Abstract
Most organelles within the exocytic and endocytic pathways typically acidify their interiors, a phenomenon that is known to be crucial for their optimal functioning in eukaryotic cells. This review highlights recent advances in our understanding of how Golgi acidity is maintained and regulated, and how its misregulation contributes to organelle dysfunction and disease. Both its biosynthetic products (glycans) and protein-sorting events are highly sensitive to changes in Golgi luminal pH and are affected in certain human disease states such as cancers and cutis laxa. Other potential disease states that are caused by, or are associated with, Golgi pH misregulation will also be discussed.
Key words::
Key messages
Golgi pH is strictly regulated.
Misregulation of Golgi pH induces glycosylation defects.
Golgi pH is misregulated in cancer cells and cutis lax.
Introduction
Eukaryotic cells have evolved multiple systems to compartmentalize their functions into membrane-bound organelles. This allows them to complete numerous reactions simultaneously with high precision and speed. A key issue in this functional optimization is the ability to generate an optimal microenvironment for each process. For example, while mitochondria are alkaline and use this pH gradient to synthesize ATP, organelles within the secretory and endocytic pathways typically are acidic (Citation1–3). Organellar acidity is used to regulate and maintain many fundamental cellular processes that range from vesicular trafficking to post-translational processing and protein sorting, import of nutrients, pathogens, and toxins, as well as degradation in lysosomes (Citation4–13). The pH dependency of these processes has traditionally been studied using organellar pH gradient-dissipating drugs, such as monensin (a Na+/H+ ionophore), chloroquine (a weak base), and bafilomycin A (a vacuolar H-ATPase inhibitor).
Organelle acidity is in general thought to be regulated by three main ion transport systems. These are ATP-mediated proton pumping, counter-ion conductance, and intrinsic proton leakage (Citation2,Citation3,Citation14–19). Protons (H+) are pumped into membranous compartments by the vacuolar H+ ATPase (V-ATPase). Counter-ion conductance is used to prevent the build-up of excess positive charge (membrane potential) via passive influx of anions (e.g. chloride) and/or efflux of another cation (e.g. potassium). Intrinsic H+ leakage across organellar membranes in turn counteracts proton import and prevents excessive over-acidification of the organelle lumen (Citation15). Therefore, it may contribute significantly to the observed variations in the steady-state pH of the organelles within the secretory and endocytic pathways (). A pertinent question that specifically concerns the Golgi apparatus is how the pH is uniquely regulated along the cis-trans axis of the organelle. Golgi pH regulation also poses another problem. This relates to its functioning as a central station between the secretory and endocytic pathways, receiving and sending vesicular carriers between these two functionally different but converging membrane systems. How the Golgi apparatus can deal with such challenges, and maintain its slightly acidic physiological pH, will be discussed next. Later we will discuss how its misregulation contributes to human disease.
Figure 1. Schematic representation of the compartmentalized secretory (biosynthetic) and endocytic (degradative) pathways and their convergence at the level of the Golgi apparatus (modified from (Citation146)). Note the different steady-state pH values of each organelle. ERGIC is a pleiomorphic membrane structure between the ER and the Golgi, where proteins are arrested at 15°C (Citation26).
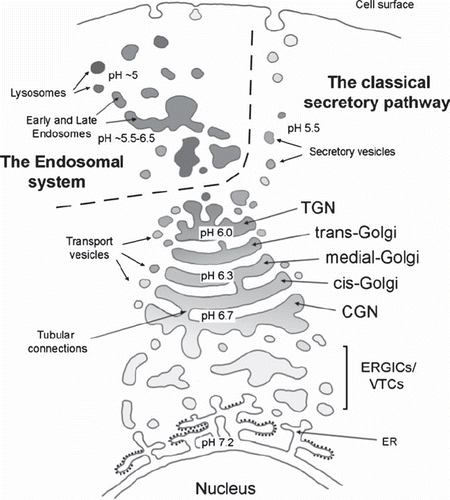
The Golgi apparatus and the secretory pathway
An Italian scientist, Camillo Golgi, initially described the Golgi apparatus in 1898 as an odd reticular structure in nerve cells (Citation20). The Golgi apparatus is now recognized as a crucial component of the secretory pathway () that also converges with the endocytotic pathway (Citation21,Citation22). Together, these two pathways are responsible for the various biosynthetic (secretory pathway) and degradative processes (endocytotic pathway). They form a dynamic, constantly fluctuating, polymorphic intracellular membrane system (Citation23–25), so that the exact borders of each organelle are sometimes difficult to define.
According to the current view, however, the Golgi apparatus begins from or after the endoplasmic reticulum–Golgi intermediate compartment (ERGIC) (Citation26,Citation27) and can be divided into three main compartments: the cis-Golgi network (CGN), the Golgi stack of cisternae (cis-Golgi, medial-Golgi, trans-Golgi), and the trans-Golgi network (TGN). Under the electron microscope the Golgi cisternae look like flattened pleiomorphic discs with dilated rims and adjacent vesicles. Tubular extrusions sometimes connect neighboring cisternae and even adjacent stacks (Citation28–33). The CGN and the TGN, in turn, consist of tubular networks, whose lateral regions are occupied by budding and fusing vesicles (Citation21,Citation22,Citation34). A typical higher eukaryotic cell contains multiple Golgi stacks that assemble into a peri- or juxtanuclear structure around the microtubule-organizing center (MTOC) (Citation35,Citation36). Each Golgi stack of cisternae is an independent unit where several post-synthetic modifications take place, such as glycosylation and sulfation of proteins and lipids (Citation37–40). It also has a role in (glyco)lipid biosynthesis (Citation41), in protein and lipid sorting, as well as in cell polarization (Citation23,Citation25,Citation41,Citation42).
Due to the dynamic nature and continuous trafficking of newly synthesized and recycling proteins and lipids, the maintenance of the Golgi architecture and functions requires constant remodeling. Although the mechanisms are not fully understood, it is known that cytoskeletal proteins and Golgi-specific matrix proteins are involved and appear to provide a protein scaffold for the cisternal assembly and architecture. Remodeling also requires the presence and correct functioning of many luminal and transmembrane proteins as well as strict control of vesicular trafficking and luminal pH. Once all these factors are under control, the Golgi apparatus can perform its many functions with astonishing precision and speed.
Golgi acidification by the vacuolar H+ ATPase
Golgi pH measurements with several different fluorescence-based approaches (Citation2,Citation3,Citation14–19) have shown that the Golgi luminal pH is mildly acidic and decreases along the cis-trans axis of the Golgi stack, from pH 6.7 in the CGN (entry face) to pH 6.0 in the TGN (exit face). As in other acidic organelles, the maintenance of the Golgi luminal pH in each of these Golgi subcompartments is thought to reflect changes in the rates between proton pumping, counter-ion conductance, and proton leakage across the organellar membranes. While all these main ion transport systems appear to be common to acidic organelles, additional components or various protein isoforms may be required in each case. In the next paragraphs, proteins that are currently known to regulate Golgi pH are described.
Golgi acidity is generated by the vacuolar H+ ATPase (V-ATPase), an ATP-powered proton pump (Citation43–45). This has been convincingly demonstrated by using V-ATPase-specific inhibitors such as bafilomycin A and concanamycin A. Treatment of cells with these drugs results in complete neutralization of the Golgi lumen within 10–20 min. Both in yeast and mammalian cells, the V-ATPase is a multisubunit protein complex that consists of a peripheral domain V1 and an integral membrane domain V0 (). The cytosolic V1 domain is composed of eight subunits (with an A3B3CDE2FG2H1–2stoichiometry), whereas the integral V0 domain has six subunits (with an adec4c′c″stoichiometry) (Citation43). Some of these subunits may differ between V-ATPases that localize in different cellular compartments. For example, the subunit a isoform Stv1p is found in Golgi-localized V-ATPases in yeast, while the Vph1p isoform is found in V-ATPases that localize to vacuoles (Citation43). These data suggest that different organelles may possess structurally and perhaps also functionally different V-ATPase variants.
Figure 2. The multisubunit structureof the V-ATPase (modified from (Citation146)). For further details, see text.
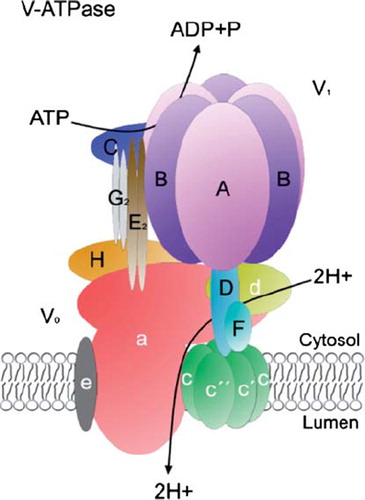
All the V-ATPases, however, are structurally and functionally similar to the mitochondrial F0F1 ATP synthase (F-ATPase), but unlike F-ATPase, which synthesizes ATP in a proton gradient-dependent manner, V-ATPase utilizes ATP to transport protons against its concentration gradient, i.e. into the organelle lumen. Hydrolysis of ATP within the V1 domain induces a rotatory movement of a c-ring in the integral V0 domain, which transports two protons, one at a time, from the cytosolic hemi-channel to a luminal hemi-channel formed by the a subunit of the V0 domain (for further details, see Jefferies et al. (Citation43) and references therein).
In the presence of excess ATP, the V-ATPase is able to acidify the Golgi lumen well below its steady-state pH (e.g. our unpublished data). This observation suggests that the density of the V-ATPase in Golgi membranes is not a limiting factor for Golgi acidity. In addition to density, the V-ATPase activity can also be regulated by other means, such as glucose-mediated reversible assembly and disassembly of the V1 and V0 domains, re-shuffling of disulfide bonds within the ATP binding A subunit, changes in coupling between ATP hydrolysis and H+ pumping (Citation43), or by regulatory proteins, such as the V-ATPase accessory subunit Ac45(Citation46). However, because the physiological role of the V-ATPase activity regulation is unclear in most cases (besides energy consumption), and the fact that V-ATPase can over-acidify the Golgi lumen, it cannot be considered as the primary factor that determines the Golgi pH (Citation1–3,Citation15,Citation17).
Counter-ion conductance
In all acidic organelles, the primary role of counter-ion conductance, i.e. influx of anions or efflux of cations, is to maintain the organelle membrane potential low despite continuous H+ import by the V-ATPase (Citation2,Citation47,Citation48). In its absence, membrane potential would increase (inside positive), which in turn would lead to inhibition of V-ATPase activity and to limited organelle acidity (Citation47). Therefore, low counter-ion conductance can modulate the extent of organelle acidification. This is the case in endosomes (Citation48–50), in which defects in the chloride channel protein 5 (ClC-5) are associated with defective endosomal acidification in the kidney (Citation51). In contrast, counter-ion conductivity of the Golgi membranes is considered to be high enough to support proton pumping (Citation1–3,Citation15,Citation17). Therefore, counter-ion conductance in the Golgi appears not to be a limiting factor for acidification. Rather, it serves as an indispensable ‘background component’ that, by preventing an increase in membrane potential, enables normal H+ pumping and Golgi acidification to or below its steady-state pH.
The transport protein that most likely mediates counter-ion conductance in the Golgi was recently reported to be an anion channel protein, called the Golgi pH regulator (GPHR)(Citation52). It was discovered by using an elegant screen of mutants with reduced secretion and altered glycosylation of cell surface constituents. Mutation of the GPHR was shown to result in delayed transport of newly synthesized proteins from the Golgi to the plasma membrane, impaired glycosylation of proteins, and structural disorganization of the Golgi apparatus. Direct measurement of the steady-state Golgi pH revealed that the mutation of GPHR decreased acidification in the Golgi by about 0.4–0.5 pH units without affecting lysosomal pH, endocytosis, or recycling. Physiological studies with recombinant GPHR showed that it may be identical to the two previously characterized Golgi-localized anion channels, GOLAC-1 and GOLAC-2 (Citation53,Citation54). All three channels have five to six conductance states that are mostly open, allowing efficient counter-ion transport. They are also weakly anion-selective (PCl/PK permeability ratio 2.8 ± 1.3), voltage and DIDS (4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid)-sensitive, and pH-insensitive. However, no data were presented to demonstrate that the GPHR alters chloride permeability of the Golgi membranes or that the lack of acidification is due to the absence of GPHR-mediated counter-ion conductance. Thus, the possibility that GPHR regulates Golgi pH by some other means cannot be excluded.
Many other ion transporters have been localized in the Golgi. These include the cystic fibrosis transmembrane conductance regulator (CFTR) (Citation55,Citation56), a mid-1-related chloride channel (MClC) (Citation57), and voltage-gated chloride channels (ClCs) (Citation51,Citation58–60). CFTR is a cAMP-regulated chloride channel which localizes to the trans-Golgi, the TGN, endosomes, and prelysosomes (Citation55). A mutation in the CFTR gene has been associated with defective acidification of these organelles (Citation55,Citation56) and will be discussed in more detail later. MClC is an anion channel (or an activator of anion channels) that localizes to the endoplasmic reticulum (ER), the Golgi, and the nucleus (Citation57). However, its role in Golgi pH regulation awaits confirmation. ClCs, on the other hand, comprise a large family of voltage-gated chloride channels, which are often required for proper acidification of intracellular organelles (Citation51,Citation59). Thus far, two ClCs, ClC-3B in mammalian cells (Citation60) and Gef-1 in yeasts(Citation58), have been detected in the Golgi membranes, but their roles in Golgi pH regulation remain unclear. Golgi membranes are also highly permeable to K+(Citation3). K+ conductance may be at least partly GPHR-mediated (Citation52,Citation53), but other K+ conductive channels may also coexist in the Golgi. An apparent role for such a K+ channel would be to expel K+ and serve as an alternative counter-ion conductance pathway during active proton pumping by the V-ATPase. Golgi membranes also contain the Na+/K+-ATPase (Citation61). Even though no clear function has been assigned to it yet, it may contribute to Golgi membrane potential via unbalanced K+/Na+ transport and thereby limit Golgi acidification by the V-ATPase. Finally, it should also be noted that the concentrations of anions or cations are interdependent and can be influenced by the transport of Ca2+, Mg2+, or Mn2+, all of which are present in the Golgi lumen at high concentrations. The latter two cations serve also as important co-factors for several Golgi glycosyltransferases (Citation62).
Golgi buffering: AE2 HCO3−/Cl− exchanger
In addition to the proteins described above, other factors such as cargo transit through the Golgi, vesicular transport, and luminal buffering may also affect Golgi pH (Citation2). Since the Golgi is located in the center of the secretory pathway, it is constantly exposed to the transit of multiple transporters and channel proteins destined for endosomes, lysosomes, and the plasma membrane. Some of the transiting proteins are fully active soon after their biosynthesis in the ER (Citation2,Citation63) and may thus affect Golgi pH. In this respect, vesicular trafficking is also a challenge, as the Golgi resides in the cross-roads between the secretory and endocytic pathways. The vesicles from the endosomal compartments bring acidic loads, whereas those from the ER bring alkaline loads to the Golgi. For the above reasons, the Golgi lumen must be able to adapt rapidly to changing pH conditions.
To confront such pH-related challenges, cells have evolved various buffering systems. One such system is thought to consist of lumenal matrix proteins that are rich in chemical groups with suitable pKa values, i.e. groups that protonate and deprotonate around physiological Golgi pH. Soluble buffering molecules, such as HCO3−, are continuously transported via specific transporters across the plasma membrane to balance cytoplasmic alkaline or acid loads and may thus also be used in the maintenance of Golgi pH (Citation1). This idea is supported by the identification of an erythrocyte anion exchanger 1 (band3, AE1, SLC4A1) homolog in the Golgi membranes of several cell types (Citation64,Citation65). More recent work has identified this transporter as the AE2a isoform (SLC4A2a) (Citation66,Citation67) of the AE gene family, members of which are all known as electroneutral HCO3−/Cl− exchangers. They normally localize in the plasma membrane where they regulate cytosolic pH, chloride concentration, and cell volume (Citation68,Citation69). Our most recent data (Rivinoja et al., unpublished) also provide direct evidenceto show that the AE2a exchanger is indeed involved in Golgi pH regulation. AE2a over-expression in COS-7 cells was found to raise Golgi pH by about 0.4 pH units. Bicarbonate also slowedATP-mediatedGolgiacidificationby the V-ATPase. Thus, by importing base (HCO3−) equivalents, the AE2a can increaseGolgi buffering capacity and prevent Golgi over-acidification. Because the exchanger resides almost exclusively in the Golgi membranes (Citation64), it seems to represent a unique system that is utilized only by this organelle.
Intrinsic proton leakage
The fourth component that is involved in Golgi pH regulation is the rate at which protons exit this compartment. As cellular membranes contain components that render them intrinsically permeable to protons, H+ exit pathways play an important role in balancing H+ pumping. Previous studies by Wu et al. (Citation15) have suggested that the decrease in pH between successive secretory compartments is likely due to decreased proton efflux and may be coupled with an increased density of the V-ATPase. Although the ‘channel’ protein itself remains to be identified, proton efflux may involve either exchange of lumenal protons for cytosolic cations, free proton conductivity, or elimination of protons by base import (Citation1,Citation2). Physiological measurements have shown that the characteristics of this putative H+ channel are reminiscent of those of the plasma membrane-localized H+ channels. Proton export from the TGN was also found to be voltage-sensitive and inhibited by Zn2+, suggesting the involvement of a regulated channel (Citation3,Citation70). However, Zn2+ inhibition was only partial. Thus, the presence of residual Zn2+ resistance may indicate the existence of another, yet unknown, proton leakage pathway. Two ubiquitously expressed and Golgi-localized Na+/H+ exchanger (NHE) isoforms, NHE7 and NHE8 (Citation71,Citation72), may also function in proton export. The Na+/H+ exchange is driven by ion gradients and may occur when H+ concentration in the Golgi lumen is high enough to drive the influx of cytosolic Na+. In support of this, over-expression of both NHE7 and NHE8 were found to increase Na+ and K+ influx to the Golgi (Citation71). Both also significantly raised luminal Golgi pH (Citation72). These data are consistent with an NHE-mediated ‘proton leakage’ pathway that involves exchange of luminal protons for cytosolic sodium or potassium ions. The existence of this pathway, however, awaits further confirmation, since NHE8 was recently reported to regulate the morphology and function of late endosomes instead of the Golgi (Citation73).
The suggested AE2-mediated import of HCO3− (in exchange for Cl− efflux) into the Golgi lumen may also provide an intriguing means to expel protons from the Golgi lumen. It is well known that HCO3−, in the presence of protons (as in the Golgi lumen), is readily converted via a short-lived carbonic acid intermediate into CO2 and H2O. These end-products can rapidly cross lipid membranes via diffusion (CO2) or aquaporin channels (H2O), thereby expelling protons. At pH 6.3, half of the HCO3− is estimated to be in the form of CO2 and H2O. This represents a sizeable H+ leak and could provide a channel-free proton exit pathway across the Golgi membranes without any extra energy cost.
An integrated model for Golgi pH regulation
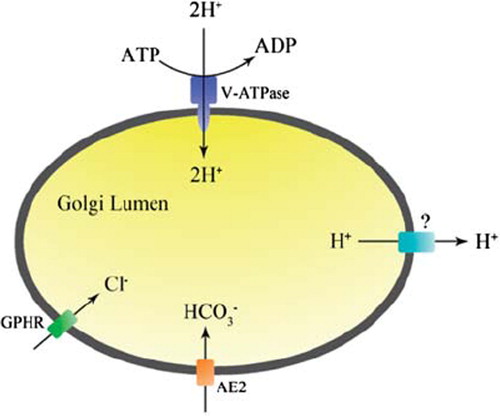
An integrated but intentionally simplified model of how Golgi pH is regulated by the various ion transport systems is presented in . In the presence of ATP, protons are continuously pumped into the Golgi lumen by the V-ATPase. This is facilitated by a simultaneous counter-anion (Cl−) influx (or alternatively via cation efflux), most likely via the GPHR ion channel. Once the Golgi pH becomes sufficiently acidic (pH < 6.3), proton efflux balances H+ pumping and prevents further acidification of the Golgi lumen. Proton efflux can be mediated by an as yet unidentified proton ‘channel’ and/or by AE2a-mediated HCO3− import. In the latter case, luminal HCO3− and H+ then can form membrane-permeable H2O and CO2via a spontaneous and reversible chemical reaction to eliminate protons. The suggested HCO3−-based model offers several benefits for the maintenance of the steady-state pH in the Golgi and, thus, also for its normal functioning. It represents an electroneutral andprotein-free proton efflux pathway across the Golgi membranes to balance H+ pumping and to prevent over-acidification of the Golgi lumen. Its end-products, H2O and CO2, can also be reutilized in the cytoplasm by converting them back to H+ and HCO3− via the activity of carbonic anhydrase II. Moreover, due to the high permeability of both end-products, the system will rarely become saturated. Future work will be needed to unravel the true nature of the H+ leakage ‘channel’ in the Golgi membranes and in other acidic organelles as well.
Figure 3. An integrated view of Golgi pH regulation with the main ion transport systems involved.
Golgi pH misregulation and diseases
Correct glycosylation, i.e. the addition of sugar structures to proteins and lipids in the ER and the Golgi apparatus, is known to be critical for a variety of basic cellular functions and biological recognition events, ranging from the regulation of gene expression to protein folding, cell signaling, immunological defense, cell motility, and the development of multicellular organisms (Citation74–77). While much progress has been made in unraveling how the highly compartmentalized, yet dynamic Golgi structure and vesicular trafficking are maintained in a mammalian cell, we still poorly understand how this organelle can faithfully construct a multitude of different sugar structures (glycans) that are often cell-, tissue-, organ-, or organism-specific; yet, if defective for some reason, a disease may develop. This is highlighted by the identification of several glycosylation-associated disease states, such as congenital diseases of glycosylation (CDGs), inflammation, cancers,and diabetes (Citation78–83). In most cases, defective glycosylation has been traced back to mutations or altered expression of Golgi glycosylation enzymes (i.e. glycosyltransferases, glycosidases) or to nucleotide sugar transporters required to provide substrates for glycan synthesis. In other cases, however, the underlying molecular defects are less apparent, and the challenge now is to identify these, which may lead to future medical interventions. One such defect is the misregulated Golgi pH, which has been shown to be directly associated in only two disease states (cancers and cutis laxa). In other cases, direct evidence is missing and warrants further investigation.
Cancer
Altered glycosylation is a hall-mark of most cancers (Citation84–88). Thesechanges include the expression of truncated and hypersialylated O-linked oligosaccharide chains (Citation89–92), reduced sulfation (Citation93), reduced O-acetylation of sialic acid (Citation94,Citation95), elevated fucosylation (Citation96), altered branching of N-glycans (Citation97,Citation98), and increased expression of blood group antigens (Citation99). Some of these glycosylation alterations, such as increased expression of truncated sialyl-Tn (Neu5Acα2-6GalNAcα1-Ser/Thr) or T-antigens (core 1 Galβ1-3GalNAcα1-Ser/Thr) (Citation92,Citation100–102), are associated with increased proliferation, invasion, and metastasis of many cancer cell types and are therefore useful prognostic markers for malignancy. Most likely, by modulatinginteractions of cells with other cells or the extracellular matrix, altered glycosylation can also modulate overall cell behavior. Consistent with this view, terminal Galβ1-3/4GalNAc and/or sialic acid may facilitateinteractions with galectins (soluble proteins that recognize N-acetyllactosamine-like (Galβ1-3/4GalNAcα1-Ser/Thr) structures (Citation144)) and siglecs (type I membrane proteins that bind to sialic acid containing glycans (Citation145)) that have established roles in cellular recognition and signaling events and are also known to modulate e.g. innate and adaptive immune responses (Citation87,Citation103).
In several cases, cancer-associated glycosylation alterations arise because of changes in the expression of the corresponding glycosyltransferases (Citation38,Citation104–106). Often, however, such strict correlations are lacking (Citation102,Citation107–111). Other causes therefore must exist and may involve e.g. increased expression of nucleotide sugar transporters, such as the UDP-Gal transporter in cases where T-antigen expression is increased (Citation112). In other cases, the absence of glycosyltransferase-specific chaperones, such asthe core 1 β1,3-galactosyl transferase-specific molecular chaperone Cosmc, has been associated with increased Tn-antigen expression, albeit in a non-cancerous specimen (Citation113). Changes in the splicing of the acceptor glycoprotein CD44 has also been shown to be associated with T-antigen expression both in cancerous and normal tissues (Citation114).
Cancer-associated glycosylation changes may also result from more general defects in Golgi function and pH homeostasis. The first observations that led to this idea came from studies in which treatment of cells with pH gradient dissipating drugs were found to increase the expression of the cancer-associated T-antigen (Citation6,Citation8,Citation10,Citation12,Citation85), most likely due to a lack of further glycosylation of this core 1 O-glycan structure. Golgi architecture was also changed, showing a fragmented morphology (Citation12). Thereafter, direct Golgi pH measurements with a fluorescent marker (GT-EGFP) protein (Citation19) in breast and colorectal cancer cells (MCF-7, HT-29, SW-48) confirmed that the Golgi pH is slightly more alkaline (∼0.2–0.4 pH units) than in non-malignant cells (Citation115). Moreover, the observed Golgi pH increase was found to correlate well with increased T-antigen expression in the cells. The observation (Citation115) that only a 0.2 pH unit increase, i.e. less than that detected in cancer cells,is sufficient to impair terminal glycosylation of both N- and O-glycans provides further support for the involvement of an altered Golgi pH in cancer-associated glycosylation abnormalities. Moreover, we have recently found that, besides T-antigen expression, other glycosylation changes typical for cancer cells are induced by raising Golgi pH only slightly (Heiskanen et al., unpublished). However, not all glycosylation events are sensitive to Golgi pH changes. For example, α2,6-sialylation (in contrast to α2,3-sialylation) is not affected by a slight Golgi pH increase. Because these two sialylation events are competitive, this observation may explain why carcinoembryonic antigen(CEA) extracted from colon cancer tissue carries α2,6-linked sialic acid (Citation97,Citation116) and not α2,3-linked sialic acid as in normal cells.
How does an altered Golgi pH then impair glycosylation? We have recently shown that, in contrast to our expectations, this is not due to a pH-dependent inactivation of Golgi glycosyltransferases, which in general have acidic pH optima,nor to the limited availability of the sugar nucleotides (Citation117). Rather, it was found that the slight Golgi pH changes induced by 20–40 μM chloroquine resulted in mislocalization of certain terminalglycosyltransferases, which accumulate in late endosomes due to their higher than normal pH (Citation118). Similar observations with other Golgi proteins and enzymes have also been demonstrated using more potent drugs such as NH4Cl and bafilomycin A(Citation10,Citation119). These observations suggest that the acidic Golgi pH is primarily required to localize correctly glycosyltransferases and glycosidases in the different Golgi subcompartments. However, this holds true only for certain enzymes, as not all are sensitive to moderate changes in Golgi pH. We anticipate that other clues such as the observed oligomerization (Citation120) may explain the differentialpH sensitivity of the enzymes.
Besides glycosylation, abnormal Golgi pH in cancer cells likely contributes also to other phenotypic changes seen in cancer cells. For example, Golgi pH changes have been shown to interfere with the apical sorting of proteins in epithelial cells (Citation7) and thus may provide a link to loss of cell polarity in cancer cells. Moreover, the extent of the acidification of the Golgi and endosomal compartments in breast cancer cells has been associated with the development of multidrug resistance (Citation121–123). Many of these chemotherapeutic drugs are weak bases and become protonated and sequestered in acidic organelles in non-malignant and multidrug-resistant cells, while in drug-sensitive cells this does not happen, because the organelles are less acidic. As a result, the drugs can bind to DNA and other cellular structures in drug-sensitive cells and eventually cause cell death. These observations emphasize how a better understanding of organelle pH regulation may help develop new strategies to combat multidrug-resistance and cancers.
Cutis laxa
Autosomal recessive cutis laxatype II is a congenital disease that is also related to Golgi pH misregulation. The disease involves excessive wrinkling of the skin at an early age and is caused by mutations in the a2 subunit of V-ATPase coded by the gene ATP6V0A2 (Citation124). Patients suffering from the related wrinkly skin syndrome carry the same mutations but without the symptoms of elastin deficiency (Citation125). The mutations result in a complete loss of V-ATPase function, thereby preventing Golgi acidification. Definitive evidence as to what extent the mutation impairs Golgi acidification, however, remains unclear, as Golgi pH was not directly measured in the study, and the Golgi also seems to co-express another isoform of the a subunit (Citation124). The mutations were also shown to impair retrograde trafficking of Golgi vesicles to the ER, most likely by inhibiting their fusion with the ER membrane. As expected, the processing of terminal glycans in the trans-Golgi was impaired. This was evidenced by the general lack of sialic acids in complex type N-glycans as well as in O-glycans in the affected individuals (Citation124,Citation126,Citation127). The terminal glycosylation defects or the general Golgi dysfunction have been linked to structural changes in ECM proteins, such as fibulin 5 and elastin, which are responsible for the elasticity of the skin (Citation125). In cutis laxa patients, defective N- and O-glycosylation also leads to a delayed development of the fetal central nervous system. Although the exact causes for the cutis laxa phenotype have remained unclear, it presents a new type of inherited disorder (CDG-II) that is almost certainly caused by misregulation of Golgi pH in the affected cells. One complicating factor is the fact that the a2 subunit has also been shown to localize in early endosomes in cells from cutix laxa patients (Citation128), which indicates that altered endosomal pH and functioning may contribute to disease etiology as well.
Cystic fibrosis
Cystic fibrosis (CF) is the most common lethal inherited disease in Caucasians. Hall-marks of CF include viscous mucus in respiratory epithelia, bacterial colonization, pancreatic dysfunction, high levels of sweat chloride, and infertility in males. The mutation that causes the CF has been traced to a cAMP-regulated chloride channel, termed the cystic fibrosis transmembrane conductance regulator (CFTR). Normally, this apical membrane channel protein allows transmembrane flow of anions, and therefore also water, into and out of cells. In CF, the CFTR-mediated anion flow is drastically reduced, resulting in the production of more viscous mucous. Altered glycosylation of the mucus is also considered to play an important role in disease etiology (Citation129,Citation130). A good indication of this is the fact that the main oligosaccharide-binding protein of Pseudomonas aeruginosa (a pathogen that typically colonizes lungs in CF) preferentially binds to altered glycans found in CF patients. These glycans are typically hyperfucosylated, hypersulfated, and hyposialylated, when compared with control samples (Citation80). However, discrepant findings, such as reduced sulfation, increased sialylation, and reduced fucosylation of CF mucins, have also been reported (Citation131). Therefore, it is currently not clear to which glycan structures the lung-colonizing bacteria predominantly bind.
An interesting link between the mutated CFTR, the observed glycosylation changes, and Golgi acidification was originally proposed by Barasch et al. (Citation55,Citation132) who reported that in CF cells the TGN was slightly more alkaline (by 0.2 pH units) than the TGN of normal counterparts. According to this scenario, mutations in the CFTR chloride channel likely impair counter-anion conductance, leading to defective acidification of the TGN and altered terminal glycosylation as observed in CF patients. However, this ‘defective Golgi acidification hypothesis’ soon turned out to be controversial, as other research groups have found no evidence that the CFTR actually affects Golgi pH (Citation130,Citation133–136). Even more confusing was the observation that the pH of the TGN in CF cells is hyperacidified relative to control cells. By using fluorescence ratio imaging, thetrans-Golgi pH in several cell lines was found to be 0.2–0.5 pH units lower in CF cells than in healthy or genetically matched rescued cells (Citation61,Citation137). Moreover, hyperacidification of the TGN and endosomal compartments was found to be Na+-dependent (Citation61), suggesting that CFTR may modulate organelle pH indirectly via other ion transporters or channels. Thus, by which mechanism, or if at all, CFTR may regulate Golgi pH remains to be clarified. The main challenge in CF, therefore, is to unravel what other roles CFTR may be playing in intracellular membranes and identify the underlying causes for altered glycosylation in CF, if not an altered Golgi pH. One possibility, as suggested by Schulz et al.(Citation131), may be accelerated degradation of CF mucin in the affected patients. How accelerated degradation would result in the increased terminal sialylation of CF mucins that was observed in the same study remains unclear.
Hailey–Hailey disease
Congenital mutations of secretory pathway Ca2+-ATPases (SPCAs) cause skin disorders that are characterized by blisters and erosions in flexural areas of the body. One of these diseases, called the Hailey–Hailey disease, is caused by a mutation in one allele of the human SPCA1 gene,ATP2C1 (Citation138,Citation139). SPCA1 has been shown to transport both Ca2+ and Mn2+ into the Golgi lumen and therefore plays an important role in the cytosolic and intra-Golgi cation homeostasis (Citation140). The decreased concentration of these cations in the Golgi lumen in affected cells is thought to lead to impaired glycosylation, proteolytic processing, folding, trafficking, or sorting of proteins that are essential for cell-to-cell adhesion (Citation141,Citation142). These defects may explain why the affected patients are unable to maintain structurally intact desmosomes and epidermis. The fact that Mn2+ is an important co-factor for many glycosyltransferases residing in the Golgi (Citation143) supports this assumption. Because Mn2+ at high concentrations is also cytotoxic, and SPCA1 is the only known Mn2+ transporter, it also plays an important role in sequestering excess Mn2+ from the cytoplasm to the extracellular space. In addition, the luminal concentrations of both Ca2+ and Mn2+ are likely to be dependent on the concentration of other ionsor may themselves be involved in counter-ion conductance, raising the possibility that Golgi pH may also be affected. Glycan analyses and direct Golgi pH measurements are required to clarify their involvement in the disease etiology.
Concluding remarks
Maintenance of a mildly acidic pH in the Golgi has turned out to be crucial for its normal functioning and, in particular, for its glycosylation potential. Therefore, any defect in glycosylation, in principle, can result from Golgi pH misregulation. Although complex, Golgi pH regulation seems to involve mainly four ion transport systems. These include V-ATPase, GPHR, AE2a, and the still poorly characterized proton leakage ‘channel’. Of these, GPHR and AE2-mediated Cl− and HCO3− transport, respectively, appear to be specific for the Golgi, given their apparent absence in other acidic organelles. The Golgi may also have a distinct V-ATPase that differs from other organellar V-ATPases by having a slightly different subunit composition or by being differently regulated. Misregulation of Golgi pH has already been shown to alter glycosylation in cancer cells and in cutis laxa, two disease states that do not seem to be related in any other way. While the defect in cutis laxa has been characterized, we currently do not know why Golgi pH is misregulated in cancer cells. Therefore, an important task in the near future is to resolve the underlying molecular defect(s) in cancer cells and to verify whether Golgi pH is also abnormal in cancer cells in vivo. This would then stimulate the development of novel diagnostic tools and glycosylation-based therapies to modulate cancer cell invasion and metastatic properties. Yet, the observations discussed above represent only the first examples of diseases that are caused by, or are associated with, misregulation ofGolgi pH. Misregulation of Golgi pH may also play a role in cystic fibrosis and Ca2+ pump-associated diseases. Other Golgi pH-associated diseases are likely to be found, and the search for these should be guided either by defects in Golgi structure or its glycosylation potential.
Acknowledgements
The authors wish to thank the Academy of Finland and the Finnish Glycoscience graduate school for financial support.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Demaurex N. pH Homeostasis of cellular organelles. News Physiol Sci. 2002;17:1–5.
- Paroutis P, Touret N, Grinstein S. The pH of the secretory pathway: measurement, determinants, and regulation. Physiology (Bethesda). 2004;19:207–15.
- Schapiro FB, Grinstein S. Determinants of the pH of the Golgi complex. J Biol Chem. 2000;275:21025–32.
- Griffiths G, Quinn P, Warren G. Dissection of the Golgi complex. I. Monensin inhibits the transport of viral membrane proteins from medial to trans Golgi cisternae in baby hamster kidney cells infected with Semliki Forest virus. J Cell Biol. 1983;96:835–50.
- Matlin KS. Ammonium chloride slows transport of the influenza virus hemagglutinin but does not cause mis-sorting in a polarized epithelial cell line. J Biol Chem. 1986; 261:15172–8.
- Thorens B, Vassalli P. Chloroquine and ammonium chloride prevent terminal glycosylation of immunoglobulins in plasma cells without affecting secretion. Nature. 1986;321:618–20.
- Caplan MJ, Stow JL, Newman AP, Madri J, Anderson HC, Farquhar MG, . Dependence on pH of polarized sorting of secreted proteins. Nature. 1987;329:632–5.
- Gawlitzek M, Ryll T, Lofgren J, Sliwkowski MB. Ammonium alters N-glycan structures of recombinant TNFR-IgG: degradative versus biosynthetic mechanism. Biotechnol Bioeng. 2000;68:637–47.
- Palokangas H, Ying M, Vaananen K, Saraste J. Retrograde transport from the pre-Golgi intermediate compartment and the Golgi complex is affected by the vacuolar H+ -ATPase inhibitor bafilomycin A1. Mol Biol Cell. 1998;9:3561–78.
- Axelsson MA, Karlsson NG, Steel DM, Ouwendijk J, Nilsson T, Hansson GC. Neutralization of pH in the Golgi apparatus causes redistribution of glycosyltransferases and changes in the O-glycosylation of mucins. Glycobiology. 2001;11:633–44.
- Campbell BJ, Rowe GE, Leiper K, Rhodes JM. Increasing the intra-Golgi pH of cultured LS174T goblet-differentiated cells mimics the decreased mucin sulfation and increased Thomsen-Friedenreich antigen (Gal beta1-3GalNac alpha-) expression seen in colon cancer. Glycobiology. 2001;11: 385–93.
- Kellokumpu S, Sormunen R, Kellokumpu I. Abnormal glycosylation and altered Golgi structure in colorectal cancer: dependence on intra-Golgi pH. FEBS Lett. 2002; 516:217–24.
- Ellis MA, Weisz OA. In vitro assays differentially recapitulate protein export from the trans-Golgi network. Anal Biochem. 2006;354:314–6.
- Wu MM, Llopis J, Adams S, McCaffery JM, Kulomaa MS, Machen TE, . Organelle pH studies using targeted avidin and fluorescein-biotin. Chem Biol. 2000;7:197–209.
- Wu MM, Grabe M, Adams S, Tsien RY, Moore HP, Machen TE. Mechanisms of pH regulation in the regulated secretory pathway. J Biol Chem. 2001;276:33027–35.
- Machen TE, Leigh MJ, Taylor C, Kimura T, Asano S, Moore HP. pH of TGN and recycling endosomes of H+/K+-ATPase-transfected HEK-293 cells: implications for pH regulation in the secretory pathway. Am J Physiol Cell Physiol. 2003;285:C205–14.
- Demaurex N, Furuya W, D'Souza S, Bonifacino JS, Grinstein S. Mechanism of acidification of the trans-Golgi network (TGN). J Biol Chem. 1998;273:2044–51.
- Kim JH, Johannes L, Goud B, Antony C, Lingwood CA, Daneman R, . Noninvasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc Natl Acad Sci USA. 1998;95:2997–3002.
- Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci USA. 1998;95:6803–8.
- Golgi C. Sur la structure des cellules nerveuses des ganglions spinaux. Arch Ital Biol 1898;30:278–86.
- Farquhar MG, Palade GE. The Golgi apparatus (complex)-(1954-1981)-from artifact to center stage. J Cell Biol. 1981;91(3 Pt 2):77s–103s.
- Farquhar MG, Palade GE. The Golgi apparatus: 100 years of progress and controversy. Trends Cell Biol. 1998;8:2–10.
- Rodriguez-Boulan E, Musch A. Protein sorting in the Golgi complex: shifting paradigms. Biochim Biophys Acta. 2005; 1744:455–64.
- Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nat Rev Mol Cell Biol. 2006;7:568–79.
- De Matteis MA, Luini A. Exiting the Golgi complex. Nat Rev Mol Cell Biol. 2008;9:273–84.
- Saraste J, Kuismanen E. Pre- and post-Golgi vacuoles operate in the transport of Semliki Forest virus membrane glycoproteins to the cell surface. Cell. 1984;38:535–49.
- Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci. 2006;119(Pt 11):2173–83.
- Tanaka K, Mitsushima A, Fukudome H, Kashima Y. Three-dimensional architecture of the Golgi complex observed by high resolution scanning electron microscopy. J Submicrosc Cytol. 1986;18:1–9.
- Cooper MS, Cornell-Bell AH, Chernjavsky A, Dani JW, Smith SJ. Tubulovesicular processes emerge from trans-Golgi cisternae, extend along microtubules, and interlink adjacent trans-golgi elements into a reticulum. Cell. 1990;61:135–45.
- Rambourg A, Clermont Y. Three-dimensional electron microscopy: structure of the Golgi apparatus. Eur J Cell Biol. 1990;51:189–200.
- Weidman PJ. Anterograde transport through the Golgi complex: do Golgi tubules hold the key? Trends Cell Biol. 1995;5:302–5.
- Mironov AA, Weidman P, Luini A. Variations on the intracellular transport theme: maturing cisternae and trafficking tubules. J Cell Biol. 1997;138:481–4.
- Griffiths G. Gut thoughts on the Golgi complex. Traffic. 2000;1:738–45.
- Kepes F, Rambourg A, Satiat-Jeunemaitre B. Morphodynamics of the secretory pathway. Int Rev Cytol. 2005;242: 55–120.
- Rogalski AA, Singer SJ. Associations of elements of the Golgi apparatus with microtubules. J Cell Biol. 1984;99: 1092–100.
- Rios RM, Bornens M. The Golgi apparatus at the cell centre. Curr Opin Cell Biol. 2003;15:60–6.
- Varki A. Factors controlling the glycosylation potential of the Golgi apparatus. Trends Cell Biol. 1998;8:34–40.
- Brockhausen I. Pathways of O-glycan biosynthesis in cancer cells. Biochim Biophys Acta. 1999;1473:67–95.
- Wopereis S, Lefeber DJ, Morava E, Wevers RA. Mechanisms in protein O-glycan biosynthesis and clinical and molecular aspects of protein O-glycan biosynthesis defects: a review. Clin Chem. 2006;52:574–600.
- Maccioni HJ. Glycosylation of glycolipids in the Golgi complex. J Neurochem. 2007;103(Suppl 1):81–90.
- De Matteis MA, D'Angelo G. The role of the phosphoinositides at the Golgi complex. Biochem Soc Symp. 2007:107–16.
- Griffiths G, Simons K. The trans Golgi network: sorting at the exit site of the Golgi complex. Science. 1986;234: 438–43.
- Jefferies KC, Cipriano DJ, Forgac M. Function, structure and regulation of the vacuolar (H+)-ATPases. Arch Biochem Biophys. 2008;476:33–42.
- Grabe M, Oster G. Regulation of organelle acidity. J Gen Physiol. 2001;117:329–44.
- Drory O, Nelson N. The emerging structure of vacuolar ATPases. Physiology (Bethesda). 2006;21:317–25.
- Jansen EJR. V-ATPase-mediated granular acidification is regulated by the V-ATPase accessory subunit Ac45 in POMC-producing cells. Mol Biol Cell. 2010;21:3330–9.
- Glickman J, Croen K, Kelly S, Al-Awqati Q. Golgi membranes contain an electrogenic H+ pump in parallel to a chloride conductance. J Cell Biol. 1983;97:1303–8.
- Rybak SL, Lanni F, Murphy RF. Theoretical considerations on the role of membrane potential in the regulation of endosomal pH. Biophys J. 1997;73:674–87.
- Hara-Chikuma M, Wang Y, Guggino SE, Guggino WB, Verkman AS. Impaired acidification in early endo-somes of ClC-5 deficient proximal tubule. Biochem Biophys Res Commun. 2005;329:941–6.
- Hara-Chikuma M, Yang B, Sonawane ND, Sasaki S, Uchida S, Verkman AS. ClC-3 chloride channels facilitate endosomal acidification and chloride accumulation. J Biol Chem. 2005;280:1241–7.
- Jentsch TJ. Chloride and the endosomal-lysosomal pathway: emerging roles of CLC chloride transporters. J Physiol. 2007;578(Pt 3):633–40.
- Maeda Y, Ide T, Koike M, Uchiyama Y, Kinoshita T. GPHR is a novel anion channel critical for acidification and functions of the Golgi apparatus. Nat Cell Biol. 2008;10:1135–45.
- Nordeen MH, Jones SM, Howell KE, Caldwell JH. GOLAC: an endogenous anion channel of the Golgi complex. Biophys J. 2000;78:2918–28.
- Thompson RJ, Nordeen MH, Howell KE, Caldwell JH. A large-conductance anion channel of the Golgi complex. Biophys J. 2002;83:278–89.
- Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, al-Awqati Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature. 1991;3526:70–3.
- Barasch J, al-Awqati Q. Defective acidification of the biosynthetic pathway in cystic fibrosis. J Cell Sci Suppl. 1993;17:229–33.
- Nagasawa M, Kanzaki M, Iino Y, Morishita Y, Kojima I. Identification of a novel chloride channel expressed in the endoplasmic reticulum, golgi apparatus, and nucleus. J Biol Chem. 2001;276:20413–8.
- Schwappach B, Stobrawa S, Hechenberger M, Steinmeyer K, Jentsch TJ. Golgi localization and functionally important domains in the NH2 and COOH terminus of the yeast CLC putative chloride channel Gef1p. J Biol Chem. 1998;273:15110–8.
- Jentsch TJ, Friedrich T, Schriever A, Yamada H. The CLC chloride channel family. Pflugers Arch. 1999;437: 783–95.
- Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH, Riordan JR. The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins. J Biol Chem. 2003;278:6440–9.
- Poschet JF, Boucher JC, Tatterson L, Skidmore J, Van Dyke RW, Deretic V. Molecular basis for defective glycosylation and Pseudomonas pathogenesis in cystic fibrosis lung. Proc Natl Acad Sci USA. 2001;98:13972–7.
- Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, . Essentials of glycobiology. 2nd. La Jolla, California: The Consortium of Glycobiology Editors; 2007.
- Ruetz S, Lindsey AE, Ward CL, Kopito RR. Functional activation of plasma membrane anion exchangers occurs in a pre-Golgi compartment. J Cell Biol. 1993;121:37–48.
- Kellokumpu S, Neff L, Jamsa-Kellokumpu S, Kopito R, Baron R. A 115-kD polypeptide immunologically related to erythrocyte band 3 is present in Golgi membranes. Science. 1988;2424:1308–11.
- Alper SL, Chernova MN, Stewart AK. Regulation of Na+-independent Cl-/HCO3- exchangers by pH. JOP. 2001;2(4 Suppl):171–5.
- Holappa K, Suokas M, Soininen P, Kellokumpu S. Identification of the full-length AE2 (AE2a) isoform as the Golgi-associated anion exchanger in fibroblasts. J Histochem Cytochem. 2001;49:259–69.
- Holappa K, Munoz MT, Egea G, Kellokumpu S. The AE2 anion exchanger is necessary for the structural integrity of the Golgi apparatus in mammalian cells. FEBS Lett. 2004;564:97–103.
- Romero MF, Fulton CM, Boron WF. The SLC4 family of HCO 3-transporters. Pflugers Arch. 2004;447:495–509.
- Alper SL. Molecular physiology of SLC4 anion exchangers. Exp Physiol. 2006;91:153–61.
- Cherny VV, DeCoursey TE. pH-dependent inhibition of voltage-gated H(+) currents in rat alveolar epithelial cells by Zn(2+) and other divalent cations. J Gen Physiol. 1999;114:819–38.
- Numata M, Orlowski J. Molecular cloning and characterization of a novel (Na+, K+)/H+ exchanger localized to the trans-Golgi network. J Biol Chem. 2001;276:17387–94.
- Nakamura N, Tanaka S, Teko Y, Mitsui K, Kanazawa H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem. 2005;280:1561–72.
- Lawrence SP, Bowers K. The sodium/proton exchanger NHE8 regulates late endosomal morphology and function. Mol Biol Cell. 2010;21:3540–51.
- Paulson JC. Glycoproteins: what are the sugar chains for? Trends Biochem Sci. 1989;14:272–6.
- Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130.
- Lowe JB, Marth JD. A genetic approach to mammalian glycan function. Ann Rev Biochem. 2003;72:643–91.
- Marth JD, Grewal PK. Mammalian glycosylation in immunity. Nat Rev Immunol. 2008;8:874–87.
- Freeze HH, Aebi M. Altered glycan structures: the molecular basis of congenital disorders of glycosylation. Curr Opin Struct Biol. 2005;15:490–8.
- Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006;7:537–51.
- Ungar D. Golgi linked protein glycosylation and associated diseases. Semin Cell Dev Biol. 2009;20:762–9.
- Reis CA, Osorio H, Silva L, Gomes C, David L. Alterations in glycosylation as biomarkers for cancer detection. J Clin Pathol. 2010;63:322–9.
- Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–67.
- Ohtsubo K. Targeted genetic inactivation of N-acetylglucosaminyltransferase-IVa impairs insulin secretion from pancreatic beta cells and evokes type 2 diabetes. Methods Enzymol. 2010;479:205–22.
- Kim YJ, Varki A. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj J. 1997;14:569–76.
- Campbell BJ, Yu LG, Rhodes JM. Altered glycosylation in inflammatory bowel disease: a possible role in cancer development. Glycoconj J. 2001;18:851–8.
- Ono M, Hakomori S. Glycosylation defining cancer cell motility and invasiveness. Glycoconj J. 2004;20:71–8.
- Yu LG. The oncofetal Thomsen-Friedenreich carbohydrate antigen in cancer progression. Glycoconj J. 2007;24:411–20.
- Zhao YY, Takahashi M, Gu JG, Miyoshi E, Matsumoto A, Kitazume S, . Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 2008; 99:1304–10.
- Boland CR, Deshmukh GD. The carbohydrate composition of mucin in colonic cancer. Gastroenterology. 1990;98(5 Pt 1):1170–7.
- Campbell BJ, Finnie IA, Hounsell EF, Rhodes JM. Direct demonstration of increased expression of Thomsen-Friedenreich (TF) antigen in colonic adenocarcinoma and ulcerative colitis mucin and its concealment in normal mucin. J Clin Invest. 1995;95:571–6.
- Karlen P, Young E, Brostrom O, Lofberg R, Tribukait B, Ost K, . Sialyl-Tn antigen as a marker of colon cancer risk in ulcerative colitis: relation to dysplasia and DNA aneuploidy. Gastroenterology. 1998;115:1395–404.
- Itzkowitz SH, Marshall A, Kornbluth A, Harpaz N, McHugh JB, Ahnen D, . Sialosyl-Tn antigen: initial report of a new marker of malignant progression in long-standing ulcerative colitis. Gastroenterology. 1995;109:490–7.
- Kuhns W, Jain RK, Matta KL, Paulsen H, Baker MA, Geyer R, . Characterization of a novel mucin sulphotransferase activity synthesizing sulphated O-glycan core 1,3-sulphate-Gal beta 1-3GalNAc alpha-R. Glycobiology. 1995;5:689–97.
- Jass JR, Allison LM, Edgar S. Monoclonal antibody TKH2 to the cancer-associated epitope sialosyl Tn shows cross-reactivity with variants of normal colorectal goblet cell mucin. Pathology. 1994;26:418–22.
- Ogata S, Ho I, Chen A, Dubois D, Maklansky J, Singhal A, . Tumor-associated sialylated antigens are constitutively expressed in normal human colonic mucosa. Cancer Res. 1995;55:1869–74.
- Parodi AJ, Blank EW, Peterson JA, Ceriani RL. Dolichol-bound oligosaccharides and the transfer of distal monosaccharides in the synthesis of glycoproteins by normal and tumor mammary epithelial cells. Breast Cancer Res Treat. 1982;2:227–37.
- Yamashita K, Totani K, Kuroki M, Matsuoka Y, Ueda I, Kobata A. Structural studies of the carbohydrate moieties of carcinoembryonic antigens. Cancer Res. 1987;47:3451.
- Roth J. Protein N-glycosylation along the secretory pathway: relationship to organelle topography and function, protein quality control, and cell interactions. Chem Rev. 2002; 102:285–303.
- Kim YS, Yuan M, Itzkowitz SH, Sun QB, Kaizu T, Palekar A, . Expression of LeY and extended LeY blood group-related antigens in human malignant, premalignant, and nonmalignant colonic tissues. Cancer Res. 1986;46:5985–92.
- Moriyama H, Nakano H, Igawa M, Nihira H. T antigen expression in benign hyperplasia and adenocarcinoma of the prostate. Urol Int. 1987;42:120–3.
- Wolf MF, Ludwig A, Fritz P, Schumacher K. Increased expression of Thomsen-Friedenreich antigens during tumor progression in breast cancer patients. Tumour Biol. 1988;9:190–4.
- Lloyd KO, Burchell J, Kudryashov V, Yin BW, Taylor-Papadimitriou J. Comparison of O-linked carbohydrate chains in MUC-1 mucin from normal breast epithelial cell lines and breast carcinoma cell lines. Demonstration of simpler and fewer glycan chains in tumor cells. J Biol Chem. 1996;271:33325–34.
- van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9:593–601.
- Whitehouse C, Burchell J, Gschmeissner S, Brockhausen I, Lloyd KO, Taylor-Papadimitriou J. A transfected sialyltransferase that is elevated in breast cancer and localizes to the medial/trans-Golgi apparatus inhibits the development of core-2-based O-glycans. J Cell Biol. 1997;137:1229–41.
- Burchell J, Poulsom R, Hanby A, Whitehouse C, Cooper L, Clausen H, . An alpha2,3 sialyltransferase (ST3Gal I) is elevated in primary breast carcinomas. Glycobiology. 1999;9:1307–11.
- Dalziel M, Whitehouse C, McFarlane I, Brockhausen I, Gschmeissner S, Schwientek T, . The relative activities of the C2GnT1 and ST3Gal-I glycosyltransferases determine O-glycan structure and expression of a tu-mor-associated epitope on MUC1. J Biol Chem. 2001;276:11007–15.
- Yang JM, Byrd JC, Siddiki BB, Chung YS, Okuno M, Sowa M, . Alterations of O-glycan biosynthesis in human colon cancer tissues. Glycobiology. 1994;4:873–84.
- Brockhausen I, Yang JM, Burchell J, Whitehouse C, Taylor-Papadimitriou J. Mechanisms underlying aberrant glycosylation of MUC1 mucin in breast cancer cells. Eur J Biochem. 1995;233:607–17.
- Hanisch FG, Stadie TR, Deutzmann F, Peter-Katalinic J. MUC1 glycoforms in breast cancer-cell line T47D as a model for carcinoma-associated alterations of 0-glycosylation. Eur J Biochem. 1996;236:318–27.
- Muller S, Hanisch FG. Recombinant MUC1 probe authentically reflects cell-specific O-glycosylation profiles of endogenous breast cancer mucin. High density and prevalent core 2-based glycosylation. J Biol Chem. 2002;277:26103–12.
- Taylor-Papadimitriou J, Burchell J, Miles DW, Dalziel M. MUC1 and cancer. Biochim Biophys Acta. 1999;1455: 301–13.
- Kumamoto K, Goto Y, Sekikawa K, Takenoshita S, Ishida N, Kawakita M, . Increased expression of UDP-galactose transporter messenger RNA in human colon cancer tissues and its implication in synthesis of Thomsen-Friedenreich antigen and sialyl Lewis A/X determinants. Cancer Res. 2001;61:4620–7.
- Schietinger A, Philip M, Yoshida BA, Azadi P, Liu H, Meredith SC, . A mutant chaperone converts a wild-type protein into a tumor-specific antigen. Science. 2006;3145: 304–8.
- Singh R, Campbell BJ, Yu LG, Fernig DG, Milton JD, Goodlad RA, . Cell surface-expressed Thomsen-Friedenreich antigen in colon cancer is predominantly carried on high molecular weight splice variants of CD44. Glycobiology. 2001;11:587–92.
- Rivinoja A, Kokkonen N, Kellokumpu I, Kellokumpu S. Elevated Golgi pH in breast and colorectal cancer cells correlates with the expression of oncofetal carbohydrate T-antigen. J Cell Physiol. 2006;208:167–74.
- Fukushima K. Carbohydrate structures of a normal counterpart of the carcinoembryonic antigen produced by colon epithelial cells of normal adults. Glycobiology. 1995;5:105.
- Waldman BC, Rudnick G. UDP-GlcNAc transport across the Golgi membrane: electroneutral exchange for dianionic UMP. Biochemistry. 1990;29:44–52.
- Rivinoja A, Hassinen A, Kokkonen N, Kauppila A, Kellokumpu S. Elevated Golgi pH impairs terminal N-glycosylation by inducing mislocalization of Golgi glycosyltransferases. J Cell Physiol. 2009;220:144–54.
- Bachert C, Lee TH, Linstedt AD. Lumenal endosomal and Golgi-retrieval determinants involved in pH-sensitive targeting of an early Golgi protein. Mol Biol Cell. 2001; 12:3152–60.
- Young WW Jr. Organization of Golgi glycosyltransferases in membranes: complexity via complexes. J Membr Biol. 2004;198:1–13.
- Simon SM. Role of organelle pH in tumor cell biology and drug resistance. Drug Discov Today. 1999;4:32–8.
- Schindler M, Grabski S, Hoff E, Simon SM. Defective pH regulation of acidic compartments in human breast cancer cells (MCF-7) is normalized in adriamycin-resistant cells (MCF-7adr). Biochemistry. 1996;35:2811–7.
- Altan N, Chen Y, Schindler M, Simon SM. Defective acidification in human breast tumor cells and implications for chemotherapy. J Exp Med. 1998;187:1583–98.
- Kornak U, Reynders E, Dimopoulou A, Van Reeuwijk J, Fischer B, Rajab A, . Impaired glycosylation and cutis laxa caused by mutations in the vesicular H-ATPase subunit ATP6V0A2. Nat Genet. 2007;40:32–4.
- Morava É, Guillard M, Lefeber DJ, Wevers RA. Autosomal recessive cutis laxa syndrome revisited. Eur J Hum Genet. 2009;17:1099–110.
- Morava E, Wopereis S, Coucke P, Gillessen-Kaesbach G, Voit T, Smeitink J, . Defective protein glyco-sylation in patients with cutis laxa syndrome. Eur J Hum Genet. 2005;13:414–21.
- Wopereis S, Morava É, Grünewald S, Mills PB, Winchester BG, Clayton P, . A combined defect in the biosynthesis of N-and O-glycans in patients with cutis laxa and neurological involvement: the biochemical characteristics. Biochim Biophys Acta. 2005;174:156–64.
- Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada G, Bourgoin S, . V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nature Cell Biol. 2006;8:124–36.
- Rhim AD, Stoykova L, Glick MC, Scanlin TF. Terminal glycosylation in cystic fibrosis (CF): a review emphasizing the airway epithelial cell. Glycoconj J. 2001;18:649–59.
- Weisz OA. Organelle acidification and disease. Traffic. 2003;4:57–64.
- Schulz BL, Sloane AJ, Robinson LJ, Prasad SS, Lindner RA, Robinson M, . Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology. 2007;17:698–712.
- Barasch J, Al-Awqati Q. Chloride channels, Golgi pH and cystic fibrosis. Trends Cell Biol. 1992;2:35–7.
- Seksek O, Biwersi J, Verkman AS. Evidence against defective trans-Golgi acidification in cystic fibrosis. J Biol Chem. 1996;271:15542–8.
- Gibson GA, Hill WG, Weisz OA. Evidence against the acidification hypothesis in cystic fibrosis. Am J Physiol Cell Physiol. 2000;279:C1088–99.
- Biwersi J, Verkman AS. Functional CFTR in endosomal compartment of CFTR-expressing fibroblasts and T84 cells. Am J Physiol. 1994;266(1 Pt 1):C149–56.
- Machen TE, Chandy G, Wu M, Grabe M, Moore HP. Cystic fibrosis transmembrane conductance regulator and H+ permeability in regulation of Golgi pH. JOP. 2001; 2(Suppl):229–36.
- Chandy G, Grabe M, Moore HP, Machen TE. Proton leak and CFTR in regulation of Golgi pH in respiratory epithelial cells. Am J Physiol Cell Physiol. 2001;281: C908–21.
- Vanoevelen J, Dode L, Raeymaekers L, Wuytack F, Missiaen L. Diseases involving the Golgi calcium pump. Subcell Biochem. 2007;45:385–404.
- Okunade GW, Miller ML, Azhar M, Andringa A, Sanford LP, Doetschman T, . Loss of the Atp2c1 secretory pathway Ca(2+)-ATPase (SPCA1) in mice causes Golgi stress, apoptosis, and midgestational death in homozygous embryos and squamous cell tumors in adult heterozygotes. J Biol Chem. 2007;282:26517–27.
- Van Baelen K, Dode L, Vanoevelen J, Callewaert G, De Smedt H, Missiaen L, . The Ca2+/Mn2+ pumps in the Golgi apparatus. Biochim Biophys Acta. 2004;174:103–12.
- Missiaen L, Raeymaekers L, Dode L, Vanoevelen J, Van Baelen K, Parys JB, . SPCA1 pumps and Hailey-Hailey disease. Biochem Biophys Res Commun. 2004;322:1204–13.
- Grice DM, Vetter I, Faddy HM, Kenny PA, Roberts-Thomson SJ, Monteith GR. Golgi calcium pump secretory pathway calcium ATPase 1 (SPCA1) is a key regulator of insulin-like growth factor receptor (IGF1R) processing in the basal-like breast cancer cell line MDA-MB-231. J Biol Chem. 2010;285:37458–66.
- Kaufman RJ, Swaroop M, Murtha-Riel P. Depletion of manganese within the secretory pathway inhibits O-linked glycosylation in mammalian cells. Biochemistry. 1994;33: 9813–9.
- Yang RY, Rabinovich GA, Liu FT. Galectins: structure, function and therapeutic potential. Expert Rev Mol Med. 2008;10:e17.
- Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–66.
- Rivinoja A. Golgi pH and glycosylation [thesis]. Acta Universitatis Ouluensis. A scientiae rerum naturalium 535. Oulu: University of Oulu; 2009.