were no data on the effect of the selected genetic variants on either plasma lutein levels or macular pigment optical density (MPOD), we were unable to perform a power analysis to calculate the number of subjects required to observe a significant effect with an 80% power. We decided to work with 30 subjects in order to divide the group into two subgroups with 15 subjects each: one took a lutein supplement; the other, a placebo (see the paragraph on lutein supplementation).
Thirty healthy, non-obese males were recruited. They had no disease history, hyperlipemia, or hyperglycemia. Their characteristics and daily nutrient intakes are reported in . One subject withdrew for personal reasons during the study. Of the 30 selected subjects, 29 were non-smokers. They were not taking any medication known to affect lutein or lipid metabolism during the month before the study started or during the study period. The study was approved by the regional committee on human experimentation (CPP Sud Est VI, France) and adhered to the tenets of the Declaration of Helsinki. The objectives and requirements of the study were fully explained to the participants, and informed written consent was obtained for each subject. The subjects’ usual diet was estimated with a 3-day food diary before the study started. Portion sizes were estimated with photographs compiled in a manual adapted from the SU.VI.MAX picture booklet (Citation31). The dietary diary was analyzed for nutrient composition with a diet analyzer software (GENI 6.5; Micro6, Villers les Nancy, France). The software database was extended for carotenoids by mean of the US Department of Agriculture (USDA) carotenoid food-composition database (Citation32). The lutein intake was close to that previously observed in French group of volunteers (Citation33–35) and close to the intake reported in a US population (Citation36).
Table I. Characteristics and nutrient intake of the 29 male subjects enrolled in the clinical study.a
Choice of candidate SNPs
Candidate SNPs in genes involved in sterol absorption were selected through an analysis of previous studies describing associations between these SNPs and digestion, transport, or metabolism of sterols. The SNP in BCDO2 was advised by Dr George Lietz (Newcastle University, UK) from an unpublished study. Characteristics of the SNPs are presented in . SNPs were validated for the oligo-ligation assay (SNPlex, see below) by several criteria: 1) genome screening, in which the SNPs may be located in a genome region that is homologous with at least one other genome region, leading to a lack of assay specificity and the potential for spurious ligation templates; 2) assay rules, in which an individual SNP assay cannot be designed due to deleterious sequence contexts or non-optimal interactions among the assay components (for example, aspects of the SNP sequence or assay components may result in secondary structure and reduce assay performance, including a series of contiguous Gs or a series of 16 weak contiguous bases (As or Ts) within 25 bases of the SNP); and 3) pooling rules, in which deleterious potential interactions may occur between specific SNP assays in the assay pools, and false signals may be generated due to components from different assays interacting with genomic DNA.
Table II. Characteristics of the studied SNPs.
The SNPs that were not validated for SNPlex were replaced by alternate SNPs, in linkage disequilibrium with the initial SNPs, or analyzed by TaqMan (see below).
DNA preparation and genotyping methods
Genomic DNA was prepared from 2 mL whole blood and purified with the NucleoSpin® Blood L kit ref 740 954 (Macherey Nagel, Hoerdt, France). A mean of 15 μg of DNA was isolated from each blood sample. The purity and quantity of DNA was checked by spectrophotometry at 260 nm and 280 nm. A total of 100 μL of DNA at a concentration of 10 ng/μL was added to a plate (Dutscher, Marseille, France) for genotyping. SNPs were genotyped with an oligo-ligation assay (SNPlex, Applied Biosystems, Foster City, CA, USA) (Citation37,Citation38) or a TaqMan method (Applied Biosystems) following the manufacturer's guidelines.
The oligo-ligation assay consists of designing 3′ specific primers for each SNP, with two primers carrying the SNP-base-specific 3′ end and one common primer that starts 5′ to the next base in the target sequence. The two allele-specific primers carry unique ZIP codes that determine each allele. Primers are annealed to the target sequence according to the manufacturer's recommendations. A ligation reaction will join the allele-specific primer with the common primer if the allele-specific 3′-base is present. A short fluorescent dye-labeled probe, homologous to the ZIP code sequence, is then hybridized to the immobilized product. Up to 48 SNPs can thus be multiplexed into one oligo-ligation reaction. Following the manufacturer's recommendations, genomic DNA was heat-fragmented. The allele-specific fluorescent probes were separated on an automated sequencer (ABI 3730; Applied Biosystems, Foster City). Alleles were binned and called with the GeneMapper software (Applied Biosystems, Foster City).
The TaqMan assays were performed when selected SNPs could not be analyzed by SNPlex. This was the case for two SCARB1 SNPs, rs4238001 and the one called ‘intron 5′. Probes were purchased from the manufacturer and used according to the manufacturer's guidelines. DNA was amplified by polymerase chain reaction (PCR) by denaturation at 95°C for 10 min, 40 cycles at 92°C for 15 s, 60°C for 1 min, and 72°C for 45 s, followed by elongation at 72°C for 5 min. TaqMan assays were then read on a 7900HT Fast Real-Time PCR System (Applied Biosystems), and alleles were called by the SDS software (Applied Biosystems).
Lutein supplementation
The 29 subjects were first asked to follow a lutein-poor diet for 3 weeks. To attain this objective they were asked to discard lutein-rich foods from their usual diet (a list of lutein-rich food was given to the volunteers). The subjects then came to a Center for Clinical Investigation (Centre de Recherche en Nutrition Humaine d'Auvergne) after an overnight fast, and a blood sample was collected. The same day, the subjects also came to a biophysics laboratory to have their MPOD measured (the right eye was used for most of the subjects). Subjects were randomly assigned to one of the two groups. One received a placebo, while the other received a lutein-rich supplement (Visiobane Protect, Pileje, France) for 6 months. The supplement was provided as two pills containing 5 mg of lutein esters each. Subjects were asked to eat the pills during their main meals. The supplement also contains Porphyra, B vitamins, vitamin C and E, fish oil, bees-wax, and gelatine. The placebo contained only refined sunflower oil. At the end of the supplementation period, all the subjects came back to the Center for Clinical Investigation for collection of a second fasting blood sample, and to the biophysics laboratory for another MPOD measurement.
Measurement of macular pigment optical density (MPOD)
The MPOD was determined as described by the van Norren team (Citation39,Citation40). In summary, the radiant flux of the pattern radiated from photoreceptors at the fovea (sample field: 2 degrees) and the radiant flux of the beam reflected specularly by the inner limiting membrane (ILM; the membrane separating the vitreous humor from the retinal nerve fiber layer) in the perifovea (used as reference) were measured. The measuring light was provided by a 75 W xenon lamp (Oriel, Statford, CT, USA) that was filtered either by an interference filter at 470 nm (FWHM 10 nm, Oriel, Statford, CT, USA), or an interference filter at 532 nm (FWHM 10 nm, Oriel, Statford, CT, USA). The retinal illuminances were 4.8 logTd and 5.5 logTd for the blue and green lights, respectively. The distribution of light in the pupil was measured with a CCD camera cooled by liquid nitrogen (Princeton Instruments Trenton, NJ, USA). This camera had a resolution of 512 × 512 pixels, with an image depth of 16 bits. The pixel size in the pupil was 0.0255 mm. We combined areas of 2 × 2 pixels, giving a resolution of 0.051 mm/pixel. Two stepping motors X and Y (Newport Irvine, CA, USA) allowed the center J of the entrance pupil (diameter 0.2 mm) to be positioned at the chosen location in the eye's pupil. The head of the subject was stabilized by a bite bar which was fixed on a three-dimensional positioner. The main optical components were mounted on a single plate which could be shifted longitudinally, thus allowing focus adjustment from −12 diopters to +12 diopters of ametropia. The pupil was dilated by application of 0.5% Mydriacyl to a minimum diameter of 7 mm. The eye was then aligned to the reflectometer. A fixation target was used to direct the subject's gaze either to the center of the sampling area or at a site 6 degrees temporal to the fovea. At the fovea the center J of the entrance pupil was aligned to photoreceptor axes. At 6 degrees eccentricity, the point J was positioned at the center of the eye's pupil. For each of these retinal eccentricities, the measuring green beam bleached the retina for a period of 15 s, then three pupil images were captured at 532 nm (integration time of 4 s), with an interval of 10 s between each one; they were followed by three pupil images at 470 nm.
Plasma lutein extraction and HPLC analysis of lutein
Plasma lutein was extracted and analyzed as previously described (Citation41). Briefly, 200 μL of plasma was deproteinized by adding one volume of ethanol containing the carotenoid echinenone (as an internal standard). Lutein was extracted twice by the addition of two volumes of hexane. The hexane phases obtained after centrifugation (500 g, 10 min, 4°C) were pooled and evaporated completely under nitrogen. The dried extract was dissolved in 200 μL of dichloromethane/methanol mixture (65/35; V/V). All extractions were performed at room temperature under yellow light to minimize light-induced damage. A volume of 80 μL was used for high-pressure liquid chromatography (HPLC) analysis. The HPLC system consisted of a 150 × 4.6 mm, RP C18, 3-μm nucleosil column (Interchim, Montluçon, France) coupled with a 250 × 4.6 mm RP C18, 5-μm vydactp 54 column (Hesperia, CA, USA) and a 10 × 4.6 mm RP C18, 5-μm hypersil guard column. The mobile phase consisted of acetonitrile/methanol containing 50 mmol/L ammonium acetate/water/dichloromethane (70/15/5/10; V/V/V/V). Solvents were HPLC grade from Carlo Erba-SDS (Peypin, France). The flow rate was 2 mL/min. The columns were kept at a constant temperature (30°C). The HPLC system consisted of a Waters system equipped with a UV-visible photodiode-array detector (Waters 996). Carotenoids were detected at 450 nm and identified by their retention time compared with pure (>95%) standards. Quantifications were performed with Millennium 32 software (version 3.05.01), comparing peak area with carotenoid standard reference curves. Carotenoid standards were a generous gift of DSM Ltd, Basel, Switzerland.
Plasma lipids and apolipoproteins
Triglyceride and total cholesterol concentrations were determined by enzymatic procedures with commercial kits (Roche, Basel, Switzerland). High-density lipoprotein (HDL) cholesterol was measured after sodium phosphotungstate–magnesium chloride precipitation. Low-density lipoprotein (LDL) cholesterol was estimated indirectly by use of the Friedewald formula.
Characteristics of the cohort of 622 subjects
In order to verify the associations observed in the study on 29 volunteers we decided to verify associations observed between genetic variants and plasma lutein on a cohort of 622 French subjects. This cohort was a subset of the French SU.VI.MAX cohort (Citation42). Characteristics of this cohort were as follows: 281 males and 341 females, 61.65 ± 0.25 years old, body mass index (BMI) 25.76 ± 0.17 kg/m2, plasma cholesterol 2.23 ± 0.01 g/L, plasma lutein 540 ± 10 nmol/L. Subjects were genotyped for SNPs in CD36 and BCMO1, and haplotype effects of these genes on plasma lutein were tested.
Statistical analysis
As a first approach, we wanted to estimate the relationships between genotypes, and both plasma lutein and MPOD in the study on 29 volunteers. For this purpose, we used partial least square (PLS) regression. PLS regression was performed by the SIMCA-P software, version 11.0 (Umetrics, Umeå, Sweden). The Y matrix was composed of the two vectors containing the initial plasma lutein concentrations and the initial MPOD. The X matrix contained the genotypes for the 20 SNPs. The values in the Y matrix were scaled with a unit variance scaling prior to the calculation of the latent vectors. After examination of the importance of each dependent variable, it appeared that only six SNPs were significant for the regression model. Therefore, a second regression, which used the genotypes from these six SNPs in the X matrix, was performed in order to minimize the noise in the model. The PLS model was calculated with the function ‘autofit’ in the SIMCA-P software in order to find the optimal number of latent vectors. Note that correction for multiple testing was not compulsory with PLS regression because independent variables are not treated independently but are used simultaneously to find a reduced number of latent variables for the model. Furthermore, model validation, testing for the robustness of the model and its generalization to new data, is performed in this model with cross-validation.
In a second approach, we performed univariate statistical analyses only in associations that were simultaneously significantly associated with the two independent markers of lutein metabolism, which were plasma lutein and MPOD. The independence of these two parameters was checked by assessing the relationship between them (lack of linear Pearson's correlation). The fact that SNPs were found associated with two independent markers of lutein metabolism by both PLS and univariate analysis reduces the risk of false positive associations. Differences between means obtained in the different genotype groups were analyzed either by ANOVA followed by the post-hoc Tukey-Kramer test, or by the Student's t test when only two genotypes were observed. Values of P < 0.05 were considered significant. All univariate statistical analyses were performed with Statview software version 5.0 (SAS Institute, Cary, NC, USA).
In a third approach we performed a haplotype-based association analysis of data which originated from a cohort of 622 French subjects (see above the characteristic of the subjects). This analysis was performed with the Thesias software, which is based on the Stochastic-EM algorithm, and which allows to infer haplotypes from genotypic data and to test their associations with phenotypes of interest (Citation43). Analyses were performed after adjustment for the following covariables: gender, age, and BMI.
Results
Effect of dietary intervention on dietary lutein intake
Dietary lutein intake of the subjects fell from 1.3 mg/d to 0.03 mg/d after the lutein-poor diet, showing a good compliance of the subjects to the recommendations, and increased to 0.4 mg/d during the dietary intervention, which was probably due to the fact that this period occurred during spring where most subjects increase their intake of fruits and vegetables rich in lutein. Note that there was no significant difference between the intakes of the two groups at any of these periods.
Effect of the dietary intervention on plasma lutein concentrations
Initial plasma lutein concentrations ranged between 110 and 470 nmol/L (290 ± 20 nmol/L). This high interindividual variability (coefficient of variation (CV) 32%) did not change when plasma lutein was corrected for plasma cholesterol (CV 36%), nor when it was corrected for lutein+zeaxanthin intake (CV 146%). Surprisingly, the group that took the placebo (placebo group) exhibited a significant (P < 0.05) rise in plasma lutein concentration after the dietary intervention (). Nevertheless, this increase was much higher in the group that took lutein supplement (+143%) than in the placebo group (+77%). Individual plasma lutein responses to the placebo and the lutein supplement are shown in and , respectively. The interindividual response (change from the initial value) varied strongly, ranging from −100 to 1,140 nmol/L. Consequently, the interindividual variability in plasma lutein increased after lutein supplementation (CV 29% and 56% before and after supplementation, respectively).
Figure 1. A: Plasma lutein concentrations, before and after supplementation, in both the group that took the placebo and the group that took the lutein supplement. White bars: values measured before supplementation. Black bars: values measured after supplementation. Means ± SEM of 15 subjects in placebo group and 14 subjects in lutein group. An asterisk indicates a significant difference (P < 0.05) between values measured before and after the supplementation period in each group (paired t test). B: Individual plasma lutein concentrations, before and after supplementation, in the placebo group. C: Individual plasma lutein concentrations, before and after supplementation, in the lutein-supplemented group.
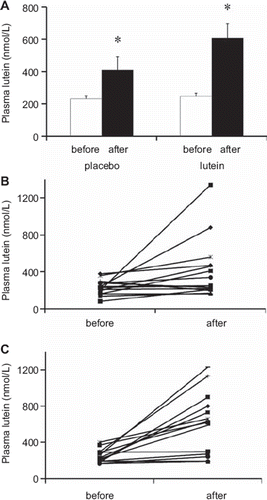
Because lutein is almost exclusively carried by lipoproteins and because it is the reason why vitamin E (α-tocopherol) is usually corrected for plasma cholesterol, we corrected plasma lutein for plasma cholesterol. This adjustment led to the observation of a significant increase (P = 0.003) in the plasma lutein/cholesterol ratio after lutein supplementation and no significant increase (P = 0.067) of this ratio after supplementation by the placebo (data not shown).
Relationships between genetic variants and biomarkers of lutein status
The PLS analysis showed that the best model to explain the variance of both plasma lutein and MPOD was the one with one latent vector only. This model explained 31.4% of the variance of the Y matrix (38.1% for MPOD and 25.1% for plasma lutein). The predictive value of the model was estimated after cross-validation. We obtained an estimated variation of Y explained by the model of 16.1% (14.6% for MPOD and 18.3% for plasma lutein). The six SNPs selected by the regression model were found in four genes: BCMO1, CD36, ABCG8, and NPC1L1. Coefficients of NPC1L1 were, however, not statistically significant. Furthermore, only the SNP in BCMO1 and two SNPs in CD36 were simultaneously significantly related to the two independent markers of lutein status. Thus association studies described thereafter were performed only with these two genes.
Associations between genetic variants and plasma lutein levels
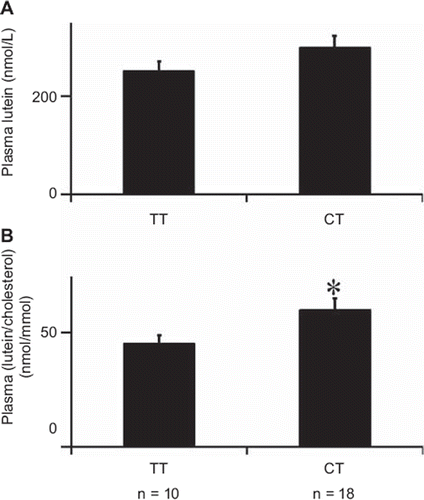
Univariate analysis of plasma lutein showed that this parameter was higher in subjects who carried the CT genotype at BCMO1 rs7501331 than in those who carried the TT genotype (), although the difference was not significant. Note that no subject homozygous for the C allele was found in the study cohort. The difference became significant (P = 0.022) when plasma lutein was corrected for plasma cholesterol: +33% in the subjects with the CT genotype (). Finally, no significant difference was observed when plasma lutein was corrected for lutein intake (data not shown).
Figure 2. Base-line plasma lutein concentrations for each BCMO1 rs7501331 genotype. A: uncorrected values. B: Values corrected for plasma total cholesterol. Data are means ± SEM of 28 subjects; n is the number of subjects in each genotype group. An asterisk indicates a significant difference (P < 0.05) between genotype groups (Student's t test).
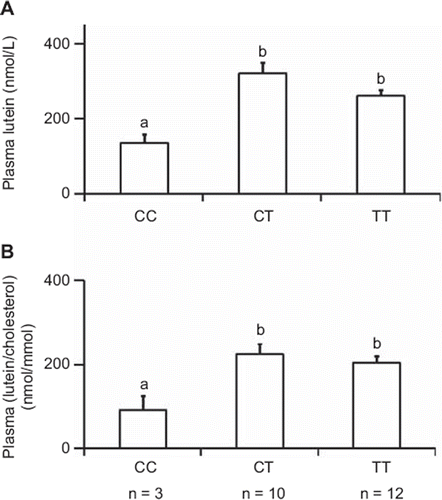
Univariate analysis also showed that subjects with the CC genotype at rs13230419 (a CD36 locus) had lower plasma lutein (P = 0.014) than subjects with a T allele at this locus (). The difference remained significant (P = 0.044) when plasma lutein was corrected for plasma cholesterol (). Adjustment for lutein intake led to a comparable figure (data not shown).
Figure 3. Base-line plasma lutein concentrations for each CD36 rs13230419 genotype. A: uncorrected values. B: Values corrected for plasma total cholesterol. Data are means ± SEM of 25 subjects; n is the number of subjects in each genotype group. In each figure, different letters indicate significant differences (P < 0.05) between genotype groups (ANOVA).
and show results of haplotype association analysis between BCMO1 and CD36 haplotypes and plasma lutein/zeaxanthin concentration in a cohort of 622 subjects. There was no significant effect of the BCMO1 haplotypes on plasma lutein, either adjusted or not adjusted for plasma cholesterol (). Conversely, a minor haplotype of CD36 (AGA) had a significant effect on plasma lutein/zeaxanthin concentrations as compared to the most frequent haplotype (AAG). Note that this effect remained significant (P = 0.048) when plasma lutein/zeaxanthin was adjusted for plasma cholesterol (data not shown).
Table III. BCMO1 haplotypes effects on plasma lutein/zeaxanthin.
Table IV. CD36 haplotypes effects on plasma lutein/zeaxanthin.
Effect of the dietary intervention on MPOD
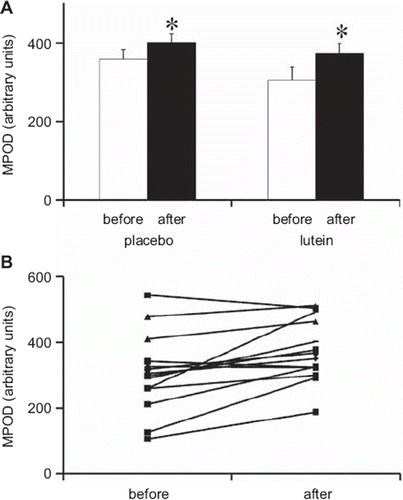
MPOD measured before supplementation was not significantly different between the two groups (). After the supplementation period, MPOD significantly increased (P < 0.05) in both groups. Again, this increase was higher in the lutein group (+22%) than in the placebo group (+11%) (). Interestingly the interindividual variability in initial MPOD (CV 32%) () was similar to that observed for plasma lutein and, as observed for plasma lutein, increased when MPOD was corrected for lutein intake (CV 142%). Conversely, opposite to what was observed for plasma lutein, the interindividual variability in MPOD decreased after supplementation with lutein (CV decreased from 39% to 25%) ().
Figure 4. A: MPOD (macular pigment optical density), before and after supplementation, in the group that took the placebo and the group that took the lutein supplement. White bars: values measured before supplementation (before). Black bars: values measured after supplementation (after). Means ± SEM of 15 subjects in placebo group and 14 subjects in lutein group. An asterisk indicates a significant difference (P < 0.05) between values measured before and after the supplementation period in each group (paired t test). B: Individual MPOD before and after supplementation in the lutein-supplemented group (n = 14).
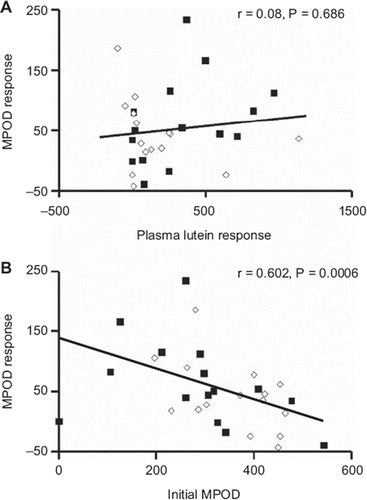
shows that the MPOD response was not related to the plasma lutein response. Furthermore, the initial MPOD was not related to the initial plasma lutein (r = 0.009; P = 0.96), when either corrected or not corrected for plasma cholesterol (r = 0.20; P = 0.30) (data not shown). Conversely, there was a significant inverse relationship between the MPOD response and the initial MPOD (). In other words, the lower the initial MPOD was, the higher the MPOD response to the dietary intervention.
Figure 5. A: MPOD response as a function of plasma lutein response. B: MPOD response as a function of initial MPOD. ‘Response’ indicates the differences between the values measured after the dietary intervention and the values measured before the intervention (n = 29). White dots: subjects in the placebo group. Black dots: subjects in the lutein-supplemented group.
Association between genetic variants and MPOD
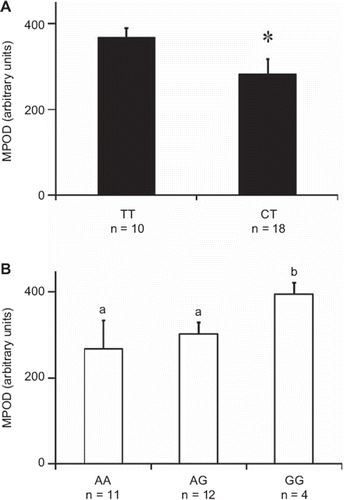
shows the results of univariate analysis between the SNPs in BCMO1 and CD36 and the initial MPOD. Subjects with CT at the BCMO1 SNP had significantly lower initial MPOD values than homozygous TT (). Interestingly the CT subjects had a higher MPOD response (delta from initial values) to the lutein supplement than the TT subjects (data not shown). Subjects bearing an A allele at rs1761667 (a CD36 locus) had significantly lower MPOD than homozygous GG (). As observed for BCMO1, the genetic groups with the lower MPOD (the AA and AG groups) had a higher MPOD response to the lutein supplement than the group with the higher MPOD (the GG group) (data not shown).
Figure 6. MPOD for each BCMO1 and CD36 genotype. A: BCMO1 rs7501331 genotypes. An asterisk indicates a significant difference (P < 0.05) between genotype groups (Student's t test). B: CD36 rs1761667 genotypes. Different letters indicate significant differences (P < 0.05) between genotype groups (ANOVA); n is the number of subjects in each genotype group.
Discussion
Initially, we verified the effect of lutein supplementation on two markers of lutein status, which were plasma lutein and MPOD. The dose of lutein provided by the supplement (10 mg/d) and the duration of the supplementation (6 months) were selected to enable the detection of significant variations in MPOD (Citation44–46). The fact that, for better compliance, lutein was provided as pills instead of foods (spinach, for example) is unlikely to have influenced the results significantly, because it has been shown that lutein bioavailability is not significantly different between spinach and lutein supplements (Citation47,Citation48). The significant increase of both markers in the lutein-supplemented group was expected, because it is in agreement with previous studies (Citation44,Citation48,Citation49). The first obvious conclusion is that these increases were due to the lutein present in the supplement. The significant increase of both markers in the placebo (control) group did, however, raise some questions about this conclusion. Nevertheless, it should be remembered that lutein was not only provided by the supplement, but it was also, of necessity, present in the subjects’ diets. We thus hypothesized that the increase of the lutein markers in the placebo group was due to an increase in dietary lutein intake. This hypothesis was supported by the fact that a second food diary, kept during the supplementation period, which occurred in spring where subjects increase their intake of lutein-rich fruit and vegetables, showed that all subjects increased their dietary intake of lutein as compared to their initial dietary intake. Given that the food diaries showed that the dietary intake of lutein during the supplementation period was not significantly different between the two groups, this demonstrated that the higher increase of both markers in the lutein-supplemented group (+143% versus +77% for plasma lutein and +22% versus +11% for MPOD, in the lutein and placebo groups, respectively) was due to the lutein supplement.
MPOD is assumed to be related to the macula lutea concentration of lutein. Because some studies showed a significant relationship between blood lutein and MPOD (Citation36,Citation50), while other studies did not find this association (Citation49,Citation51), we aimed to re-assess this relationship. Our results show that MPOD was not related to plasma lutein, whatever the parameter used, such as plasma lutein, plasma lutein corrected for plasma cholesterol, or plasma lutein corrected for lutein intake. In fact, this association, when observed, was weak and found only with a large number of subjects (Citation36,Citation52). Thus, considering the small number of subjects in our study, our result is not surprising. Interestingly, and in agreement with a recent study (Citation53), we found that the MPOD response was inversely and significantly related to the initial MPOD. This shows that the subjects who respond the most to dietary lutein are those who have the lowest initial MPOD. This should be taken into account in future studies of the effect of different parameters on MPOD.
The main objective of this study was to check the hypothesis that genetic variants can explain, at least in part, the interindividual variability in blood lutein concentrations and MPOD observed in this study ( and ), as well as in previous studies. To that aim, we compared plasma lutein concentrations and MPOD in volunteers bearing different SNPs in genes potentially involved in lutein metabolism. Because we wanted to predict blood lutein concentrations and MPOD (dependent variables) from a large set of SNPs (independent variables), we used PLS regression. As stated in the Materials and method section this analysis has the advantage of avoiding false positive results that could be obtained in multiple testing and small sample size. This analysis showed that a significant part of the variability (38% for MPOD and 25% for plasma lutein) can be assigned to genetic variants in CD36, BCMO1, and ABCG8. Furthermore, three SNPs were simultaneously and significantly related to plasma lutein and MPOD: one in BCMO1 and two in CD36. Since plasma lutein and MPOD were not related, this double association further supports the involvement of these genes in lutein metabolism. However, in order to reinforce the validity of these associations we decided to perform a haplotype-based association analysis of data which originated from a cohort of 622 subjects. This analysis confirmed the association of plasma lutein with CD36 but failed to find a significant association between plasma lutein and BCMO1. This is likely due to insufficient statistical power because a recent genome-wide association study supported a role of BCMO1 in plasma lutein levels (Citation54).
The association of plasma lutein concentration with BCMO1 suggests that this gene, which encodes an enzyme responsible for the cleavage of provitamin A carotenoids into retinal (Citation29), is also involved in the metabolism of lutein, which is not a provitamin A carotenoid. This association is probably indirect because zeaxanthin (a xanthophyll which exhibits a chemical structure very close to lutein) was not cleaved in vitro by BCMO1 (Citation55), supporting the assumption that BCMO1 is not involved in lutein cleavage. One hypothesis might be that vitamin A status, related to BCMO1 activity, can modulate lutein metabolism by modulating expression of transporters of lutein. This is supported by a publication showing that all-trans retinoic acid strongly up-regulates CD36 (Citation56). The association between BCMO1 and MPOD was observed for the first time and suggests that BCMO1 activity modulates the concentration of lutein in the retina. The effect of this SNP in BCMO1 on plasma and retina concentration of lutein is probably related to an effect on BCMO1 activity. Indeed this SNP leads to a change in an amino acid (A379V), and this change is functional, as a recombinant 267S + 379V double mutant showed a reduced BCMO1 catalytic activity and carriers of the 379V genotype had a reduced ability to convert β-carotene (Citation57).
The second gene that is apparently involved in lutein metabolism is the scavenger receptor CD36, also known as FAT. This is not very surprising, as SR-BI, a scavenger receptor like CD36, has been involved in the uptake of lutein by both intestinal (Citation13) and retinal cells (Citation28). Because CD36 is expressed in the intestine and in several other tissues, we hypothesize that functional variants in its gene may affect lutein absorption efficiency, or tissue uptake of lutein, or both, leading to lower concentrations of plasma and retina lutein. Unfortunately, there are no functional data available about the effect of the studied variants in CD36 on the activity, or expression, of CD36.
The lack of association between the two lutein biomarkers and the SNPs in SCARB1, the gene that encodes SR-BI, was rather unexpected, since this transporter has been involved in lutein absorption (Citation13) and retinal cell uptake (Citation28). Furthermore, two out of the three genotyped SNPs (rs5888 and rs4238001) have been associated with lower SR-BI protein expression and function (Citation58,Citation59). Thus, the lack of association between these SNPs and plasma lutein and MPOD, together with the fact that CD36 (another scavenger receptor which shares many structural characteristics with SR-BI) was associated with plasma and retina lutein, suggests that CD36 is probably more important for lutein uptake by cells than SR-BI and that it can compensate for lower expression/function of SR-BI.
The fact that two genes were associated with the studied biomarkers of lutein status raises questions about interactions between these genes. Unfortunately, the number of subjects enrolled in this study was too low to detect a significant interaction between BCMO1 and CD36 (two factors ANOVA).
In summary, our results suggest that genetic variants in BCMO1 and CD36 modulate plasma and retina lutein concentrations. This observation may have consequences with regard to the recommended dietary intake of lutein in groups of subjects bearing unfavorable genetic variants in these genes.
Declaration of interest: Work on the cohort of 29 subjects was funded by Pileje. Pileje participated in the design of the study and approval of the manuscript. Work on the subset cohort of SU.VI.MAX was granted by ANR (n°ANR-05-PNRA-010), DGS (Ministry of Health), and supported by Médéric, Ipsen, MGEN, SODEXHO, and Pierre Fabre. None of these organizations or companies participated in the design or conducted of this research.
S. Baudry, S. Drouault-Holowacz, and S. Bieuvelet all worked for Pileje which provided the supplement (Visiobane protect) used in the study.
References
- Evans JR. Risk factors for age-related macular degeneration. Prog Retin Eye Res. 2001;20:227–53.
- Krinsky NI, Landrum JT, Bone RA. Biologic mechanisms of the protective role of lutein and zeaxanthin in the eye. Annu Rev Nutr. 2003;23:171–201.
- Landrum JT, Bone RA. Lutein, zeaxanthin, and the macular pigment. Arch Biochem Biophys. 2001;385:28–40.
- Johnson EJ, Neuringer M, Russell RM, Schalch W, Snodderly DM. Nutritional manipulation of primate retinas, III: Effects of lutein or zeaxanthin supplementation on adipose tissue and retina of xanthophyll-free monkeys. Invest Ophthalmol Vis Sci. 2005;46:692–702.
- Tyssandier V, Reboul E, Dumas JF, Bouteloup-Demange C, Armand M, Marcand J, . Processing of vegetable-borne carotenoids in the human stomach and duodenum. Am J Physiol Gastrointest Liver Physiol. 2003;284:G913–23.
- Reboul E, Richelle M, Perrot E, Desmoulins-Malezet C, Pirisi V, Borel P. Bioaccessibility of carotenoids and vitamin E from their main dietary sources. J Agric Food Chem. 2006; 54:8749–55.
- Borel P, Grolier P, Armand M, Partier A, Lafont H, Lairon D, . Carotenoids in biological emulsions: solubility, surface-to-core distribution, and release from lipid droplets. J Lipid Res. 1996;37:250–61.
- Tyssandier V, Lyan B, Borel P. Main factors governing the transfer of carotenoids from emulsion lipid droplets to micelles. Biochim Biophys Acta. 2001;1533:285–92.
- Borel P. Factors affecting intestinal absorption of highly lipophilic food microconstituents (fat-soluble vitamins, carotenoids and phytosterols). Clin Chem Lab Med. 2003;41: 979–94.
- Hollander D, Muralidhara KS. Vitamin A1 intestinal absorption in vivo: influence of luminal factors on transport. Am J Physiol Gastrointest Liver Physiol. 1977;232:E471–7.
- Hollander D. Intestinal absorption of vitamin A, E, D, and K. J Lab Clin Med. 1981;97:449–62.
- Hollander D, Ruble PE. beta-carotene intestinal absorption: bile, fatty acid, pH, and flow rate effects on transport. Am J Physiol. 1978;235:E686–91.
- Reboul E, Abou L, Mikail C, Ghiringhelli O, Andre M, Portugal H, . Lutein transport by Caco-2 TC-7 cells occurs partly by a facilitated process involving the scavenger receptor class B type I (SR-BI). Biochem J. 2005;387(Pt 2): 455–61.
- van Bennekum A, Werder M, Thuahnai ST, Han CH, Duong P, Williams DL, . Class B scavenger receptor-mediated intestinal absorption of dietary beta-carotene and cholesterol. Biochemistry. 2005;44:4517–25.
- During A, Dawson HD, Harrison EH. Carotenoid transport is decreased and expression of the lipid transporters SR-BI, NPC1L1, and ABCA1 is downregulated in Caco-2 cells treated with ezetimibe. J Nutr. 2005;135:2305–12.
- During A, Harrison EH. Mechanisms of provitamin A (carotenoid) and vitamin A (retinol) transport into and out of intestinal Caco-2 cells. J Lipid Res. 2007;48:2283–94.
- Moussa M, Landrier JF, Reboul E, Ghiringhelli O, Comera C, Collet X, . Lycopene absorption in human intestinal cells and in mice involves scavenger receptor class B type I but not niemann-pick C1-like 1. J Nutr. 2008;138:1432–6.
- , Davis HR JrZhu LJ, Hoos LM, Tetzloff G, Maguire M, Liu J, . Niemann-Pick C1 Like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J Biol Chem. 2004;279:33586–92.
- Narushima K, Takada T, Yamanashi Y, Suzuki H. Niemann-pick C1-like 1 mediates alpha-tocopherol transport. Mol Pharmacol. 2008;74:42–9.
- Reboul E, Klein A, Bietrix F, Gleize B, Malezet-Desmoulins C, Schneider M, . Scavenger receptor class B type I (SR-BI) is involved in vitamin E transport across the enterocyte. J Biol Chem. 2006;281:4739–45.
- Lee MH, Lu K, Patel SB. Genetic basis of sitosterolemia. Curr Opin Lipidol. 2001;12:141–9.
- Oram JF. Tangier disease and ABCA1. Biochim Biophys Acta. 2000;1529:321–30.
- Oram JF, Vaughan AM, Stocker R. ATP-binding cassette transporter A1 mediates cellular secretion of alpha-tocopherol. J Biol Chem. 2001;276:39898–902.
- Reboul E, Trompier D, Moussa M, Klein A, Landrier JF, Chimini G, . ATP-binding cassette transporter A1 is significantly involved in the intestinal absorption of alpha- and gamma-tocopherol but not in that of retinyl palmitate in mice. Am J Clin Nutr. 2009;89:177–84.
- Tyssandier V, Choubert G, Grolier P, Borel P. Carotenoids, mostly the xanthophylls, exchange between plasma lipoproteins. Int J Vitam Nutr Res. 2002;72:300–8.
- Borel P, Moussa M, Reboul E, Lyan B, Defoort C, Vincent-Baudry S, . Human plasma levels of vitamin E and carotenoids are associated with genetic polymorphisms in genes involved in lipid metabolism. J Nutr. 2007;137:2653–9.
- Borel P, Moussa M, Reboul E, Lyan B, Defoort C, Vincent-Baudry S, . Human fasting plasma concentrations of vitamin E and carotenoids, and their association with genetic variants in apo C-III, cholesteryl ester transfer protein, hepatic lipase, intestinal fatty acid binding protein and microsomal triacylglycerol transfer protein. Br J Nutr. 2009; 101:680–7.
- During A, Doraiswamy S, Harrison EH. Xanthophylls are preferentially taken up compared with beta-carotene by retinal cells via a SRBI-dependent mechanism. J Lipid Res. 2008;49:1715–24.
- von Lintig J, Vogt K. Vitamin A formation in animals: molecular identification and functional characterization of carotene cleaving enzymes. J Nutr. 2004;134:251S–6S.
- Vage DI, Boman IA. A nonsense mutation in the beta-carotene oxygenase 2 (BCO2) gene is tightly associated with accumulation of carotenoids in adipose tissue in sheep (Ovis aries). BMC Genet. 2010;11:10.
- Hercberg S, Deheeger M, Preziosi P. SU.VI.MAX: Portions alimentaires. Manuel photos pour l'estimation des quantités. SU.VI.MAX. Economica. Paris: 2002. Portions alimentaires - Manuel photos pour l'estimation des quantités.
- Chug-Ahuja JK, Holden JM, Forman MR, Mangels AR, Beecher GR, Lanza E. The development and application of a carotenoid database for fruits, vegetables, and selected multicomponent foods. J Am Diet Assoc. 1993;93: 318–23.
- Cardinault N, Tyssandier V, Grolier P, Winklhofer-Roob BM, Ribalta J, Bouteloup-Demange C, . Comparison of the postprandial chylomicron carotenoid responses in young and older subjects. Eur J Nutr. 2003;42:315–23.
- Tyssandier V, Feillet-Coudray C, Caris-Veyrat C, Guilland JC, Coudray C, Bureau S, . Effect of tomato product consumption on the plasma status of antioxidant microconstituents and on the plasma total antioxidant capacity in healthy subjects. J Am Coll Nutr. 2004;23:148–56.
- Caris-Veyrat C, Amiot MJ, Tyssandier V, Grasselly D, Buret M, Mikolajczak M, . Influence of organic versus conventional agricultural practice on the antioxidant microconstituent content of tomatoes and derived purees; consequences on antioxidant plasma status in humans. J Agric Food Chem. 2004;52:6503–9.
- Curran-Celentano J, Hammond BR Jr, Ciulla TA, Cooper DA, Pratt LM, Danis RB. Relation between dietary intake, serum concentrations, and retinal concentrations of lutein and zeaxanthin in adults in a Midwest population. Am J Clin Nutr. 2001;74:796–802.
- De la Vega FM, Lazaruk KD, Rhodes MD, Wenz MH. Assessment of two flexible and compatible SNP genotyping platforms: TaqMan SNP Genotyping Assays and the SNPlex Genotyping System. Mutat Res. 2005;573:111–35.
- Tobler AR, Short S, Andersen MR, Paner TM, Briggs JC, Lambert SM, . The SNPlex genotyping system: a flexible and scalable platform for SNP genotyping. J Biomol Tech. 2005;16:398–406.
- Zagers NPA, van Norren D. Absorption of the eye lens and macular pigment derived from the reflectance of cone photoreceptors. J Opt Soc Am. 2004;21:2257–68.
- van de Kraats J, van Norren D. Directional and nondirectional spectral reflection from the human fovea. J Biomed Opt. 2008;13:024010.
- Lyan B, Azais-Braesco V, Cardinault N, Tyssandier V, Borel P, Alexandre-Gouabau MC, . Simple method for clinical determination of 13 carotenoids in human plasma using an isocratic high-performance liquid chromatographic method. J Chromatogr B Biomed Appl. 2001;751:297–303.
- Hercberg S, Galan P, Preziosi P, Bertrais S, Mennen L, Malvy D, . The SU.VI.MAX Study: a randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch Intern Med. 2004;164: 2335–42.
- Tregouet DA, Escolano S, Tiret L, Mallet A, Golmard JL. A new algorithm for haplotype-based association analysis: the Stochastic-EM algorithm. Ann Hum Genet. 2004;68(Pt 2): 165–77.
- Johnson EJ, Chung HY, Caldarella SM, Snodderly DM. The influence of supplemental lutein and docosahexaenoic acid on serum, lipoproteins, and macular pigmentation. Am J Clin Nutr. 2008;87:1521–9.
- Richer S, Stiles W, Statkute L, Pulido J, Frankowski J, Rudy D, . Double-masked, placebo-controlled, randomized trial of lutein and antioxidant supplementation in the intervention of atrophic age-related macular degeneration: the Veterans LAST study (Lutein Antioxidant Supplementation Trial). Optometry. 2004;75:216–30.
- Bone RA, Landrum JT, Guerra LH, Ruiz CA. Lutein and zeaxanthin dietary supplements raise macular pigment density and serum concentrations of these carotenoids in humans. J Nutr. 2003;133:992–8.
- Riso P, Brusamolino A, Ciappellano S, Porrini M. Comparison of lutein bioavailability from vegetables and supplement. Int J Vitam Nutr Res. 2003;73:201–5.
- Chung HY, Rasmussen HM, Johnson EJ. Lutein bioavailability is higher from lutein-enriched eggs than from supplements and spinach in men. J Nutr. 2004;134: 1887–93.
- Cardinault N, Gorrand JM, Tyssandier V, Grolier P, Rock E, Borel P. Short-term supplementation with lutein affects biomarkers of lutein status similarly in young and elderly subjects. Exp Gerontol. 2003;38:573–82.
- Broekmans WM, Berendschot TT, Klopping-Ketelaars IA, de Vries AJ, Goldbohm RA, Tijburg LB, . Macular pigment density in relation to serum and adipose tissue concentrations of lutein and serum concentrations of zeaxanthin. Am J Clin Nutr. 2002;76:595–603.
- Johnson EJ, Hammond BR, Yeum KJ, Qin J, Wang XD, Castaneda C, . Relation among serum and tissue concentrations of lutein and zeaxanthin and macular pigment density. Am J Clin Nutr. 2000;71:1555–62.
- Bone RA, Landrum JT, Dixon Z, Chen Y, Llerena CM. Lutein and zeaxanthin in the eyes, serum and diet of human subjects. Exp Eye Res. 2000;71:239–45.
- Richer S, Devenport J, Lang JC. LAST II: Differential temporal responses of macular pigment optical density in patients with atrophic age-related macular degeneration to dietary supplementation with xanthophylls. Optometry. 2007; 78:213–9.
- Ferrucci L, Perry JR, Matteini A, Perola M, Tanaka T, Silander K, . Common variation in the beta-carotene 15,15′-monooxygenase 1 gene affects circulating levels of carotenoids: a genome-wide association study. Am J Hum Genet. 2009;84:123–33.
- Grolier P, Duszka C, Borel P, Alexandre-Gouabau MC, Azais-Braesco V. In vitro and in vivo inhibition of beta-carotene dioxygenase activity by canthaxanthin in rat intestine. Arch Biochem Biophys. 1997;348:233–8.
- Langmann T, Liebisch G, Moehle C, Schifferer R, Dayoub R, Heiduczek S, . Gene expression profiling identifies retinoids as potent inducers of macrophage lipid efflux. Biochim Biophys Acta. 2005;1740:155–61.
- Leung WC, Hessel S, Meplan C, Flint J, Oberhauser V, Tourniaire F, . Two common single nucleotide polymorphisms in the gene encoding beta-carotene 15,15′-monoxygenase alter beta-carotene metabolism in female volunteers. FASEB J. 2009;23:1041–53.
- West M, Greason E, Kolmakova A, Jahangiri A, Asztalos B, Pollin TI, . Scavenger receptor class B type I protein as an independent predictor of high-density lipoprotein cholesterol levels in subjects with hyperalphalipoproteinemia. J Clin Endocrinol Metab. 2009;94:1451–7.
- Constantineau J, Greason E, West M, Filbin M, Kieft JS, Carletti MZ, . A synonymous variant in scavenger receptor, class B, type I gene is associated with lower SR-BI protein expression and function. Atherosclerosis. 2010;210: 177–82.