Abstract
The present review first summarizes the complex chain of events, in endothelial and vascular smooth muscle cells, that leads to endothelium-dependent relaxations (vasodilatations) due to the generation of nitric oxide (NO) by endothelial nitric oxide synthase (eNOS) and how therapeutic interventions may improve the bioavailability of NO and thus prevent/cure endothelial dysfunction. Then, the role of other endothelium-derived mediators (endothelium-derived hyperpolarizing (EDHF) and contracting (EDCF) factors, endothelin-1) and signals (myoendothelial coupling) is summarized also, with special emphasis on their interaction(s) with the NO pathway, which make the latter not only a major mediator but also a key regulator of endothelium-dependent responses.
| Abbreviations | ||
| ADMA | = | asymmetric dimethyl-L-arginine |
| ADP | = | adenosine diphosphate |
| AT1 | = | angiotensin receptor type 1 |
| AT2 | = | angiotensin receptor type 2 |
| BH2 | = | dihydrobiopterin |
| BH4 | = | tetrahydrobiopterin |
| BKCa | = | calcium-activated potassium channels of large conductance |
| cAMP | = | cyclic adenosine monophosphate |
| CaV | = | voltage-activated calcium channel |
| cGKI | = | cGMP-dependent protein kinase I |
| cGMP | = | cyclic guanosine monophosphate |
| COX | = | cyclo-oxygenase |
| DDAH-2 | = | dimethylarginine dimethylaminohydrolase-2 |
| DHET | = | dihydroxyeicosatrienoic acids |
| DHFR | = | dihydrofolate reductase |
| DOCA-salt | = | deoxycorticosterone acetate-salt-induced rats hypertensive rats |
| ECE | = | endothelin-converting enzyme |
| EDCF | = | endothelium-derived contracting factor |
| EDHF | = | endothelium-derived hyperpolarizing factor |
| EDRF | = | endothelium-derived relaxing factor |
| EETs | = | epoxyeicosatrienoic acids |
| ET-1 | = | endothelin-1 |
| ETA | = | endothelin-receptors subtype A |
| ETB | = | endothelin-receptors subtype B |
| GTP | = | guanosine triphosphate |
| GTPCH | = | guanosine triphosphate cyclohydrolase |
| H2O2 | = | hydrogen peroxide |
| H2S | = | hydrogen sulfide |
| HETE | = | hydroxyeicosatetraenoic acids |
| IKCa | = | calcium-activated potassium channels of intermediate conductance |
| IP | = | prostacyclin receptor |
| KCa | = | calcium-activated potassium channels |
| KIR | = | inwardly rectifying potassium channel |
| KLF-2 | = | Krupper-like factor-2 |
| L-NMMA | = | N(G)-monomethyl L-arginine |
| LOX | = | lipoxygenase |
| M3 | = | muscarinic receptor type 3 |
| NADPH | = | nicotinamide adenine dinucleotide phosphate |
| NO | = | nitric oxide |
| NOX | = | NAD(P)H oxidases |
| eNOS | = | endothelial nitric oxide synthase |
| iNOS | = | inducible nitric oxide synthase |
| nNOS | = | neuronal nitric oxide synthase |
| NO2- | = | nitrite ion |
| NO3- | = | nitrate ion |
| O2.- | = | superoxide anion |
| ONOO- | = | peroxynitrite |
| P450 | = | cytochrome P450 mono-oxygenases |
| PDE-5 | = | phosphodiesterase-5 |
| PGD2 | = | prostaglandin D2 |
| PGE2 | = | prostaglandin E2 |
| PGF2α | = | prostaglandin F2α |
| PGI2 | = | prostaglandin I2 or prostacyclin |
| PGH2 | = | prostaglandin H2, endoperoxide |
| PKG | = | protein kinase G |
| PLA2 | = | phospholipase A2 |
| PPAR | = | peroxisome proliferator-activated receptor |
| PRMT-I | = | protein arginine N-methyltransferase type I |
| SERCA | = | sarcoplasmic reticulum calcium ATPase |
| SHR | = | spontaneously hypertensive rat |
| SHRSP | = | stroke-prone spontaneously hypertensive rat |
| ROS | = | reactive oxygen species |
| sEH | = | soluble epoxide hydrolase |
| sGC | = | soluble guanylyl cyclase |
| SKCa | = | calcium-activated potassium channels of small conductance |
| THETA | = | trihydroxyeicosatrienoic acids |
| TP | = | thromboxane/endoperoxide receptor |
| TRP | = | transient receptor potential channel |
| TXA2 | = | thromboxane A2 |
Key messages
NO, an endothelium-derived powerful vasodilator and a potent anti-thrombotic agent is also an important regulator of the other endothelium-mediated responses (EDHF, EDCF, endothelin).
Multiple potential therapeutic targets have been identified along the L-arginine–NOS pathway, in particular at the levels of the substrate (L-arginine), the NO-generating enzyme (eNOS), the soluble guanylyl cyclase (the main target of NO), and cGMP (the main effector of its action).
Additional therapeutic opportunities have been identified in the regulation of the other endothelium-mediated responses in various cardiovascular diseases such as hypertension, diabetes, or atherothrombosis.
Introduction
The endothelium plays a major role as local regulator of vascular tone by releasing vasodilator (endothelium-derived relaxing factors (EDRF)) and vasoconstrictor (endothelium-derived contracting factors (EDCF)) that diffuse to the underlying vascular smooth muscle to cause moment-to-moment changes in their degree of contraction. Of those endothelium-derived mediators, nitric oxide (NO) is the key player as it not only induces vasodilatation but also greatly protects the vascular wall by inhibiting platelet aggregation (in synergy with prostacyclin), expression of adhesion molecules (and thus penetration of white cells in the intima and media), and oxidation of lipids. These actions converge in healthy arteries to prevent the unwanted coagulation of the blood and the inflammatory response that leads to the atherosclerotic process. When the endothelial cells lose the ability to release enough NO or when the bioavailability of the gaseous mediator is reduced, endothelial dysfunction ensues, a hallmark of vascular disease, and indeed a prognostic marker of severe cardiovascular events (Citation1–5). Besides its pivotal role in modulating the function of blood components and vascular smooth muscle directly, NO also appears to interact closely with the other EDRFs (endothelium-derived hyperpolarizing factors (EDHFs)) and to control the production/action of endothelium-derived vasoconstrictors (endothelin-1 (ET-1) and vasoconstrictor prostanoids (EDCFs)), making it the gate-keeper of endothelial function in terms of vasomotor control (Citation6–9). This review summarizes the current knowledge concerning the role of NO in vascular homeostasis, focusing on its intrinsic effect but also discussing its modulatory role in controlling the other endothelium-derived mediators. Because of space limitations, we have selected to refer only to key or recent original articles and to reviews by others and us, to which the reader is referred for a more complete coverage of the literature.
Nitric oxide
Furchgott and Zawadzki (Citation10) first showed that the presence of the endothelial lining is required to observe the relaxation of isolated strips of rabbit aorta in response to acetylcholine. They attributed the relaxation to the calcium-dependent release of an endothelial factor that was later identified as NO (Citation4,Citation11–13). Subsequently, Moncada and colleagues demonstrated that the enzymatic origin of NO production in the endothelial cells involves the L-arginine–NO-synthase pathway (Citation14). NO is not only a powerful vasodilator but is also a potent anti-thrombotic agent, and it plays important biological roles in the gastrointestinal, respiratory, nervous, and immune systems (Citation15,Citation16).
Nitric oxide synthases
The production of NO is associated with three different subtypes of biosynthetic enzymes. These were cloned, characterized, and termed according to the chronology of their characterization: NO-synthase I (or neuronal, nNOS, NOS-1) (Citation17,Citation18), NO-synthase II (or inducible, iNOS, NOS-2) (Citation19,Citation20), and NO-synthase III (or endothelial, eNOS, NOS-3) (Citation21,Citation22). The two constitutive isoforms (nNOS and eNOS) are stimulated by an increase in intracellular calcium concentration, while iNOS is a calcium-independent enzyme (Citation23). In fact, the expression of the various NOS isoforms is much more diverse than their name would suggest. For instance, the three NOS can be present in the vascular wall. Thus, nNOS is expressed not only in perivascular nerves but can also be detected in endothelial and smooth muscle cells (Citation24–26). The expression of iNOS has been documented in all nucleated cells in the cardiovascular system. In the blood vessel wall, this includes endothelial and smooth muscle cells as well as fibroblasts, leukocytes, and mast cells (Citation27). Furthermore, eNOS is expressed not only in endothelial cells but also in cardiac myocytes and platelets (Citation28,Citation29). Nevertheless, the preponderant NOS isoform in healthy endothelial cells is the calcium-dependent eNOS, and eNOS is predominantly expressed in endothelial cells (Citation30).
The three NOS enzymes contain a reductase and an oxygenase domain. In the presence of molecular oxygen, they catalyze a five-electron oxidation of one of the guanido nitrogens of L-arginine. They all require the co-factors NADPH, flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), heme, and tetrahydrobiopterin (BH4) in order to catalyze this reaction (Citation31). An essential feature of NOS, despite the ability of the reductase or the oxygenase domain to function independently, is that the NO synthase activity requires dimerization. The heme group of the enzyme plays an essential role in the formation of the homodimer, and BH4 stabilizes this dimer once formed. The activation of eNOS requires also the calcium-dependent binding of calmodulin (Citation32) ().
Figure 1. NO biosynthesis and vascular smooth muscle targets. In endothelial cells, the L-arginine–endothelial nitric oxide synthase (eNOS) system and the synthesis via the de-novo synthesis and salvage pathways of tetrahydrobiopterin (BH4), an essential co-factor of eNOS, are shown. ([Ca2+]i = intracellular calcium concentration; Ca2+-sensitivity = sensitivity to calcium of contractile proteins; cGMP = cyclic guanosine monophosphate; cGKI = cGMP-dependent protein kinase I; Cyt-C = cytochrome C; DHFR = dihydrofolate reductase; FAD = flavin adenine dinucleotide; FMN = flavin mononucleotide; GTP = guanosine triphosphate; NADPH = nicotinamide adenine dinucleotide phosphate; NO2- = nitrite ion; NO3- = nitrate ion; O2.- = superoxide anion; ONOO- = peroxynitrite; P450 = cytochrome P450 mono-oxygenases; SERCA = sarcoplasmic reticulum calcium ATPase; sGC = soluble guanylyl cyclase; XO = xanthine oxidase).
![Figure 1. NO biosynthesis and vascular smooth muscle targets. In endothelial cells, the L-arginine–endothelial nitric oxide synthase (eNOS) system and the synthesis via the de-novo synthesis and salvage pathways of tetrahydrobiopterin (BH4), an essential co-factor of eNOS, are shown. ([Ca2+]i = intracellular calcium concentration; Ca2+-sensitivity = sensitivity to calcium of contractile proteins; cGMP = cyclic guanosine monophosphate; cGKI = cGMP-dependent protein kinase I; Cyt-C = cytochrome C; DHFR = dihydrofolate reductase; FAD = flavin adenine dinucleotide; FMN = flavin mononucleotide; GTP = guanosine triphosphate; NADPH = nicotinamide adenine dinucleotide phosphate; NO2- = nitrite ion; NO3- = nitrate ion; O2.- = superoxide anion; ONOO- = peroxynitrite; P450 = cytochrome P450 mono-oxygenases; SERCA = sarcoplasmic reticulum calcium ATPase; sGC = soluble guanylyl cyclase; XO = xanthine oxidase).](/cms/asset/a16fd03e-aac8-4a72-9bb1-e8cd98a18391/iann_a_585658_f0001_b.gif)
Transcriptional and post-translational regulation of eNOS in endothelial cells
Since NO is a gaseous freely diffusible radical with a high biological reactivity, its synthesis has to be tightly controlled in order to be delivered at the right time, the right place, and in the right amount (Citation23).
The transcription of eNOS is under epigenetic control and can be regulated by various physical, chemical, and pharmacological stimuli. For instance, laminar shear stress is a major stimulus not only for the transcription of eNOS but also for the co-ordinated expression of many other genes that not only control vascular tone but also promote an atheroprotective, anti-thrombotic, and anti-inflammatory phenotype. This phenomenon involves the up-regulation of an endothelial transcription factor KLF-2 (Citation33–35). Since eNOS is only one component of a large signaling complex that, besides calmodulin and caveolin-1, involves many other different proteins, its activity can be altered by several overlapping post-translational regulatory modifications, including lipidation, phosphorylations (at various serine, threonine, and tyrosine residues), S-nitrosylations, acetylation, and protein–protein interactions (Citation34,Citation36,Citation37). The subcellular location of eNOS is critical for its state of activation, and the trafficking of the enzyme is under multiple levels of regulation (Citation38).
NO storage and NO-synthase-independent production of nitric oxide
NO is a reactive radical with a half-life of a few seconds in tissues and physiological fluids, since it reacts with superoxide anion to form peroxynitrite or is rapidly inactivated by oxyhemoglobin to form nitrate and methemoglobin (Citation39–41). However, NO can also exert remote and long-lasting effects. Indeed, it can be either transported by protein carriers or stored locally. S-nitrosylation/denitrosylation of protein cysteine residues is a dynamic process than can affect hundreds of proteins in intact cells and albumin and hemoglobin in the blood. Not only can S-nitrosylation affect the activity of these proteins, but the generation of S-nitrosoglutathione allows the transfer of NO activity at distance from the site where it has been generated (Citation42). Besides S-nitrosylation of local proteins, NO can also be stored by forming protein-bound dinitrosyl iron complexes to be displaced later by low-molecular weight thiols (Citation43) ().
In addition, NO can also be oxidized into nitrite, either spontaneously in the presence of oxygen or enzymatically by the copper storage plasma enzyme, ceruloplasmin (Citation44,Citation45). Furthermore, nitrite can originate directly from ingested food or indirectly from the reduction of nitrates either by reductase enzymes from commensal bacteria colonizing the mouth and the digestive track or by xanthine oxidase (Citation46). When nitrite is present, deoxyhemoglobin in particular and heme-globins in general, as well as xanthine oxidase, cytochrome P450 mono-oxygenase, cytochrome C, or eNOS itself, can act as nitrite reductases and produce NO (Citation46–48) ().
Multiple targets of nitric oxide
Most of the physiological effects of NO are associated with the activation of soluble guanylyl cyclase (Citation49,Citation50). In addition, NO forms S-nitrosylated proteins (see above) (Citation42) and can interact with the heme iron moiety of various proteins. For instance, the reduction in the intracellular concentration of calcium, which contributes to the inhibition of platelet activation and initiates vascular relaxation, involves the uptake of calcium by sarcoplasmic reticulum ATPase (SERCA) (Citation51,Citation52). This guanylyl cyclase-independent effect is attributed to the redox regulation of SERCA via the NO-dependent S-glutathiolation of a reactive cysteine (Citation53,Citation54). NO binds reversibly to cytochrome C oxidase and competes with oxygen. The NO-mediated inhibition of cytochrome C oxidase activity plays a fundamental role in the regulation of mitochondrial signaling and the cellular response to hypoxia (Citation55,Citation56) ().
Soluble guanylyl cyclase is a heterodimer, consisting of an α- and a β-subunit, the latter containing the prosthetic ferrous heme group (Fe2+) that catalyzes the conversion of guanosine-5′ triphosphate (GTP) to the second messenger cyclic 3′,5′-guanosine monophosphate (cGMP) (Citation57). The latter predominantly activates cGMP-dependent protein kinases, cGKI being the preferential isoform expressed in vascular smooth muscle cells, platelets, and to a lesser extent endothelial cells (Citation50,Citation58). Furthermore, cGMP interacts with phosphodiesterases and regulates the concentration of cyclic 3′,5′-adenosine monophosphate (cAMP) (Citation59), and activates cyclic nucleotide-gated cation channels (Citation60). In vascular smooth muscle cells, activation of cGKI produces relaxation by multiple mechanisms, which either reduce the intracellular calcium concentration or affect the calcium sensitivity of the contractile proteins (Citation50) ().
L-arginine–NOS pathway and potential therapeutic targets in cardiovascular diseases
Various potential therapeutic targets have been identified along the L-arginine–NOS pathway, in particular at the levels of the substrate (L-arginine), the NO-generating enzyme (eNOS), soluble guanylyl cyclase (the main target of NO), and cGMP (the main effector of its action).
Availability of the substrate L-arginine. When L-arginine is deficient, eNOS can generate both superoxide anions and NO, leading to the detrimental production of peroxynitrite (Citation61) ( and ). A few clinical studies have shown that L-arginine supplementation could bring some benefit under pathophysiological conditions, including hypercholesterolemia and hypertension, although the elevated doses required are not appropriate for a standard therapy.
Figure 2. L-arginine–NO-synthase–soluble guanylyl cyclase pathway and potential sites of therapeutic intervention. The circled numbers (from 1 to 14) indicate potential sites of therapeutic intervention. 1) L-arginine supplementation. 2) Inhibition of protein arginine N-methyltransferase type I (PRMT-I) to prevent the formation of asymmetric dimethyl-L-arginine (ADMA). 3) Increasing the expression and/or the activity of dimethylarginine dimethylaminohydrolase-2 (DDAH-2) to facilitate ADMA catabolism. 4) Inhibition of arginase-2 to prevent L-arginine metabolism. 5) Increasing the expression and/or activity of endothelial nitric oxide synthase (eNOS). 6) Designing drugs that stimulate endothelium-derived nitric oxide release. 7) Enhancing the expression and/or activity of guanosine triphosphate cyclohydrolase (GTPCH), the rate-limiting enzyme for tetrahydrobiopterin synthesis (BH4), or direct supplementation with BH4, or with its precursor sepiapterin. 8) Enhancing the expression and/or activity of dihydrofolate reductase (DHFR), involved in BH4 regeneration. 9) Scavengers of reactive oxygen species (ROS), anti-oxidants. 10) Inhibition of the activity and/or expression of enzymes that generate ROS such as NAD(P)H oxidases (NOX), cyclo-oxygenases (COX), lipoxygenases (LOX), or cytochrome P450 mono-oxygenases (P450). 11) Enhancing the expression and/or activity of enzymes that metabolized ROS such as superoxide dismutase (SOD) or catalase (or alternatively synthesis of mimetics). 12) Stimulators of soluble guanylyl cyclase (sGC). 13) Activators of sGC. 14) Inhibitors of phosphodiesterase-5 (PDE-5). (BH2 = dihydrobiopterin; CAT-1 = cationic amino acid transporters; CaV = voltage-activated calcium channel; cGMP = cyclic guanosine monophosphate; EC = endothelial cell; FAD = flavin adenine dinucleotide; FMN = flavin mononucleotide; O2.- = superoxide anion; ONOO- = peroxynitrite; PKG = protein kinase G; VSMC = vascular smooth muscle cell).
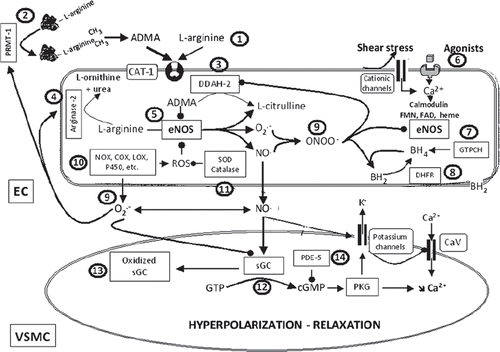
In vivo, true L-arginine deficiency is unlikely to occur (Citation62). However, L-arginine can be locally submitted to competition with endogenously produced analogs such as symmetric dimethyl-L-arginine, asymmetric dimethyl-L-arginine (ADMA), and N(G)-monomethyl L-arginine (L-NMMA). L-arginine is transported through the plasma membrane by the system Y+, a member of the family of cationic amino acid transporters, and is in competition with other amino acids such as L-lysine but also with the endogenous analogs mentioned above. Furthermore, ADMA and L-NMMA directly compete with L-arginine at the NOS site and are inhibitors of the enzyme (Citation63). ADMA represents a novel independent predictor for all causes of cardiovascular mortality (Citation62). Free dimethylarginines are the products of proteolytic degradation of arginine-methylated proteins following the preferential action of protein arginine N-methyltransferase type I (PRMT-I). ADMA is mainly metabolized by dimethylarginine dimethylaminohydrolase (DDAH), whereby the DDAH-2 isoform is the preponderant enzyme expressed in endothelial cells. Endothelial cells also express and contain arginases (mainly arginase-II), which metabolize L-arginine to L-ornithine and urea. Arginase-II competes with eNOS for substrate, and its expression and/or activity are enhanced in cardiovascular diseases. Therefore, PRMT-1, arginase II (inhibition), and DDAH-2 (activation) are promising therapeutic targets for the treatment of cardiovascular disease (Citation64–66) ().
Proper expression and functionality of eNOS. A reduced expression of eNOS could be responsible for a decreased NO production. However, in most situations associated with endothelial dysfunction the expression of eNOS is increased paradoxically. The augmented endothelial level of reactive oxygen species, in particular those of hydrogen peroxide, which increases the expression of eNOS at the transcriptional and translational levels, likely explains this paradox. An endothelial dysfunction associated with an increased expression of eNOS shows that the ability to generate NO is reduced or/and that its bioavailability is decreased. The reduction in NO generation can be attributed to the eNOS uncoupling phenomenon whereby NOS itself becomes a source of superoxide anions exacerbating endothelial dysfunction. The decrease in NO bioavailability is associated with an oxidizing environment, i.e. superoxide anion generation from different sources, and the formation of peroxynitrite (Citation67) ( and ).
The two independent enzymatic steps required for the eNOS-dependent conversion of L-arginine to NO necessitate the presence of tetrahydrobiopterin (BH4). Decreased endothelial levels of this essential pteridine co-factor are not only responsible for a reduction in NO production but also for the uncoupling of eNOS. In such an uncoupled state, electrons normally flowing from the reductase domain of one subunit to the oxygenase domain of the other subunit are diverted to molecular oxygen rather than to L-arginine, resulting in production of superoxide anion rather than NO (Citation68). Homocysteine is a potent risk factor for endothelial dysfunction and atherosclerosis, and some of its effects may be due to inhibition of BH4 de-novo synthesis (Citation69) ().
Direct supplementation with BH4, or with its precursor sepiapterin, improves endothelial function in numerous animal models of cardiovascular disease. Controversial findings have been published concerning the therapeutic efficacy of BH4, although some positive observations have been reported in patients with hypertension, diabetes, hypercholesterolemia, coronary insufficiency and in smokers (Citation69). Enhancing the expression or the activity of GTP-cyclohydrolase (GTPCH-I), the first and rate-limiting step of de-novo BH4 synthesis, or that of dihydrofolate reductase (DHFR), involved in BH4 regeneration, may prevent the occurrence of endothelial dysfunction. Alternative strategies include the administration of folic acid, which enhances the binding affinity of BH4 to NOS, stabilizes BH4 chemically, and by stimulating DHFR enhances its regeneration. Preventing peroxynitrite formation and therefore BH4 oxidation or facilitating the recycling of BH4 regeneration with vitamin C may also be considered (Citation67,Citation70) ().
In various animal models, inhibition or genetic deletion of eNOS causes accelerated atherosclerosis, abdominal aortic aneurysms, and ischemic heart disease (Citation71–74), indicating that endogenous eNOS plays an important anti-atherosclerotic role. However, apoE-/- mice over-expressing eNOS develop paradoxically larger atherosclerotic lesions than control apoE-/- mice (Citation75). This paradox is explained by the fact that in the presence of oxidative stress, reactive oxygen species from various sources (kindling radicals) and the formation of peroxynitrite will lead to eNOS uncoupling and the subsequent eNOS contribution to superoxide production (bonfire radicals). Therefore, up-regulation of eNOS in itself is a double-edged sword strategy. However, if the up-regulation of eNOS is associated with an increased availability of essential co-factors such as BH4, eNOS uncoupling can be prevented. Some compounds, such as AVE9488 and AVE3085, which enhance eNOS promoter activity, possess such a co-ordinated activity and validate the concept that enhanced transcriptional expression of eNOS can be beneficial in atherosclerosis and possibly in other cardiovascular diseases (Citation76) ().
Furthermore, statins (3-hydroxy-3-methylglutaryl-CoA reductase inhibitors), by their ‘pleiotropic’ (independent of their lipid-lowering action) effects, and the polyphenol, resveratrol, which activates SIRT-1, increase the expression of the transcription factor KLF-2 (Citation77,Citation78), which is also stimulated by laminar shear stress (Citation34). KLF-2 is described as a master regulator of the transcription of anti-inflammatory, anti-thrombotic, and vasoactive genes, including eNOS, leading to a vasoprotective phenotype (Citation35). Additionally, statins and polyphenols as well as drugs such as fibrates, metformin, and peroxisome proliferator-activated receptor-gamma (PPARγ) agonists activate AMP-kinase. This could lead to an increased expression of eNOS and to post-translational modifications, in particular phosphorylations, that enhance the protein expression and the enzymatic activity of eNOS (Citation79–81) ().
Increasing the levels of NO and/or its bioavailability. Besides preventing eNOS uncoupling (see above), different therapeutic strategies can be designed to restore proper NO levels. These include drugs that stimulate the release of NO by endothelial eNOS, NO donor drugs, anti-oxidant compounds, drugs that boost the anti-oxidant defense mechanisms, and inhibitors of the enzyme(s) involved in the generation of reactive oxygen species ().
Some anti-hypertensive drugs can stimulate the release of endothelial NO. This is the case for nebivolol, a third-generation β-blocker (Citation82,Citation83), and various dihydropyridines, by a mechanism that is unrelated to the calcium channel-blocking properties of these compounds (Citation84–86). Inhibitors of angiotensin-converting enzyme indirectly produce endothelium-dependent relaxations in vitro or vasodilatations, in vivo, by preventing the degradation of endogenous bradykinin, a potent stimulator of eNOS (Citation87–89). Independently of the metabolism of bradykinin, inhibitors of angiotensin-converting enzyme can also enhance endothelium-dependent relaxations by regulating the sequestration and the sensitivity of the bradykinin B2 receptors (Citation90,Citation91). Similarly, in various arteries, including human coronary arteries, antagonists of the angiotensin AT1 receptor unmask endothelium-dependent, NO-dependent vasodilator effects due to the preferential activation of AT2 receptor subtype by the high levels of circulating angiotensin-II (Citation92–94). Additionally, chronic treatments with inhibitors of angiotensin-converting enzyme or antagonists of the angiotensin AT1 receptor increase eNOS expression in both animals with high blood pressure and in hypertensive humans (Citation95,Citation96). The limited information available in animals with the renin inhibitor, aliskiren, suggests that this way of inhibiting the renin–angiotensin system is also likely to improve endothelium-dependent relaxations and to increase eNOS expression (Citation97,Citation98).
Besides uncoupled eNOS, potential sources of superoxide anions in the vascular wall include NAD(P)H oxidases, xanthine oxidase, cyclo-oxygenases, lipoxygenases, cytochrome P450 mono-oxygenases, and the mitochondrial respiratory chain. The production of reactive oxygen species is increased in patients with essential hypertension, renovascular hypertension, malignant hypertension, and pre-eclampsia. In essential hypertension, an increase in the levels and/or activity of vascular NAD(P)H oxidase is a predominant source of excess reactive oxygen species (Citation99). Superoxide anions not only decrease NO bioavailability, but the peroxynitrite generated exacerbates vascular injury by oxidation of sulfhydryl groups as well as nitration and hydroxylation of aromatic groups, including tyrosine, tryptophan, and guanine (Citation1,Citation100). Thus, the scavenging of reactive oxygen species with anti-oxidants and inactivation of superoxide anions with superoxide dismutase mimetics seem obvious ways of increasing NO bioavailability (). However, if such a strategy has been successful in various animal models of hypertension, large clinical trials in hypertensive patients showed that chronic anti-oxidant therapy does not produce a major reduction in arterial blood pressure and does not improve the associated morbidity and mortality, with the possible exception of the long-term intake of polyphenols that are present in red wine, fruits, and vegetables, i.e. the so-called beneficial effect of a Mediterranean diet (Citation101,Citation102). The reasons underlying these failures are still not completely understood.
Direct inhibition of the generation of reactive oxygen species might be a better alternative. Major efforts toward the synthesis of NADPH oxidase inhibitors have been performed in academic institutions and pharmaceutical companies. However, it is not yet clear which isoform(s) of NADPH should be inhibited, and the inhibitors available are still not specific, not very potent, and possess poor pharmacokinetics properties (Citation103) ().
The activation of the renin–angiotensin system and the resulting stimulation of AT1 receptors are a major stimulus for NAD(P)H oxidase and reactive oxygen species (ROS) production, also in hypertensive humans (Citation99,Citation104). Preventing the generation of reactive oxygen species, for instance by deleting a subunit of the NADPH oxidase, causes resistance to angiotensin II-induced hypertension and markedly reduces the associated endothelial generation of superoxide anions (Citation105). Statins, by another of their ‘pleiotropic’ actions, decrease the expression of NADPH subunits, improving, therefore, the balance between NO and reactive oxygen species. These endothelial effects may contribute to the therapeutic benefice exerted by these compounds, although no clinical data exist demonstrating that the reversal of endothelial dysfunction is associated with a reduction in cardiovascular events (Citation95,Citation106–109).
Substituting NO with NO donor drugs started empirically in the late nineteenth century with the nitrate compound nitroglycerin to alleviate the symptoms of angina. However, the development of nitrate tolerance, a complex multifactorial phenomenon, limits their continuous clinical use and is associated with the generation of oxidative stress and endothelial dysfunction (Citation110). Different chemical classes of NO donors exist. Some of those release NO spontaneously and may be of therapeutic interest beyond coronary disease and heart failure (Citation111). S-nitrosothiols are not prone to tolerance and are not cross-tolerant with nitrates. They possess vasodilator and anti-aggregating properties and may boost the anti-oxidant capacity of the plasma and the vascular wall (Citation111).
Alternatively, hybrid compounds possessing dual activities may be of therapeutic interest to treat endothelial dysfunction in hypertension and the ensuing end-organ damage. Earlier compounds such as nicorandil, an organic nitrate that also opens potassium channels, or nipradilol, a non-selective adrenoceptor blocker with weak β-adrenoceptor-antagonistic and NO-releasing properties are still predominantly prescribed for angina and glaucoma, respectively (Citation107,Citation111). However, among the new hybrid compounds that have been designed, i.e. NO-releasing anti-adrenergic drugs, NO-releasing-dihydropyridines, NO-releasing statins, NO-releasing antagonists of the angiotensin AT1 receptor, or NO-releasing inhibitors of angiotensin-converting enzyme, some may present a therapeutic interest in hypertension and other cardiovascular diseases (Citation112). For instance, a NO-releasing derivative of the angiotensin-converting enzyme inhibitor enalapril, NCX-899, has demonstrated a superior activity when compared to enalapril alone in animal models of heart failure and hypertension (Citation113,Citation114). Proper clinical trials must be undertaken in order to demonstrate whether or not this strategy brings a therapeutic benefit when compared to the original therapeutic entity.
Soluble guanylyl cyclase and cGMP production. When NO binds to the heme moiety of soluble guanylyl cyclase, the activity of the enzyme, leading to the production of the signaling molecule cGMP, is enhanced 400-fold. Thus, regulation of NO bioactivity is achieved not only by modulation of eNOS and curtailing oxidative stress but also by affecting soluble guanylyl cyclase (Citation115) or the half-life of cGMP, which is readily hydrolyzed by phosphodiesterases (Citation116) ().
NO cannot activate soluble guanylyl cyclase when the enzyme is heme-free (Citation117) or when the heme moiety is oxidized (Fe3+), following oxidative stress (Citation118,Citation119). However, some pharmacological agents can interact with soluble guanylyl cyclase either under the native form or when oxidized. They generate cGMP independently of NO and are potent vasodilators and inhibitors of platelet aggregation (Citation120,Citation121). Some of these compounds are undergoing clinical trials in acute decompensated heart failure, peripheral arterial occlusive diseases, and pulmonary hypertension. In addition, the potential therapeutic indications of these agents may include hypertension, myocardial ischemia, erectile dysfunction, atherosclerosis, and thrombosis (Citation120,Citation121) ().
Phosphodiesterases are a diverse family of enzymes that hydrolyze cyclic nucleotides. In the cardiovascular system, phosphodiesterase 5 (PDE5) was the first identified selective cGMP esterase and is the major isoform involved in the hydrolysis of the cGMP pools generated by the activation of soluble guanylyl cyclase (Citation122,Citation123). Specific PDE5 inhibitors are currently approved for erectile dysfunction but, in the future, their therapeutic indications may also include pulmonary hypertension, heart failure, and essential hypertension (Citation124–126) ().
In conclusion: The NO pathway is a major regulator of cardiovascular homeostasis. A decrease in NO bioavailability or function is associated with most of endothelial dysfunctions observed in virtually all cardiovascular diseases. However, the mechanisms underlying the deficiencies in the NO pathway are multiple and thus so are the new potential therapeutic targets. Only oncoming clinical trials will define which of these new targets and in which indication(s) they will fulfill their therapeutic potential.
NO and EDHF
Nature and action of EDHF
Endothelium-dependent vasodilatations resistant to inhibitors of NO and prostacyclin synthesis are mediated by hyperpolarization leading to the closure of voltage-gated calcium channels in the vascular smooth muscle cells (Citation127,Citation128). The resulting decrease in intracellular calcium concentration leads to relaxation. This hyperpolarization has been proposed initially to be caused by a factor, of at that time unknown chemical nature, released by the endothelium and termed endothelium-derived hyperpolarizing factor (EDHF). Structurally very different factors including eicosanoids (Citation129,Citation130), hydrogen peroxide (Citation131), C-type natriuretic peptide (Citation132), potassium ions released by endothelial KCa channels (Citation133), NO itself (Citation134), and H2S (Citation135) have been proposed to act as diffusible EDHFs that cause opening of K+ channels of the underlying smooth muscle (for extensive review and debate see (Citation8,Citation136)). However, it soon appeared that the term EDHF may be misleading because endothelium-dependent hyperpolarization can also be caused by non-chemical, electrotonical coupling of endothelial and vascular smooth muscle cells via myoendothelial gap junctions without the need of the generation of a chemical diffusible mediator (Citation137). In aggregate, EDHF(s) pathways are presumably multiform, and their roles differ between species and within the vascular tree (). In any case, EDHF-signaling is a powerful vasodilator system, particularly in small-caliber arteries and arterioles in which NO-signaling is not the predominant pathway (Citation138–140). However, in several cardiovascular pathologies (hypertension, hypercholesterolemia, atherosclerosis, and diabetes) disturbances in EDHF-signaling contribute to endothelial dysfunction and thus arterial malfunction (for in-depth review see (Citation128,Citation141)). This section introduces the major candidate molecules and pathways leading to endothelium-dependent hyperpolarizations and their interactions with NO. From a more medical perspective, it focuses on how EDHF(s) systems fit into the complex setting of arterial malfunction and discusses whether or not drug-targetable EDHF(s) systems can theoretically be exploited for novel endothelium-specific treatments of cardiovascular disease involving endothelial dysfunction.
Figure 3. NO and EDHF. Endothelium-dependent relaxations resistant to inhibitors of NO synthases and cyclo-oxygenases, leading to the hyperpolarization of the underlying smooth muscle cells, are associated with different mechanisms: the metabolism of arachidonic acid via the lipoxygenases (LOX) or cytochrome P450 mono-oxygenases (P450) pathways and the non-chemical EDHF pathways, which, following the calcium-calmodulin-dependent activation of endothelial potassium channels, involve either the direct transfer of negative charges (e−) via the myoendothelial gap junction or the accumulation of potassium ions in the intercellular space and the subsequent activation of the smooth muscle Na+/K+-ATPase and/or the inwardly rectifying potassium channel. The circled numbers (from 1 to 7) indicate the principal steps in the development of endothelium-dependent hyperpolarizations and the potential sites of therapeutic intervention. The privileged therapeutic options, inhibitors of soluble epoxide hydrolase (sEH) and activators of endothelial potassium channels, are indicated by a broad arrow. 1) Agonists interacting with their receptor and physical forces such as shear stress increase endothelial intracellular calcium concentration. 2) Activation of transient receptor potential channels (TRP) contributes to calcium entry. 3) Activation of endothelial calcium-activated potassium channels of small (SKCa or KCa2.3) and intermediate conductance (IKCa or KCa3.1); the endothelial cell hyperpolarization increases the driving force for calcium entry. 4) Calcium-dependent activation of the endothelial nitric oxide synthase (eNOS); epoxyeicosatrienoic acids, P450 metabolites of arachidonic acid (AA), enhance NO production. 5) EETs and LOX metabolites, such as hydroxyeicosatetraenoic acids (HETE) and trihydroxyeicosatrienoic acids (THETA), can be released by endothelial cells and activate potassium channels of the smooth muscle cells to produce their hyperpolarization and relaxation. EETs are metabolized by sEH to form the generally less active dihydroxyeicosatrienoic acids (DHET). 6) Electrotonical coupling between endothelial and vascular smooth muscle cells via myoendothelial gap junctions clustered at specific endothelial projections toward the vascular smooth muscle cells. 7) Vascular smooth muscle hyperpolarization inhibits the activation of voltage-gated calcium channels (CaV) and decreases the intracellular calcium concentration.
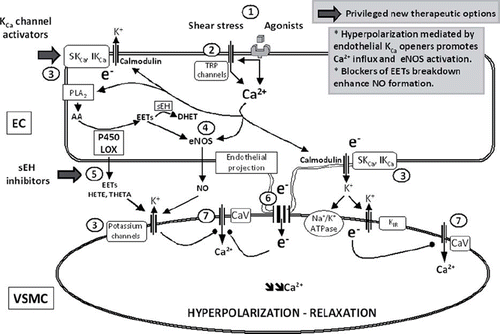
Eicosanoids. Endothelial lipoxygenases and cytochrome P450 enzymes metabolize the polyunsaturated fatty acid, arachidonic acid, to eicosanoids which stimulate the activity of the large-conductance KCa channels (BKCa or maxi K) of vascular smooth muscle and thus could act as diffusible endothelium-derived hyperpolarizing factors (Citation129,Citation130).
Epoxygenases. The cytochrome P450 epoxygenases 2C and 2J and their subforms produce epoxyeicosatrienoic acids (EETs; in varying amounts 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET), all of which open BK channels of vascular smooth muscle and cause relaxation (Citation129) (). They are considered to contribute predominantly to endothelium-dependent dilatations attributed to EDHF in porcine and bovine coronary arteries (Citation142,Citation143) and in the human forearm microcirculation (Citation144). However, they are not equally effective in all human vascular beds (e.g. the cutaneous microcirculation (Citation145)). In patients with essential hypertension (Citation144) and primary hyperparathyroidism (Citation146), the production of EETs appears to take over, at least partially, during endothelial dysfunction related to NO deficiency.
EETs may contribute to the systemic hemodynamic control. Indeed, a lower arterial blood pressure has been observed in male mice, which lack the EET-degrading enzyme soluble epoxide hydrolase (sEH) (Citation147). Transgenic mice with endothelial over-expression of the human CYP2J2 and CYP2C8 epoxygenases, and resulting enhanced EET synthesis, exhibit augmented dilator responses to acetylcholine. Their arterial blood pressure tends to be lower than in wild-type animals and increases less upon administration of a NO synthase inhibitor (Citation148), suggesting a particular relevance and efficacy of this system during systemic hypertension. Moreover, pharmacological inhibition of sEH ensuing higher EET levels leads to a reduction in arterial blood pressure in experimental angiotensin II hypertension (Citation149,Citation150). The specific inhibitor of sEH, AR9281 (Citation149), when tested in clinical trials for efficacy in mild to moderate hypertension, failed to have blood pressure-lowering efficacy, presumably because of the low in-vivo potency of the compound at the dosage used. However, AR9281 and analogs with an improved profile (Citation151) are effective in experimental pulmonary hypertension (Citation152,Citation153), atherosclerosis (Citation154–156), and have additional vascular protective effects in brain (Citation157,Citation158) and heart (Citation159,Citation160) and in diabetes (Citation161). Moreover, genetic sEH deficiency in mice and pharmacological inhibition of the enzyme with the improved sEH inhibitor t-AUCB prevent hyperglycemia, favor insulin secretion, and protect pancreatic islets against apoptosis (Citation161). As to atheroprotective effects, both pharmacological inhibition and genetic sEH deficiency reduce neointima formation in hyperlipidemic mice (Citation162). The former decreases neointima formation in the presence but not in the absence of the endothelium (Citation163). In addition, t-AUCB protects against ischemia–reperfusion injury (Citation164). In deoxycorticosterone acetate (DOCA)-salt hypertensive mice, genetic sEH deficiency and t-AUCB have also beneficial effects on the kidneys by reducing inflammation and injury (Citation165), although they augment albuminuria in mice with progressive renal disease (Citation166). Thus, these results highlight the efficacy of sEH inhibitors in renal hypertension and inflammation but perhaps question their therapeutic utility in cardiovascular disease associated with chronic renal failure. Besides the potential cardiovascular protective actions of sEH inhibitors, they may also be considered anti-inflammatory drugs because of their ability to augment the anti-inflammatory actions of aspirin and of inhibitors of synthesis of pro-inflammatory 5-lipoxygenase-derived leukotrienes (Citation167) and to reduce inflammatory pain (Citation151).
Lipoxygenases. 12-HETE, a product of the lipoxygenase isoform 12-LO expressed in the arterial wall, is a vasodilator and enhances BK channel activity in vascular smooth muscle (Citation130). Another isoform, 15-LO, generates HEETA and hydroxyepoxyeicosatrienoic acid (THETA) which activate a KCa-like channel (Citation168). However, HETE, derived from lipoxygenase but also from cytochrome P450, and cysteinyl-leukotrienes can evoke contractions of vascular smooth muscle cells. Endothelium-dependent contractions involving a lipoxygenase pathway have been reported in various arteries and veins in response to neuro-humoral mediators, mechanical stretch, and ischemia (Citation1–5). Therefore, their true contribution as vasodilator molecules in vivo is still uncertain but they are not likely to contribute to the control of basal arterial blood pressure since the administration of lipoxygenase inhibitor BW755C to rats and rabbits or deletion of the 12/15-LO gene in mice does not result in marked hemodynamic alterations (Citation130). Nonetheless, the insertion of human 15-LO-1 gene in rabbits enhances the depressor responses to the classical endothelial agonist, acetylcholine (Citation169). In angiotensin-II-hypertensive animals, lipoxygenase-inhibitors also lower arterial blood pressure due to a diminished production of vasoconstrictor HETE(s) (Citation130). An enhanced production of 5-HETE, 12-HETE, and 15-HETE metabolites has been observed in placental tissue of pre-eclamptic women, suggesting either a compensatory or a pathophysiological role of these eicosanoids in pregnancy-induced hypertension (Citation170). Moreover, the 12/15-LO inhibitor baicalein and its derivatives have favorable effects in animal models of ischemic stroke (Citation130). In keeping with this on first sight Janus-faced vasoactive profile of HETE(s), it remains uncertain whether HETE(s) are beneficial or detrimental in hypertension ().
Non-chemical EDHF pathways. Electrotonical coupling between endothelial and vascular smooth muscle cells is a simple but effective route for direct transfer of negative charges and may help to explain why EDHF-signaling is particularly prominent in smaller arteries (Citation139,Citation140,Citation171), since their wall, consisting of only one or few layers of smooth muscle cells, represents a small electrical sink. Here endothelial hyperpolarization mediated by small/intermediate conductance Ca2+-activated potassium channels (KCa3.1 and KCa2.3) appears to be the starting-point (Citation133,Citation172,Citation173). In addition, K+-release mediated by perhaps KCa3.1 (Citation133), which is located at myoendothelial junctions (Citation174,Citation175), increases the local concentration of the ion in the inter-myoendothelial space and activates inwardly rectifying (KIR) channels and the Na+/K+ -pump to elicit hyperpolarization of the smooth muscle (Citation133). This EDHF-signaling involving electrotonical coupling—with, as initial trigger, endothelial hyperpolarization mediated by KCa3.1 and KCa2.3—is a Ca2+ -dependent process and involves calmodulin which serves as the Ca2+ -sensing channel subunit (Citation141,Citation176) (). KCa3.1 channels sense preferentially Ca2+ originating from stores after receptor stimulation. By contrast, KCa2.3 channels sense Ca2+ influx through ‘osmo/mechanosensitive’ transient receptor potential (TRP) channels (Citation177,Citation178), as suggested by the reduced flow-induced vasodilatation observed in genetic deficiencies of both KCa2.3 (Citation172) and TRPV4 (Citation179,Citation180), and appear required to produce hyperemia in working skeletal muscle in order to meet the metabolic demands (Citation181). The role of KCa3.1 and KCa2.3 in systemic cardiovascular homeostasis is indicated by the higher arterial blood pressure in mice lacking one or both of these channels (Citation172,Citation182,Citation183).
In several models of hypertension, hypercholesterolemia, atherosclerosis and diabetes as well as in some human cardiovascular pathologies, alterations of the KCa3.1/KCa2.3-EDHF system appear to contribute to endothelial dysfunction (184-187). For example, mesenteric arteries of spontaneously hypertensive mice exhibit compromised endothelium-dependent hyperpolarization and relaxations resulting from defective expression/activity of KCa2.3 and inwardly-rectifying K+ channels and KIR as well as from a change in caveolin-1 oligomers (Citation188). By contrast, EDHF-mediated responses involving KCa3.1 are intact. Similar results are obtained in mesenteric arteries of stroke-prone spontaneously hypertensive rats (SHRSP), in which actually the expression of KCa3.1 is enhanced, possibly to compensate for the KCa2.3 deficiency (Citation189).
Although the roles of KCa3.1 and KCa2.3 in human cardiovascular regulation are not properly defined yet, there is evidence for endothelial defects related to alterations of KCa3.1 functions in isolated coronary arterioles after cardioplegic arrest and reperfusion (Citation184) and in the skeletal muscle microvasculature (Citation185). The downstream electrotonic transfer of hyperpolarization via myoendothelial gap junctions may also be impaired in small myometrial arteries from women with pre-eclampsia (Citation186).
Small molecule activators of these channels may provide a way to improve endothelial function. Indeed, NS309, an activator of KCa3.1, re-establishes EDHF-responses in mesenteric arteries from type 2 diabetic rats (Citation190). Furthermore, the KCa3.1/KCa2.3 opener SKA-31, with a 5-fold higher potency on KCa3.1 over KCa2.3, lowers arterial blood pressure in angiotensin-II hypertensive mice and improves the defective endothelium-dependent vasodilatation in carotid arteries of eNOS-deficient mice (Citation191–193). Moreover, inhibition of endothelial KCa promotes endothelium-dependent contractions (Citation194), which further augments the potential of KCa3.1/KCa2.3 activators to counteract endothelial dysfunction. Thus, the KCa3.1/KCa2.3–EDHF-dilator system provides promising perspectives in terms of therapeutic improvement of endothelial function and/or arterial blood pressure. However, compounds like NS309 and SKA-31 are not optimized, and their bioavailability is still poor (Citation192).
Modulation by NO
In a number of blood vessels, EDHF-mediated responses become obvious only when the synthesis of NO is inhibited (Citation128,Citation195). Thus, EDHF(s) are able to take over in the case of ‘classical’ endothelial dysfunction associated with a loss of NO synthesis (e.g. in arteries with regenerated endothelium (Citation196) or in eNOS-deficient mice (Citation197)), thus demonstrating strong compensatory efficiency of EDHF(s). Conversely, activation of EDHF pathways can impact on the production and/or action of endothelium-derived NO:
EETs. Pharmacological inhibition of sEH ensuing higher EET levels leads to a reduction of arterial blood pressure in experimental angiotensin-II hypertension (Citation149,Citation150). This blood pressure-lowering action depends on intact NO-synthesis as it is prevented by systemic administration of L-NAME (Citation150), a finding that further supports the notion that, besides being EDHF(s), higher EET levels improve NO-formation, and thus that EET and NO are part of the same pathways and/or act synergistically.
In addition to its direct EDHF actions on BK channels, 5,6-EET improves endothelial Ca2+ - signaling (Citation198) and likely also NO synthesis in an autocrine fashion (Citation150). In fact, 5,6-EET, 11,12-EET, 14,15-EET, and, surprisingly, their degradation products dihydroxyeicosatrienoic acids (DHETs), which have been suggested to have no biological activity whatsoever, cause vasodilatation by stimulating endothelial NO formation and NO-dependent vasodilatation (Citation150) ().
KCa3.1/KCa2.3 channels. Endothelium-dependent hyperpolarization, in response to agonists that stimulate G protein-coupled receptors, are associated with an increase in endothelial [Ca2+]i. Furthermore, substances that increase endothelial [Ca2+]i in a receptor-independent manner, such as calcium ionophore, also produce endothelium-dependent hyperpolarization of the smooth muscle cells. The principal mechanism that sustains the opening of endothelial KCa channels, following stimulation, is the capacitive calcium entry elicited by the depletion of calcium stores (Citation7,Citation129,Citation180). KCa3.1/KCa2.3 activity may be required for proper eNOS activation or NO-signaling (Citation172,Citation199). This idea is based on the assumption that endothelial hyperpolarization mediated by the KCa3.1/KCa2.3 channels also increases the driving force for Ca2+ influx through TRP channels or, and perhaps also, through store-operated channels after receptor activation and Ca2+ release from the endoplasmic reticulum. This Ca2+ influx-promoting action of endothelial hyperpolarization thus also increases Ca2+ -dependent eNOS activation and NO formation (). Likewise, prevention of any endothelial hyperpolarization by non-vasospasmogenic K+ levels impairs endothelium-dependent and NO-mediated vasodilatations (Citation200). However, there are also other reports challenging the biophysical dogma according to which membrane hyperpolarization promotes Ca2+ influx in endothelial cells (Citation199). Irrespectively, the KCa3.1 activator, NS309, not only elicits EDHF responses by producing strong endothelial and smooth muscle hyperpolarization but also increases NO-signaling in porcine retinal arterioles (Citation199) and NO formation in a human endothelial cell line (Citation201). Moreover, deficiency of KCa2.3 channels in mice appears to impair the large NO-mediated vasodilatation in carotid arteries in response to acetylcholine and to shear stress stimulation, while the EDHF-mediated vasodilatation to the muscarinic agonist is still intact in KCa2.3-deficient mice (Citation172). Mechanistically, this role of KCa2.3 for proper NO synthesis can be explained by the coupled compartmentalization of KCa2.3, eNOS, and Ca2+ influx channels of the TRP-type in caveolae (small Ω-shaped membrane invaginations) and/or intracellular caveosomes which—when shuttled to the cell membrane—may constitute a functional signalosome for synergistic NO/EDHF vasodilatation (Citation141) ().
The combined improvement of NO- and EDHF-signaling by endothelial KCa-activators may be useful considering the regionally different and vessel size-dependent contribution of the two vasodilator systems. Where EDHF-signaling is weak and that of NO strong, i.e. in large conduit arteries, an improvement of NO-signaling could be advantageous and could also be relevant in terms of preventing platelet aggregation and thrombosis. Where EDHF is strong and NO less dominant, i.e. in the arterioles of the microcirculation, the synergistic activation of both systems may produce a decrease in total peripheral resistance and exert anti-thrombotic actions.
In conclusion: Endothelial key enzyme and ion channels driving EDHF-signaling also stimulate NO synthesis by amplifying intracellular Ca2+ -signaling and eNOS activation. Thus, EDHF signals, besides acting as vasodilator stimuli per se, converge at least in part at the level of the NO pathway. From a medical and pharmacological perspective, pharmacologic stimulators of EDHF(s) systems (e.g. channel modulators with an improved selectivity and bioavailability profile compared to the known KCa3.1/KCa2.3 activators NS309 and SKA-31) could be useful to improve endothelial dysfunction in cardiovascular disease, not only by virtue of their own vasodilator potential, but also by improving NO formation. However, whether this principle applies to the treatment of human endothelial dysfunction warrants further translational research.
NO and endothelin-1
Release and action of endothelin-1
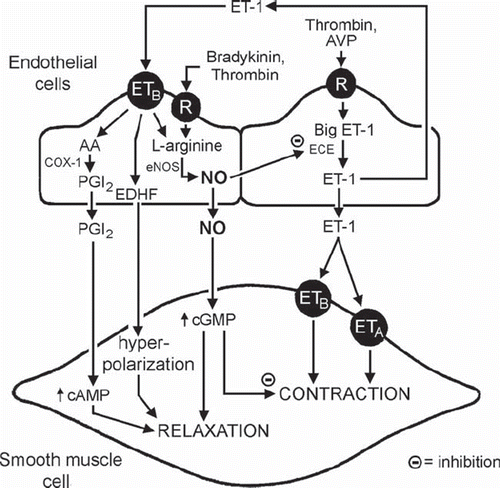
Endothelial cells can produce the potent vasoconstrictor peptide endothelin-1 (ET-1), one of the most potent vasoconstrictor substances known, and the release of the peptide may contribute to the long-term regulation of arterial blood pressure (Citation202–209). However, its contribution to moment-to-moment changes in vascular tone under physiological conditions is not established (Citation6,Citation210), except in arteries of aged rats (Citation211). Although under normal physiological conditions the production and/or the action of ET-1 cannot proceed unmatched (Citation6,Citation9), its generation can be up-regulated by a number of factors believed to play a role in vascular disease (Citation203,Citation206,Citation208,Citation211–213). Thus, the peptide may become progressively more important with aging, contribute to the abnormalities of pulmonary hypertension, and play a role in the terminal stages of vascular disease (Citation206,Citation208,Citation211,Citation213,Citation214).
Modulation by NO
Augmenting the production of NO and/or increasing the intracellular concentration of cGMP reduce the expression and the production of ET-1 (Citation6,Citation9,Citation215,Citation216). Both exogenous and endothelium-derived NO reduces, in a cGMP-dependent manner, the contraction of vascular smooth muscle cells in response to stimulation of ETA (and ETB) receptors by ET-1 (Citation217–221). The peptide also activates endothelial ETB receptors, linked to the activation of eNOS by Gi proteins (Citation6,Citation9,Citation222–228). Hence, augmented production of ET-1 by endothelial cells under physiological conditions will be offset by an augmented release of NO, which not only curtails the further generation of the peptide but also prevents its vasoconstrictor effects (). The unleashing of the production of ET-1 by the absence of NO is exemplified best by the sustained increase in arterial blood pressure caused by eNOS inhibitors, which is nearly prevented by antagonists of ET receptors (Citation6,Citation9). The feedback inhibition of the production and/or action of ET-1 is not limited to NO, as the peptide also increases the release of prostacyclin and EDHF, both of which further reduce any activation of the vascular smooth muscle that it may cause. Thus, the endothelial response to ET-1 provides an explanation for the minor contribution of the peptide to the moment-to-moment regulation of vascular tone under physiological conditions. As the endothelial cells age and/or regenerate and thus lose their Gi protein-mediated responsiveness, the moderation of the production and action of ET-1 declines, and the peptide contributes more and more to the dysfunction characteristic of the terminal stages of vascular disease (Citation9,Citation229) ().
Figure 4. NO and endothelin. Release and actions of endothelin-1 (ET-1) in the vascular wall. (AA = arachidonic acid; AVP = arginine vasopressin; cAMP = cyclic AMP; cGMP = cyclic GMP; COX = cyclo-oxygenases; ECE = endothelin-converting enzyme; ETA and ETB = endothelin-receptors; NO = nitric oxide; NOS = nitric oxide synthase; PGI2 = prostacyclin; R = cell membrane receptor) (from Feletou et al., 2008, with permission). (Reference 9)
NO and EDCF
Release and action of EDCF
Increases in intracellular calcium concentration in the endothelial cells not only evoke the release of relaxing (NO and EDHF) but also that of endothelium-derived contracting factors (EDCF). The mechanical consequence of these concomitant releases depends on the balance between the resulting tendencies toward relaxation or contraction. The EDCFs contributing to moment-to-moment changes in vascular tone are the products of endothelial cyclo-oxygenase, mainly its COX-1 isoform (Citation230–232). Indeed, increases in the endothelial intracellular calcium concentration activate the enzyme phospholipase A2, which liberates arachidonic acid from the cell membrane providing the substrate for COX-1 (Citation233). The breakdown of arachidonic acid by cyclo-oxygenase generates prostanoids (mainly endoperoxides and prostacyclin, but in certain cases also thromboxane A2 and prostaglandin F2α) and reactive oxygen species (Citation234–239). These mediators activate TP receptors of the vascular smooth muscle to evoke their contraction () (Citation240–242). The release of EDCF, and the resulting activation of TP receptors, contributes to the vasoconstrictor responses to adenine nucleotides and ET-1 (Citation243–246).
Figure 5. Endothelium-derived contracting factors (EDCF). Endothelium-dependent contraction is likely to be comprised of two components: generation of prostanoids and ROS. Each component depends on the activity of endothelial COX-1 and the stimulation of the TP receptors located on the smooth muscle to evoke contraction. In the SHR aorta, there is an increased expression of COX-1 and EP3 receptors, increased release of calcium, ROS, endoperoxides, and other prostanoids, which facilitates the greater occurrence of endothelium-dependent contraction in the hypertensive rat. The necessary increase in intracellular calcium can be triggered by receptor-dependent agonists, such as acetylcholine or ADP, or mimicked with calcium-increasing agents, such as the calcium ionophore A23187. The abnormal increase in intracellular ROS can be mimicked by the exogenous addition of H2O2 or the generation of extracellular ROS by incubation of xanthine with xanthine oxidase. The circled numbers indicate potential sites of therapeutic intervention: 1) inhibition of cyclo-oxygenases; and 2) antagonists of TP receptors. (AA = arachidonic acid; ACh = acetylcholine; ADP = adenosine diphosphate; H2O2 = hydrogen peroxide; m = muscarinic receptors; P2Y = purinergic receptor isoform 2Y; PGS, prostaglandin synthases; PGD2 = prostaglandin D2; PGE2 = prostaglandin E2; EP3, prostaglandin E2 receptor subtype 3; PGF2α = prostaglandin F2α; PGI2 = prostacyclin; PGIS = prostacyclin synthase; PLA2 = phospholipase A2; ROS = reactive oxygen species; TXA2 = thromboxane A2; TXAS = thromboxane synthase; X + XO = xanthine plus xanthine oxidase) (from Tang and Vanhoutte, 2009, with permission). (Reference 239)
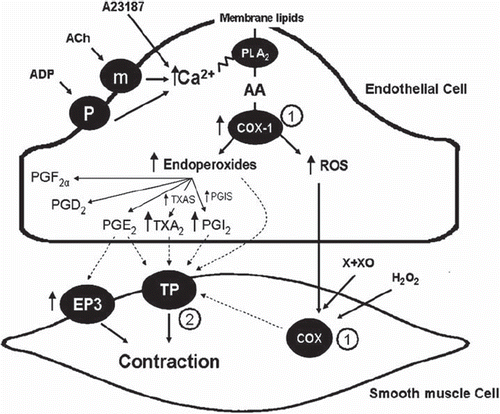
In certain arteries, ROS produced by the endothelium diffuse to the underlying vascular smooth muscle cells and cause them to contract, presumably by activating the cyclo-oxygenase that they contain (Citation247). However, in most cases, ROS mainly facilitate rather than induce EDCF-mediated responses (Citation238). Obviously, since superoxide anions scavenge NO (Citation40), resulting in the formation of peroxynitrite, oxidative stress impairs endothelium-dependent relaxations, which per se favors the occurrence of endothelium-dependent contractions (Citation239,Citation248–250).
Under physiological conditions, the releases of NO (and/or EDHF) predominates to maintain a normal vascular tone. However, with age and diseases such as hypertension, diabetes, and atherosclerosis, the balance between the endothelium-derived relaxing and contracting mediators is lost and the generation of EDCF becomes prominent (Citation5,Citation230,Citation251–254). This preponderance becomes a hallmark of endothelial dysfunction, in particular in hypertension ().
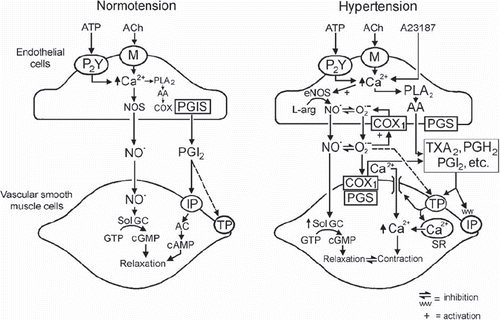
Figure 6. Endothelium-dependent effects of acetylcholine in rat aorta. Left: endothelium-dependent relaxations in normotensive rats. Right: cyclo-oxygenase-dependent, endothelium-dependent contractions to acetylcholine in SHR aorta. (AA = arachidonic acid; AC = adenylyl cyclase; COX1 = cyclo-oxygenase 1; IP = PGI2 receptor; M = muscarinic receptor; PGH2 = endoperoxides; PGI2 = prostacyclin; PGIS = prostacyclin synthase; PLA2 = phospholipase A2; R = receptor; sGC = soluble guanylyl cyclase; SR = sarcoplasmic reticulum; TP = TP receptor; + = activation; - = inhibition) (from Félétou and Vanhoutte 2006, with permission from the American Physiological Society). (Reference 1)
Modulation by NO
Acute inhibition. In the aorta of the spontaneously hypertensive rat (SHR), endothelium-dependent contractions, whether induced by acetylcholine or a calcium ionophore, are immediately potentiated by inhibitors of eNOS, intra- or extracellular NO scavengers, and selective inhibitors of soluble guanylyl cyclase (Citation255,Citation256). Since the combination of eNOS inhibitors or NO scavengers with inhibitors of soluble guanylyl cyclase has no additive potentiating effects on endothelium-dependent contractions (Citation256), the inhibition of EDCF-mediated responses by NO must result from a functional antagonism at the level of the vascular smooth muscle cells. This acute functional antagonism by endothelium-derived NO explains why most studies on endothelium-dependent contractions are performed in the presence of eNOS inhibitors, NO scavengers, and/or inhibitors of soluble guanylyl cyclase to optimize the EDCF-mediated responses ().
Long-term inhibition. In the SHR aorta, previous exposure to NO, as provided by an exogenous donor (sodium nitroprusside), causes a time- and concentration-dependent subsequent inhibition of EDCF-mediated contractions (obtained in the presence of an eNOS inhibitor), which persists long after the disappearance of NO from the biophase (Citation257). Thus, NO not only exerts an acute functional antagonism on endothelium-dependent contractions but also exerts a long-term inhibition of EDCF-mediated responses (Citation257). The mechanism underlying the prevention by previous exposure to NO of subsequent endothelium-dependent contractions is identical for acetylcholine (which activates M3 muscarinic receptors) and the calcium ionophore, A23187 (which opens pores in the cell membrane and permits the entry of extracellular calcium in cells). Indeed, previous exposure to the exogenous NO donor caused a comparable reduction in subsequent EDCF-mediated responses evoked by both agonists (Citation257), suggesting that sodium nitroprusside interferes with an event downstream of the surge in calcium required to initiate endothelium-dependent contractions. The long-term inhibition by previous exposure to the NO donor of subsequent endothelium-dependent contractions is reduced by an inhibitor of soluble guanylyl cyclase, demonstrating the involvement of cyclic GMP (Citation257). Since direct contractions of the vascular smooth muscle evoked by a specific TP receptor agonist were not blunted by a previous exposure to the NO donor (Citation257), the cyclic GMP-dependent long-term inhibition exerted by NO takes place within the endothelial cells.
In conclusion: NO and EDCF behave acutely as functional antagonists at the level of vascular smooth muscle. In addition, NO exerts a long-term negative modulation of the bioavailability of EDCF and thus curtails the occurrence of endothelium-dependent contractions. Under conditions of reduced NO production, for example due to eNOS uncoupling or augmented release of the endogenous eNOS inhibitor asymmetric dimethylarginine (ADMA) (Citation258), or in the case of a reduced NO bioavailability resulting from enhanced formation of superoxide anions (Citation40), the emergence of EDCF-mediated responses becomes a prominent sign of endothelial dysfunction (Citation9,Citation254).
General conclusions
In conclusion, the available experimental evidence highlights endothelial NO as the major orchestrating vasodilator molecule on which other vasodilator systems (EDHFs) and also detrimental EDCFs converge in an integrative fashion. The therapeutic utility of pharmacologic manipulations of the synthesis and action of NO to treat hypertension and cardiovascular disease remains likely but is still puzzling. Indeed, it appears that an improvement of eNOS function cannot be simply achieved by supplementation of substrate and co-factors for the enzyme. This may be explained by the more complex nature of arterial malfunction in cardiovascular disease states, involving disintegration/inefficacies at the level of gene transcription, enzyme function, and fate of NO. Moreover, NO actions are likely to be curtailed at the smooth muscle level. From the pharmacotherapeutic perspective, the question whether endothelial dysfunction, malfunction of vascular smooth muscle, and the resulting pathological vessel remodeling can be reversed or at least improved is of on-going interest. Results from experimental pharmacological interventions targeting the complex interconnections between NO, EDHFs, and EDCFs systems provide new venues for improved NO-signaling and thus for novel treatments and are an incentive for further laboratory and clinical research and for pharmaceutical discovery.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Félétou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol. 2006;291:H985–1002.
- Vanhoutte PM. COX-1 and vascular disease. Clin Pharmacol Ther. 2009;86:212–15.
- Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circulation J. 2009;73: 595–601.
- Vanhoutte PM, Tang E, Félétou M, Shimokawa H. Endothelial dysfunction and vascular disease. Acta Physiol. 2009;196:193–222.
- Tang EHC, Vanhoutte PM. Endothelial dysfunction: a strategic target in the treatment of hypertension? Pflugers Arch. 2010;459:995–1004.
- Vanhoutte PM. Say NO to ET. J Auto Nerv Sys. 2000;81:271–7.
- Félétou M, Vanhoutte PM. Endothelium-dependent hyperpolarizations: past beliefs and present facts. Ann Med. 2007;39:495–516.
- Félétou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond). 2009;117:139–55.
- Feletou M, Tang EHC, Vanhoutte PM. Nitric oxide the gatekeeper of endothelial vasomotor control. Front Biosci. 2008;13:4198–217.
- Furchgott RF, Zawadzki JV. The obligatory role of the endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–6.
- Palmer RMJ, Ferridge AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–6.
- Furchgott RF. Studies on relaxation of rabbit aorta by sodium nitrite: The basis for the proposal that the acid-activable inhibitory factor from bovine retractor penis is inorganic nitrite and the endothelium-derived relaxing factor is nitric oxide. Vanhoutte PM. Mechanism of vasodilatation. New York, NY: Raven Press; 1988. 401–14.
- Ignarro LJ, Byrns RE, Wood KS. Biochemical and pharmacological properties of EDRF and its similarity to nitric oxide radical. Vanhoutte PM. Mechanism of vasodilatation. New York, NY: Raven Press; 1988. 427–35.
- Palmer RMJ, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–6.
- Moncada S, Palmer RJM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–42.
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424.
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a camodulin requiring enzyme. Proc Natl Acad Sci U S A. 1990;87:682–86.
- Bredt TB, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351:714–7.
- Yui Y, Hattori R, Kosuga K, Eizawa H, Hiki K, Kawai C. Purification of nitric oxide synthase from rat macrophages. J Biol Chem. 1991;266:12544–8.
- Lyons CR, Orloff GJ, Cunningham JM. Molecular cloning and functional expression of an inducible nitric oxide synthase from a murine macrophage cell line. J Biol Chem. 1992;267:6370–4.
- Pollock JS, Fostermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, . Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine endothelial cells. Proc Natl Acad Sci U S A. 1991;88:10480–4.
- Sessa WC, Harrison JK, Barber CM, Zeng D, Durieux ME, D'Angelo DD, . Molecular cloning and expression of a cDNA encoding endothelial cell nitric oxide synthase. J Biol Chem. 1992;267:15274–8.
- Kone BC, Kuncewicz T, Zhang W, Yu ZY. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am J Physiol Renal Physiol. 2003;285:F178–90.
- Gonzales C, Barosso C, Martin C, Gulbenkian S, Estrada C. Neuronal nitric oxide synthase activation by vasoactive intestinal peptide in bovine cerebral arteries. J Cereb Blood Flow Metab. 1997;17:977–84.
- Papapetropoulos A, Desai KM, Rudic RD, Mayer B, Zhang R, Ruiz-Torres MP, . Nitric oxide synthase inhibitors attenuate transforming-growth-factor-beta1-stimulated capillary organization in vitro. Am J Pathol. 1997;150:1835–44.
- Boulanger CM, Heymes C, Benessiano J, Geske RS, Levy BI, Vanhoutte PM. Neuronal nitric oxide synthase is expressed in rat vascular smooth muscle cells: activation by angiotensin II in hypertension. Circ Res. 1998;83:1271–8.
- Papapetropoulos A, Rudic RD, Sessa WC. Molecular control of nitric oxide synthases in the cardiovascular system. Cardiovasc Res. 1999;43:509–20.
- Balligand JL, Kelly RA, Marsden PA, Smith TW, Michel T. Control of cardiac muscle cell function by an endogenous nitric oxide signalling system. Proc Natl Acad Sci USA. 1993;90:347–51.
- Sase K, Michel T. Expression of constitutive endothelial nitric oxide synthase in human blood platelets. Life Sci. 1995;57:2049–55.
- Matouk CC, Marsden PA. Epigenetic regulation of vascular endothelial gene expression. Circ Res. 2008;102:873–87.
- Bredt DS, Snyder SH. Nitric oxide: a physiological messenger molecule. Annu Rev Biochem. 1994;63:175–95.
- Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. 1999;43:521–31.
- Groenendijk BC, Van der Heiden K, Hierck BP, Poelmann RE. The role of shear stress on ET-1, KLF2, and NOS-3 expression in the developing cardiovascular system of chicken embryos in a venous ligation model. Physiology (Bethesda). 2007;22:380–9.
- Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89:481–534.
- Gimbrone MA. The Gordon Wilson lecture: Understanding vascular endothelium: a pilgrim's progress: endothelial dysfunction, biomechanical forces and the pathobiology of atherosclerosis. Trans Am Clin Climatol Assoc. 2010;121: 115–27.
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363: 15–23.
- Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247–60.
- Oess S, Icking A, Fulton D, Govers R, Müller-Esterl W. Subcellular targeting and trafficking of nitric oxide synthases. Biochem J. 2006;396:401–9.
- Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–6.
- Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor(s). Am J Physiol. 1986;250:H822–7.
- Wennmalm A, Benthin G, Edlund A, Jungersten L, Kieler-Jensen N, Lundin S, . Metabolism and excretion of nitric oxide in humans. An experimental and clinical study. Circ Res. 1993;73:1121–7.
- Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010; 106:633–46.
- Muller B, Kleschyov AL, Alencar JL, Vanin A, Stoclet JC. Nitric oxide transport and storage in the cardiovascular system. Ann N Y Acad Sci. 2002;962:131–9.
- Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, . Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci U S A. 2001;98:12814–9.
- Shiva S, Wang X, Ringwood LA, Xu X, Yuditskaya S, Annavajjhala V, . Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2:486–93.
- van Faassen EE, Bahrami S, Feelisch M, Hogg N, Kelm M, Kim-Shapiro DB, . Nitrite as regulator of hypoxic signaling in mammalian physiology. Med Res Rev. 2009;29:683–741.
- Basu S, Azarova NA, Font MD, King SB, Hogg N, Gladwin MT, . Nitrite reductase activity of Cytochrome C. J Biol Chem. 2008;283:32590–7.
- Gladwin MT, Kim-Shapiro DB. The functional nitrite reductase activity of the heme-globins. Blood. 2008;112: 2636–47.
- Rapoport RM, Murad F. Agonist induced endothelium-dependent relaxation in rat thoracic aorta may be mediated though cyclic GMP. Circ Res. 1983;52:352–7.
- Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23.
- Trepakova ES, Cohen RA, Bolotina VM. Nitric oxide inhibits capacitative cation influx in human platelets by promoting sarcoplasmic/endoplasmic reticulum Ca2+ -ATPase-dependent refilling of Ca2+ stores. Circ Res. 1999;84: 201–9.
- Cohen RA, Weisbrod RM, Gericke M, Yaghoubi M, Bierl C, Bolotina VM. Mechanism of nitric oxide-induced vasodilatation: refilling of intracellular stores by sarcoplasmic reticulum Ca2+ ATPase and inhibition of store-operated Ca2+ influx. Circ Res. 1999;84: 210–9.
- Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schöneich C, . S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–7.
- Tong X, Evangelista A, Cohen RA. Targeting the redox regulation of SERCA in vascular physiology and disease. Curr Opin Pharmacol. 2010;10:133–8.
- Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:2524–31.
- Taylor CT, Moncada S. Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia. Arterioscler Thromb Vasc Biol. 2010;30:643–7.
- Ignarro LJ. Heme-dependent activation of guanylate cyclase by nitric oxide: a novel signal transduction mechanism. Blood Vessels. 1991;28:67–73.
- Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122:216–38.
- Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280–91.
- Biel M, Zong X, Ludwig A, Sautter A, Hofmann F. Structure and function of cyclic nucleotide-gated channels. Rev Physiol Biochem Pharmacol. 1999;135:151–71.
- Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci USA. 1996;93: 6770–4.
- Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–14.
- Chin-Dusting JP, Willems L, Kaye DM. L-arginine transporters in cardiovascular disease: a novel therapeutic target. Pharmacol Ther. 2007;116:428–36.
- Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. 2007;293:H3227–45.
- Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol. 2007;34:906–11.
- Ryoo S, Gupta G, Benjo A, Lim HK, Camara A, Sikka G, . Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ Res. 2008;102:923–32.
- Schulz E, Jansen T, Wenzel P, Daiber A, Münzel T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal. 2008;10:1115–26.
- Vasquez-Vivar J, Kalyanaraman B, Martasek P. The role of tetrahydrobiopterin in superoxide generation from eNOS: enzymology and physiological implications. Free Radical Res. 2003;37:121–7.
- Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2006;26: 2439–44.
- Schmidt TS, Alp NJ. Mechanisms for the role of tetrahydrobiopterin in endothelial function and vascular disease. Clin Sci (Lond). 2007;113:47–63.
- Cayatte AJ, Palacino JJ, Horten K, Cohen RA. Chronic inhibition of nitric oxide production accelerates neointima formation and impairs endothelial function in hypercholesterolemic rabbits. Arterioscler Thromb. 1994;14:753–9.
- Kauser K, da Cunha V, Fitch R, Mallari C, Rubanyi GM. Role of endogenous nitric oxide in progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Physiol Heart Circ Physiol. 2000;278:H1679–85.
- Chen J, Kuhlencordt PJ, Astern J, Gyurko R, Huang PL. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation. 2001;104:2391–4.
- Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, . Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–54.
- Ozaki M, Kawashima S, Yamashita T, Hirase T, Namiki M, Inoue N, . Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J Clin Invest. 2002;110:331–40.
- Wohlfart P, Xu H, Endlich A, Habermeier A, Closs EI, Hübschle T, . Antiatherosclerotic effects of small-molecular-weight compounds enhancing endothelial nitric-oxide synthase (eNOS) expression and preventing eNOS uncoupling. J Pharmacol Exp Ther. 2008;325:370–9.
- Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, . Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–6.
- Gracia-Sancho J, Villarreal G Jr, Zhang Y, García-Cardeña G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc Res. 2010;85:514–9.
- Kukreja RC, Xi L. eNOS phosphorylation: a pivotal molecular switch in vasodilation and cardioprotection? J Mol Cell Cardiol. 2007;42:280–2.
- Fisslthaler B, Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res. 2009;105:114–27.
- Wong AK, Howie J, Petrie JR, Lang CC. AMP-activated protein kinase pathway: a potential therapeutic target in cardiometabolic disease. Clin Sci (Lond). 2009;116:607–20.
- Gao YS, Nagao T, Bond RA, Janssens WJ, Vanhoutte PM. Nebivolol induces endothelium-dependent relaxations of canine coronary arteries. J Cardiovasc Pharmacol. 1991; 17:964–9.
- Münzel T, Gori T. Nebivolol: the somewhat-different beta-adrenergic receptor blocker. J Am Coll Cardiol. 2009;54: 1491–9.
- Vilaine JP, Biondi ML, Villeneuve N, Feletou M, Peglion JL, Vanhoutte PM. The calcium channel antagonist S 11568 causes endothelium-dependent relaxation in canine arteries. Eur J Pharmacol. 1991;197:41–8.
- Zhang X, Hintze TH. Amlodipine releases nitric oxide from canine coronary microvessels: an unexpected mechanism of action of a calcium channel-blocking agent. Circulation. 1998;97:576–80.
- Dhein S, Salameh A, Berkels R, Klaus W. Dual mode of action of dihydropyridine calcium antagonists: a role for nitric oxide. Drugs. 1999;58:397–404.
- Félétou M, Teisseire B. Converting enzyme inhibition in isolated porcine resistance artery potentiates bradykinin relaxation. Eur J Pharmacol. 1990;190:159–66.
- Mombouli JV, Illiano S, Nagao T, Vanhoutte PM. The potentiation of bradykinin-induced relaxations by perindoprilat in canine coronary arteries involves both nitric oxide and endothelium-derived hyperpolarizing factor. Circ Res. 1992;71:137–44.
- Enseleit F, Hurlimann D, Luscher TF. Vascular protective effects of angiotensin converting enzyme inhibitors and their relation to clinical events. J Cardiovasc Pharmacol. 2001;37:S21–30.
- Benzing T, Fleming I, Blaukat A, Müller-Esterl W, Busse R. Angiotensin-converting enzyme inhibitor ramiprilat interferes with the sequestration of the B2 kinin receptor within the plasma membrane of native endothelial cells. Circulation. 1999;99:2034–40.
- Mombouli JV, Ballard KD, Vanhoutte PM. Kininase-independent potentiation of endothelium-dependent relaxations to kinins by converting enzyme inhibitor perindoprilat. Acta Pharmacol Sin. 2002;23:203–7.
- Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, . Effects of angiotensin-converting enzyme inhibitors and angiotensin II type I receptor antagonists in rats with heart failure. Role of kinins and angiotentensin II type 2 receptors. J Clin Invest. 1997;99:1926–35.
- Katada J, Majima M. AT(2) receptor-dependent vasodilation is mediated by activation of vascular kinin generation under flow conditions. Br J Pharmacol. 2002;136:484–91.
- Batenburg WW, Garrelds IM, Bernasconi CC, Juillerat-Jeanneret L, van Kats JP, Saxena PR, . Angiotensin II type 2 receptor-mediated vasodilation in human coronary microarteries. Circulation. 2004;109:2296–301.
- Li H, Wallerath T, Münzel T, Förstermann U. Regulation of endothelial-type NO synthase expression in pathophysiology and in response to drugs. Nitric Oxide. 2002;7: 149–64.
- Thai H, Wollmuth J, Goldman S, Gaballa M. Angiotensin subtype 1 rReceptor (AT1) blockade improves vasorelaxation in heart failure by up-regulation of endothelial nitric-oxide synthase via activation of the AT2 receptor. J Pharmacol Exp Ther. 2003;307:1171–8.
- Imanishi T, Tsujioka H, Ikejima H, Kuroi A, Takarada S, Kitabata H, . Renin inhibitor aliskiren improves impaired nitric oxide bioavailability and protects against atherosclerotic changes. Hypertension. 2008;52:563–72.
- Dong YF, Liu L, Kataoka K, Nakamura T, Fukuda M, Tokutomi Y, . Aliskiren prevents cardiovascular complications and pancreatic injury in a mouse model of obesity and type 2 diabetes. Diabetologia. 2010;53:180–91.
- Xu S, Touyz RM. Reactive oxygen species and vascular remodelling in hypertension: still alive. Can J Cardiol. 2006;22:947–51.
- Sedeek M, Hébert RL, Kennedy CR, Burns KD, Touyz RM. Molecular mechanisms of hypertension: role of Nox family NADPH oxidases. Curr Opin Nephrol Hypertens. 2009;18:122–7.
- Pechanova O, Simko F. Chronic antioxidant therapy fails to ameliorate hypertension: potential mechanisms behind. J Hypertens. 2009;27 Suppl 6:S32–36.
- Simonsen U, Rodriguez-Rodriguez R, Dalsgaard T, Buus NH, Stankevicius E. Novel approaches to improving endothelium-dependent nitric oxide-mediated vasodilatation. Pharmacol Rep. 2009;61:105–15.
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313.
- Lassègue B, Griendling KK. Reactive oxygen species in hypertension. Am J Hypertens. 2004;17:852–60.
- Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, . Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–5.
- Taddei S, Salvetti A. Endothelial dysfunction in essential hypertension: clinical implications. J Hypertens. 2002;20: 1671–4.
- Herman AG, Moncada S. Therapeutic potential of nitric oxide donors in the prevention and treatment of atherosclerosis. Eur Heart J. 2005;26:1945–55.
- Lerman A, Zeiher AM. Endothelial function: cardiac events. Circulation. 2005;111:363–8.
- Spieker LE, Flammer AJ, Lüscher TF. The vascular endothelium in hypertension. Handb Exp Pharmacol. 2006;(176 Pt 2):249–83.
- Mayer B, Beretta M. The enigma of nitroglycerin bioactivation and nitrate tolerance: news, views and troubles. Br J Pharmacol. 2008;155:170–84.
- Megson IL, Webb DJ. Nitric oxide donor drugs: current status and future trends. Expert Opin Investig Drugs. 2002;11:587–601.
- Martelli A, Breschi MC, Calderone V. Pharmacodynamic hybrids coupling established cardiovascular mechanisms of action with additional nitric oxide releasing properties. Curr Pharm Des. 2009;15:614–36.
- Iwanaga Y, Gu Y, Dieterle T, Presotto C, Del Soldato P, Peterson KL, . A nitric oxide-releasing derivative of enalapril, NCX 899, prevents progressive cardiac dysfunction and remodeling in hamsters with heart failure. FASEB J. 2004;18:587–8.
- Okuyama CE, Mendes GD, Faro R, Rezende VM, Lagos RM, Astigarraga RE, . Pharmacokinetics and pharmacodynamics of a nitric oxide-releasing derivative of enalapril in male beagles. Clin Exp Pharmacol Physiol. 2007;34:290–5.
- Evgenov OV, Pacher P, Schmidt PM, Haskó G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–68.
- Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR, . Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol. 2003;64:533–46.
- Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, . Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414.
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995; 48:184–8.
- Stasch JP, Hobbs AJ. NO-independent, haem-dependent soluble guanylate cyclase stimulators. Handb Exp Pharmacol. 2009;191:277–308.
- Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, Arun Kumar HS, Meurer S, . Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116: 2552–61.
- Schmidt HH, Schmidt PM, Stasch JP. NO- and haem-independent soluble guanylate cyclase activators. Handb Exp Pharmacol. 2009;191:309–39.
- Kass DA, Champion HC, Beavo JA. Phosphodiesterase type 5: expanding roles in cardiovascular regulation. Circ Res. 2007;101:1084–95.
- Zaccolo M, Movsesian MA. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res. 2007;100:1569–78.
- Boswell-Smith V, Spina D, Page CP. Phosphodiesterase inhibitors. Br J Pharmacol. 2006;147 Suppl 1:S252–7.
- Kass DA, Takimoto E, Nagayama T, Champion HC. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res. 2007;75:303–14.
- Ghiadoni L, Versari D, Taddei S. Phosphodiesterase 5 inhibition in essential hypertension. Curr Hypertens Rep. 2008;10:52–7.
- Edwards G, Feletou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch. 2010;459:863–79.
- Feletou M, Köhler R, Vanhoutte PM. Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep. 2010;12:267–75.
- Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 2010; 59:881–95.
- Chawengsub Y, Gauthier KM, Campbell WB. Role of arachidonic acid lipoxygenase metabolites in the regulation of vascular tone. Am J Physiol Heart Circ Physiol. 2009; 297:H495–507.
- Shimokawa H, Morikawa K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Mol Cell Cardiol. 2005;39:725–32.
- Chauhan SD, Nilsson H, Ahluwalia A, Hobbs AJ. Release of C-type natriuretic peptide accounts for the biological activity of endothelium-derived hyperpolarizing factor. Proc Natl Acad Sci U S A. 2003;100:1426–31.
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–72.
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–3.
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, . H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322: 587–90.
- Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol. 2006;26:1215–25.
- Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol. 2004;141:881–903.
- Koeppen M, Feil R, Siegl D, Feil S, Hofmann F, Pohl U, . cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension. 2004;44:952–5.
- Nagao T, Illiano S, Vanhoutte PM. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-L-arginine in rats. Am J Physiol. 1992;263: H1090–4.
- Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, . The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–11.
- Grgic I, Kaistha BP, Hoyer J, Köhler R. Endothelial Ca2+ -activated K+ channels in normal and impaired EDHF-dilator responses—relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol. 2009;157: 509–26.
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, . Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–7.
- Gauthier KM, Edwards EM, Falck JR, Reddy DS, Campbell WB. 14,15-Epoxyeicosatrienoic acid represents a transferable endothelium-dependent relaxing factor in bovine coronary arteries. Hypertension. 2005;45:666–71.
- Taddei S, Versari D, Cipriano A, Ghiadoni L, Galetta F, Franzoni F, . Identification of a cytochrome P450 2C9-derived endothelium-derived hyperpolarizing factor in essential hypertensive patients. J Am Coll Cardiol. 2006; 48:508–15.
- Lenasi H. The role of nitric oxide- and prostacyclin-independent vasodilatation in the human cutaneous microcirculation: effect of cytochrome P450 2C9 inhibition. Clin Physiol Funct Imaging. 2009;29:263–70.
- Virdis A, Cetani F, Giannarelli C, Banti C, Ghiadoni L, Ambrogini E, . The sulfaphenazole-sensitive pathway acts as a compensatory mechanism for impaired nitric oxide availability in patients with primary hyperparathyroidism. Effect of surgical treatment. J Clin Endocrinol Metab. 2010;95:920–7.
- Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–10.
- Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, . Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010;24:3770–81.
- Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol. 2007;50:225–37.
- Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, . Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol. 2009;29:54–60.
- Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, . 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J Med Chem. 2010;53: 7067–75.
- Loot AE, Fleming I. Cytochrome P450-derived epoxyeicosatrienoic acids and pulmonary hypertension: central role of transient receptor potential (TRP) C6 channels. J Cardiovasc Pharmacol. 2011;57:140–7.
- Revermann M, Barbosa-Sicard E, Dony E, Schermuly RT, Morisseau C, Geisslinger G, . Inhibition of the soluble epoxide hydrolase attenuates monocrotaline-induced pulmonary hypertension in rats. J Hypertens. 2009;27: 322–31.
- Ulu A, Davis BB, Tsai HJ, Kim IH, Morisseau C, Inceoglu B, . Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol. 2008;52:314–23.
- Wang YX, Ulu A, Zhang LN, Hammock B. Soluble epoxide hydrolase in atherosclerosis. Curr Atheroscler Rep. 2010; 12:174–83.
- Zhang LN, Vincelette J, Cheng Y, Mehra U, Chen D, Anandan SK, . Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb Vasc Biol. 2009;29:1265–70.
- Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, . Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol. 2009;174:2086–95.
- Zhang W, Otsuka T, Sugo N, Ardeshiri A, Alhadid YK, Iliff JJ, . Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39:2073–8.
- Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gosele C, . Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat Genet. 2008;40:529–37.
- Motoki A, Merkel MJ, Packwood WH, Cao Z, Liu L, Iliff J, . Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am J Physiol Heart Circ Physiol. 2008;295: H2128–34.
- Luo P, Chang HH, Zhou Y, Zhang S, Hwang SH, Morisseau C, . Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther. 2010; 334:430–8.
- Revermann M, Schloss M, Barbosa-Sicard E, Mieth A, Liebner S, Morisseau C, . Soluble epoxide hydrolase deficiency attenuates neointima formation in the femoral cuff model of hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2010;30:909–14.
- Simpkins AN, Rudic RD, Roy S, Tsai HJ, Hammock BD, Imig JD. Soluble epoxide hydrolase inhibition modulates vascular remodeling. Am J Physiol Heart Circ Physiol. 2010;298:H795–806.
- Chaudhary KR, Abukhashim M, Hwang SH, Hammock BD, Seubert JM. Inhibition of soluble epoxide hydrolase by trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is protective against ischemia-reperfusion injury. J Cardiovasc Pharmacol. 2010;55:67–73.
- Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, . Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009;297:F740–8.
- Jung O, Jansen F, Mieth A, Barbosa-Sicard E, Pliquett RU, Babelova A, . Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PLoS One. 2010;5:e11979.
- Liu JY, Yang J, Inceoglu B, Qiu H, Ulu A, Hwang SH, . Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem Pharmacol. 2010;79:880–7.
- Gauthier KM, Chawengsub Y, Goldman DH, Conrow RE, Anjaiah S, Falck JR, . 11®,12(S),15(S)-trihydroxyeicosa-5(Z),8(Z),13(E)-trienoic acid: an endothelium-derived 15-lipoxygenase metabolite that relaxes rabbit aorta. Am J Physiol Heart Circ Physiol. 2008;294:H1467–72.
- Aggarwal NT, Chawengsub Y, Gauthier KM, Viita H, Yla-Herttuala S, Campbell WB. Endothelial 15-lipoxygenase-1 overexpression increases acetylcholine-induced hypotension and vasorelaxation in rabbits. Hypertension. 2008; 51:246–51.
- Pearson T, Zhang J, Arya P, Warren AY, Ortori C, Fakis A, . Measurement of vasoactive metabolites (hydroxyeicosatetraenoic and epoxyeicosatrienoic acids) in uterine tissues of normal and compromised human pregnancy. J Hypertens. 2010;28:2429–37.
- Wölfle SE, Schmidt VJ, Hoyer J, Köhler R, de Wit C. Prominent role of KCa3.1 in endothelium-derived hyperpolarizing factor-type dilations and conducted responses in the microcirculation in vivo. Cardiovasc Res. 2009;82: 476–83.
- Brähler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, . Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. 2009;119:2323–32.
- Zygmunt PM, Hogestatt ED. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br J Pharmacol. 1996;117:1600–6.
- Dora KA, Gallagher NT, McNeish A, Garland CJ. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res. 2008;102: 1247–55.
- Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, . Functional architecture of inositol 1,4, 5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008; 105:9627–32.
- Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+ -activated K + channel complexed with Ca2 + /calmodulin. Nature. 2001;410:1120–4.
- Liedtke W, Kim C. Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci. 2005;62:2985–3001.
- Sharif-Naeini R, Dedman A, Folgering JH, Duprat F, Patel A, Nilius B, . TRP channels and mechanosensory transduction: insights into the arterial myogenic response. Pflugers Arch. 2008;456:529–40.
- Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, . Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One. 2007;2:e827.
- Zhang DX, Gutterman DD. TRP channel activation and endothelium-dependent dilation in the systemic circulation. J Cardiovasc Pharmacol. 2011;57:133–9.
- Milkau M, Köhler R, de Wit C. Crucial importance of the endothelial K + channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J. 2010;24:3572–9.
- Köhler R, Ruth P. Endothelial dysfunction and blood pressure alterations in K + -channel transgenic mice. Pflugers Arch. 2010;459:969–76.
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, . Altered expression of small-conductance Ca2+ -activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–31.
- Feng J, Liu Y, Clements RT, Sodha NR, Khabbaz KR, Senthilnathan V, . Calcium-activated potassium channels contribute to human coronary microvascular dysfunction after cardioplegic arrest. Circulation. 2008;118:S46–51.
- Liu Y, Sellke EW, Feng J, Clements RT, Sodha NR, Khabbaz KR, . Calcium-activated potassium channels contribute to human skeletal muscle microvascular endothelial dysfunction related to cardiopulmonary bypass. Surgery. 2008;144:239–44.
- Luksha L, Luksha N, Kublickas M, Nisell H, Kublickiene K. Diverse mechanisms of endothelium-derived hyperpolarizing factor-mediated dilatation in small myometrial arteries in normal human pregnancy and preeclampsia. Biol Reprod. 2010;83:728–35.
- Feletou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol. 2009;156:545–62.
- Weston AH, Porter EL, Harno E, Edwards G. Impairment of endothelial SK(Ca) channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br J Pharmacol. 2010;160: 836–43.
- Giachini FR, Carneiro FS, Lima VV, Carneiro ZN, Dorrance A, Webb RC, . Upregulation of intermediate calcium-activated potassium channels counterbalance the impaired endothelium-dependent vasodilation in stroke-prone spontaneously hypertensive rats. Transl Res. 2009; 154:183–93.
- Brondum E, Kold-Petersen H, Simonsen U, Aalkjaer C. NS309 restores EDHF-type relaxation in mesenteric small arteries from type 2 diabetic ZDF rats. Br J Pharmacol. 2009;159:154–65.
- Hasenau AL, Nielsen G, Morisseau C, Hammock BD, Wulff H, Köhler R. Improvement of endothelium-dependent vasodilations by SKA-31 and SKA-20, activators of small- and intermediate-conductance Ca2 + -activated K + -channels. Acta Physiol (Oxf). 2011 Mar 1 (Epub ahead of print).
- Köhler R, Kaistha BP, Wulff H. Vascular KCa-channels as therapeutic targets in hypertension and restenosis disease. Expert Opin Ther Targets. 2010;14:143–55.
- Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, . Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol. 2009;75:281–95.
- Michel FS, Man GS, Man RY, Vanhoutte PM. Hypertension and the absence of EDHF-mediated responses favour endothelium-dependent contractions in renal arteries of the rat. Br J Pharmacol. 2008;155:217–26.
- Olmos L, Mombouli JV, Illiano S, Vanhoutte PM. cGMP mediates the desensitization to bradykinin in isolated canine coronary arteries. Am J Physiol. 1995;268:H865–70.
- Thollon C, Fournet-Bourguignon MP, Saboureau D, Lesage L, Reure H, Vanhoutte PM, . Consequences of reduced production of NO on vascular reactivity of porcine coronary arteries after angioplasty: importance of EDHF. Br J Pharmacol. 2002;136:1153–61.
- Brandes RP, Schmitz-Winnenthal FH, Feletou M, Godecke A, Huang PL, Vanhoutte PM, . An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc Natl Acad Sci U S A. 2000;97:9747–52.
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–8.
- Dalsgaard T, Kroigaard C, Misfeldt M, Bek T, Simonsen U. Openers of small conductance calcium-activated potassium channels selectively enhance NO-mediated bradykinin vasodilatation in porcine retinal arterioles. Br J Pharmacol. 2010;160:1496–508.
- Schmidt K, Dubrovska G, Nielsen G, Fesüs G, Uhrenholt TR, Hansen PB, . Amplification of EDHF-type vasodilatations in TRPC1-deficient mice. Br J Pharmacol. 2010;161:1722–33.
- Sheng JZ, Ella S, Davis MJ, Hill MA, Braun AP. Openers of SKCa and IKCa channels enhance agonist-evoked endothelial nitric oxide synthesis and arteriolar vasodilation. FASEB J. 2009;23:1138–45.
- Yanagisawa M, Kurihara H, Kimura S, Tomobe TY, Kobayashi M, Mitsui Y, . A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–15.
- Schini VB, Vanhoutte PM. Endothelin-1: A potent vasoactive peptide. Pharmacol Toxicol. 1991;69:1–7.
- Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology and pathophysiology. Pharmacol Rev. 1994;6:325–415.
- Nava E, Lüscher TF. Endothelium-derived vasoactive factors in hypertension: nitric oxide and endothelin. J Hypertens Suppl. 1995;13:S39–48.
- Schiffrin EL. Endothelin and endothelin antagonists in hypertension. J Hypertension. 1998;1891–5.
- Miyauchi T, Masaki T. Pathophysiology of endothelin in the cardiovascular system. Annu Rev Physiol. 1999;61: 391–415.
- Iglarz M, Clozel M. Mechanisms of ET-1-induced endothelial dysfunction. J Cardiovasc Pharmacol. 2007;50:621–8.
- Kisanuki YY, Emoto N, Ohuchi T, Widyantoro B, Yagi K, Nakayama K, . Low blood pressure in endothelial cell-specific endothelin 1 knockout mice. Hypertension. 2010;56:121–8.
- Vanhoutte PM. Is endothelin involved in the pathogenesis of hypertension? Hypertension. 1993;21:747–51.
- Goel A, Su B, Flavahan S, Lowenstein CJ, Berkowitz DE, Flavahan NA. Increased endothelial exocytosis and generation of endothelin-1 contributes to constriction of aged arteries. Circ Res. 2010;107:242–51.
- de Andrade CR, Leite PF, Montezano AC, Casolari DA, Yogi A, Tostes RC, . Increased endothelin-1 reactivity and endothelial dysfunction in carotid arteries from rats with hyperhomocysteinemia. Br J Pharmacol. 2009; 157:568–80.
- Raja SG. Endothelin receptor antagonists for pulmonary arterial hypertension: an overview. Cardiovasc Ther. 2010;28:e65–71.
- Schiffrin EL. Endothelin: role in experimental hypertension. J Cardiovasc Pharmacol. 2000;35:S33–5.
- Boulanger C, Lüscher TF. Release of endothelium from the porcine aorta: inhibition of endothelium-derived nitric oxide. J Clin Invest. 1990;85:587–90.
- Lüscher TF, Boulanger CM, Yang Z, Dohi Y. Interaction between endothelin and endothelium-derived relaxing factor(s). Rubanyi GM. Endothelin. New York, NY: Oxford University Press; 1992.
- Goligorsky MS, Tsukahara H, Magazine H, Andersen TT, Malik AB, Bahou WF. Termination of endothelin signaling: role of nitric oxide. J Cell Physiol. 1994;158:485–94.
- Miller VM, Komori K, Burnett JC Jr, Vanhoutte PM. Differential sensitivity to endothelin in canine arteries and veins. Am J Physiol. 1989;257:H1127–31.
- Chen Y, McCarron RM, Golech S, Bembry J, Ford B, Lenz FA, . ET-1- and NO-mediated signal transduction pathway in human brain capillary endothelial cells. Am J Physiol Cell Physiol. 2003;284:C243–9.
- Deuchar GA, Docherty A, MacLean MR, Hicks MN. Pulmonary hypertension secondary to left ventricular dysfunction: the role of nitric oxide and endothelin-1 in the control of pulmonary vascular tone. Br J Pharmacol. 2002; 135:1060–8.
- Matsumoto T, Ishida K, Nakayama N, Kobayashi T, Kamata K. Involvement of NO and MEK/ERK pathway in enhancement of endothelin-1-induced mesenteric artery contraction in later-stage type 2 diabetic Goto-Kakizaki rat. Am J Physiol Heart Circ Physiol. 2009;296:H1388–97.
- Flavahan NA, Vanhoutte PM. G-proteins and endothelial responses. Blood Vessels. 1990;27:218–29.
- Flavahan NA, Vanhoutte PM. Endothelial cell signaling and endothelial dysfunction. Am J Hypertens. 1995;8: S28–41.
- Schini VB, Kim ND, Vanhoutte PM. The basal and stimulated release of EDRF inhibits the contractions evoked by endothelin-1 and endothelin-3 in aortae of normotensive and spontaneously hypertensive rats. J Cardiovasc Pharmacol. 1991;17:S266–70.
- Bagnall AJ, Kelland NF, Gulliver-Sloan F, Davenport AP, Gray GA, Yanagisawa M, . Deletion of endothelial cell endothelin B receptors does not affect blood pressure or sensitivity to salt. Hypertension. 2006;48:286–93.
- Bagnall A, Webb D, Kotelevtsev Y. A transgenic strategy for analysis of the function of the endothelin-B-receptor. J Cardiovasc Pharmacol. 2000;36(5 Suppl 1):S90–2.
- Tykocki NR, Gariepy CE, Watts SW. Endothelin ET(B) receptors in arteries and veins: multiple actions in the vein. J Pharmacol Exp Ther. 2009;329:875–81.
- Tian J, Wong WT, Tian XY, Zhang P, Huang Y, Wang N. Rosiglitazone attenuates endothelin-1-induced vasoconstriction by upregulating endothelial expression of endothelin B receptor. Hypertension. 2010;56:129–35.
- Taddei S, Virdis A, Ghiadoni L, Sudano I, Notari M, Salvetti A. Vasoconstriction to endogenous endothelin-1 is increased in the peripheral circulation of patients with essential hypertension. Circulation. 1999;100:1680–83.
- Vanhoutte PM, Félétou M, Taddei S. Endothelium-dependent contractions in hypertension. Br J Pharmacol. 2005;144:449–58.
- Vanhoutte PM, Tang EHC. Endothelium-dependent contractions: when a good guy turns bad! J Physiol. 2008;586.22:5295–304.
- Félétou M, Huang Y, Vanhoutte PM. Vasoconstrictor prostanoids. Pflugers Arch. 2010;459:941–50.
- Wong MSK, Vanhoutte PM. COX-mediated endothelium-dependent contractions: from the past to recent discoveries. Acta Pharmacol Sin. 2010;31:1095–102.
- Ge T, Hughes H, Junquero DC, Wu KK, Vanhoutte PM, Boulanger CM. Endothelium-dependent contractions are associated with both augmented expression of prostaglandin H synthase-1 and hypersensitivity to prostaglandin H2 in the SHR aorta. Circ Res. 1995;76:1003–10.
- Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Félétou M. Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol. 2005;146:834–45.
- Gluais P, Paysant J, Badier-Commander C, Verbeuren T, Vanhoutte PM, Félétou M. In SHR aorta, calcium ionophore A-23187 releases prostacyclin and thromboxane A2 as endothelium-derived contracting factors. Am J Physiol Heart Circ Physiol. 2006;291:H2255–64.
- Zhou Y, Varadharaj S, Zhao X, Parinandi N, Flavahan NA, Zweier JL. Acetylcholine causes endothelium-dependent contraction of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289:H1027–32.
- Tang EH, Leung FP, Huang Y, Félétou M, So KF, Man RY, . Calcium and reactive oxygen species increase in endothelial cells in response to releasers of endothelium-derived contracting factor. Br J Pharmacol. 2007;151: 15–23.
- Tang EHC, Vanhoutte PM. Prostanoids and reactive oxygen species: team players in endothelium-dependent contractions. Pharmacol Ther. 2009;122:140–9.
- Auch-Schwelk W, Katusic ZS, Vanhoutte PM. Thromboxane A2 receptor antagonists inhibit endothelium-dependent contractions. Hypertension. 1990;15:699–703.
- Félétou M, Verbeuren TJ, Vanhoutte PM. Endothelium-dependent contractions in SHR: a tale of prostanoid TP and IP receptors. Br J Pharmacol. 2009;156:563–74.
- Félétou M, Vanhoutte PM, Verbeuren TJ. The thromboxane/endoperoxide receptor (TP): the common villain. J Cardiovasc Pharmacol. 2010;55:317–32.
- Taddei S, Vanhoutte PM. Role of endothelium in endothelin-evoked contractions in the rat aorta. Hypertension. 1993;21:9–15.
- Mombouli JV, Vanhoutte PM. Purinergic endothelium-dependent and independent contractions in rat aorta. Hypertension. 1993;22:577–83.
- Park S-J, Lee JL, Vanhoutte PM. Endothelin-1 releases endothelium-derived endoperoxides and thromboxane A2 in porcine coronary arteries with regenerated endothelium. Zhongguo Yao Lixue Bao. 1999;20:872–8.
- Desjardins F, Aubin MC, Carrier M, Perrault LP. Decrease of endothelin receptor subtype ETB and release of COX-derived products contribute to endothelial dysfunction of porcine epicardial coronary arteries in left ventricular hypertrophy. J Cardiovasc Pharmacol. 2005;45:499–508.
- Katusic ZS, Vanhoutte PM. Superoxide anion is an endothelium-derived contracting factor. Am J Physiol. 1989;257:H33–7.
- Cosentino F, Sill JC, Katusic ZS. Role of superoxide anions in the mediation of endothelium-dependent contractions. Hypertension. 1994;23:229–35.
- Katusic ZS. Superoxide anion and endothelial regulation of arterial tone. Free Radic Biol Med. 1996;20:443–8.
- Shi Y, Vanhoutte PM. Reactive oxygen-derived free radicals are key to the endothelial dysfunction of diabetes. J Diabetes. 2009;1:151–62.
- Cohen RA. Does EDCF contribute to diabetic endothelial cell dysfunction? Dialog Cardiovasc Med. 2002;7:225–31.
- Medow MS, Taneja I, Stewart JM. Cyclooxygenase and nitric oxide synthase dependence of cutaneous reactive hyperemia in humans. Am J Physiol Heart Circ Physiol. 2007;293:H425–32.
- Signori LU, da Silva AM, Plentz RD, Moreno H Jr, Irigoyen MC, Schaan BD. Reversal of postprandial endothelial dysfunction by cyclooxygenase inhibition in healthy volunteers. J Cardiovasc Pharmacol. 2009;54:90–3.
- Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br J Pharmacol. 2009; 157:527–36.
- Auch-Schwelk W, Katusic ZS, Vanhoutte PM. Nitric oxide inactivates endothelium-derived contracting factor in the rat aorta. Hypertension. 1992;19:442–5.
- Yang D, Gluais P, Zhang JN, Vanhoutte PM, Feletou M. Nitric oxide and inactivation of the endothelium-dependent contracting factors released by acetylcholine in SHR. J Cardiovasc Pharm. 2004;43:815–20.
- Tang EH, Félétou M, Huang Y, Man RY, Vanhoutte PM. Acetylcholine and sodium nitroprusside cause long-term inhibition of EDCF-mediated contractions. Am J Physiol Heart Circ Physiol. 2005;289:H2434–40.
- Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler Suppl. 2003;4:33–40.