Abstract
The annual incidence of sudden cardiac death (SCD) is estimated at 1 per 1,000 for adults over the age of 35 years, and 1 per 100,000 for adolescents and young adults. Although the overall incidence of unexpected SCD among previously healthy persons is small, the emotional impact of these events is devastating. The 12-lead electrocardiogram (ECG) has been used as a risk assessment tool from healthy occupational applicants and athletes to patients with cardiovascular disorders. The ECG is also routinely recorded in the majority of patients hospitalized for non-cardiovascular causes. Thus, it is a widely used tool intended for identification of unsuspected heart disease generally, as well as for diagnosing specific disorders predisposing to fatal arrhythmias in subjects who have not experienced such events but who are at increased risk. Recognition of specific ECG features is of importance for prevention of SCD in asymptomatic persons. The purpose of this review is to catalog the disorders associated with SCD that may be reflected in 12-lead ECGs seen in office or hospital practices and to discuss their prevalence and the magnitude of risks. The focus is on ECG findings suggesting increased SCD risk among the asymptomatic subjects without previously diagnosed cardiac disease.
Key messages
The purpose of this review is to catalog the disorders associated with sudden cardiac death (SCD) risk that may be reflected in routine 12-lead electrocardiograms (ECGs) seen in office or hospital practices and to discuss their prevalence and the magnitude of risks.
The primary focus is on ECG findings suggesting increased SCD risk among the asymptomatic subjects without previously diagnosed cardiac disease.
Introduction
The annual incidence of sudden cardiac death (SCD) in the United States is estimated at 1 per 1,000 for adults over the age of 35 years, and 1 per 100,000 for adolescents and young adults (Citation1). In many victims, an increased risk of SCD is identifiable based on the nature of an underlying disease, but in middle-aged and older adults at least 30% and perhaps up to 50% of all SCDs occur among previously asymptomatic subjects (Citation2). The proportion is likely even greater among adolescents and young adults (Citation3). Although the overall incidence of unexpected SCD among previously healthy persons among the general population is small, the emotional impact of these events is devastating.
The 12-lead electrocardiogram (ECG) has been used as a risk assessment tool for population subgroups ranging from healthy occupational applicants and athletes to patients with cardiovascular disorders. The ECG is also routinely recorded in the majority of patients before surgery beginning at middle age, as well as in patients hospitalized for non-cardiovascular causes. Thus, it is a widely used tool intended for identification of possible unsuspected heart disease generally, as well as for diagnosing specific disorders predisposing to fatal arrhythmias in subjects who have not experienced such events but who are at increased risk. Recognition of specific ECG features is of importance for prediction and prevention of SCD in asymptomatic persons.
ECG manifestations of cardiomyopathies
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is common among the causes of SCD in young adults, particularly among competitive athletes (Citation2). It is a rare cause of SCD among middle-aged adults. HCM is characterized by myocardial hypertrophy in the absence of hemodynamic stress that would cause the degree of hypertrophy, and has the further feature of histopathological myocyte disarray (Citation4–6). The HCM phenotype occurs in four patterns: HCM with asymmetrical septal hypertrophy (ASH); outflow left ventricular (LV) obstruction (HOCM); diffuse HCM with LV systolic cavity obliteration but no outflow obstruction; and apical HCM.
The prevalence of HCM has been estimated to be 1 in 500 (Citation7). The annual SCD incidence is ∼1% in non-obstructive forms, and among patients with LV outflow tract obstruction caused by the hypertrophy the incidence is ∼2% (Citation7–11). SCDs have also been reported in apical HCM (Citation12), but the apical form conveys a smaller risk, with an annual mortality rate of 0.1% (Citation13).
The ECG changes in HCM primarily reflect voltage patterns associated with LV hypertrophy. The ECG differences between secondary hypertrophy (i.e. hypertension, valvular aortic stenosis) and HCM are often difficult to distinguish, but typically HCM patients demonstrate evidence of left atrial enlargement, left ventricular enlargement, and marked strain patterns characterized by prominent ST-T wave changes in the absence of hypertension (). In the obstructive form with asymmetrical septal hypertrophy, prominent inferior Q waves and/or tall R waves in V1, reflecting septal hypertrophy, may be present (Citation14–17). Sometimes HCM patients also have slow, delta wave-like notched R wave up-strokes without the typical short PR interval of the Wolff–Parkinson–White (WPW) syndrome (see below) (Citation18). The ECG presentation of apical HCM differs dramatically from that of classical HCM, with the possibility of absence of increased voltage, but with prominent T wave inversions in the antero-lateral leads (). These ECG changes can be confused with ischemia.
Figure 1. The ECG in hypertrophic cardiomyopathy. High QRS voltage in the absence of hypertension with repolarization changes (strain pattern) in the ST segments of inferior and lateral leads, and inversion of terminal P wave in lead V1. Notching of the QRS up-stroke in inferior leads, giving a delta wave-like appearance, is also detected. Paper speed 25 mm/s, gain 10 mm/mV.
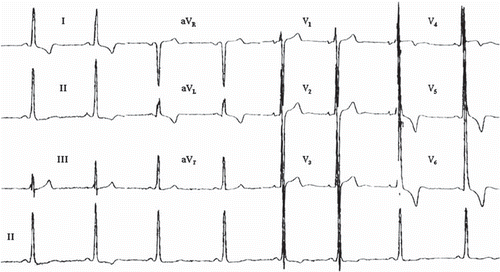
Figure 2. ECG manifestation of apical hypertrophic cardiomyopathy. ST segment and T wave abnormalities similar to repolarization abnormalities seen in ischemia are commonly observed in the antero-apical leads in apical hypertrophic cardiomyopathy. QRS voltage may not be as prominent as in obstructive or diffuse HCM. Paper speed 25 mm/s, gain 10 mm/mV.
ECG changes that may mimic obstructive and diffuse HCM are common among young athletes, particularly males, with so-called athlete's hearts (), but ECGs in normal athletes commonly show less prominent repolarization changes and absence of atrial enlargement patterns (Citation19). Patients who present ECG changes of hypertrophy without known underlying cardiac disease, hypertension, or an athletic background should undergo echocardiographic examinations. HCM in adolescents and young adults can cause SCD in recreational athletes, as well as in competitive athletes participating in organized sports, which should not be overlooked (Citation3,Citation19). In some occasions, patients presenting with ‘tall and narrow’ QRS complexes without hypertrophy could be at risk for SCD as described by Wolpert et al., but this finding needs further studies on prevalence in the general population and associated SCD risk (Citation20).
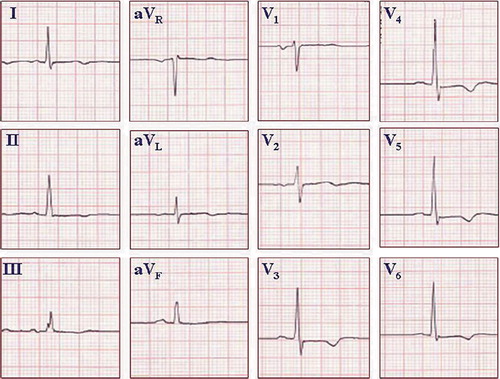
Figure 3. ECG from a healthy young athlete. The pattern of an ‘athlete heart’ may mimic the ECG changes seen in HCM. QRS voltage criteria for LVH based on the Sokolow–Lyon criteria, ST segment abnormalities similar to strain pattern, and inversion of terminal P wave in lead V1 may all be present. An echocardiogram may be needed to exclude the possibility of HCM. If uncertain, serial echocardiograms may be repeated over time, because the anatomic pattern of HCM may lag behind the ECG changes. Paper speed 25 mm/s, gain 10 mm/mV.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) usually presents with various symptoms of heart failure but may be discovered incidentally on the basis of an ECG abnormality or an unexpected finding on an echocardiogram. In its pre-symptomatic stage, or among patients who are Functional Class I after treatment of heart failure, the mortality rate is low, but the probability that a death will be sudden is proportionately higher than in the general population (Citation21).
DCM generates a variety of non-specific ECG abnormalities—commonly ST-T wave abnormalities and abnormal intraventricular conduction. Patients with lamin A/C mutation-related cardiomyopathy have been shown to present conduction abnormalities (prolonged PR interval) with only slightly reduced left ventricular function in the early stages of the disease (Citation22). Premature ventricular and atrial contractions and atrial fibrillation are commonly observed before the occurrence of life-threatening arrhythmias (Citation23) but do not necessarily predict cardiac arrest. In rare cases, SCD is the first manifestation of the disease (Citation24). In the advanced stage of DCM, ventricular arrhythmias are common (Citation21). The ECG manifestations are concomitant with the structural changes related to DCM, such as atrial enlargement and ventricular dilatation, with thin walls due to loss of myocardium. Atrial strain can be suspected from biphasic P wave with prominent inversion of its terminal portion in lead V1. Additionally, ST abnormalities, particularly in the lateral leads, are common, as are T wave inversions. Heart failure symptoms and ultimately echocardiogram will differentiate the ECG features from other cardiac conditions.
Arrhythmogenic right ventricular dysplasia/cardiomyopathy
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD, ARVC) is characterized by fibro-fatty replacement of the cardiac muscle in the right ventricle (RV) (Citation25,Citation26). The fibro-fatty deposits tend to occur in three regions of the RV: the RV outflow tract, subtricuspid area, and apex. In advanced cases, it may extend into the left ventricle, and isolated involvement of the LV has been observed rarely. The fibro-fatty replacement has been proposed to be the basis for delayed conduction and non-uniform depolarization in the involved areas, predisposing to re-entry-mediated ventricular tachycardia.
The prevalence of ARVD is estimated to be 1 in 5,000 (Citation26–28). However, this number may be an under-estimation due to the difficulties in the diagnosis of ARVD. Early clinical investigations suggested that ventricular tachycardia (VT) associated with ARVD was commonly a benign form of symptomatic monomorphic VT, but a subsequent study on post-mortem examinations after SCD indicated that many ARVD deaths occurred as first major clinical events, and the cumulatively the condition is high-risk (Citation29). The annual incidence of mortality is 3% without treatment and 1% with treatment, but in some cases the mode of death could be other than SCD (Citation30).
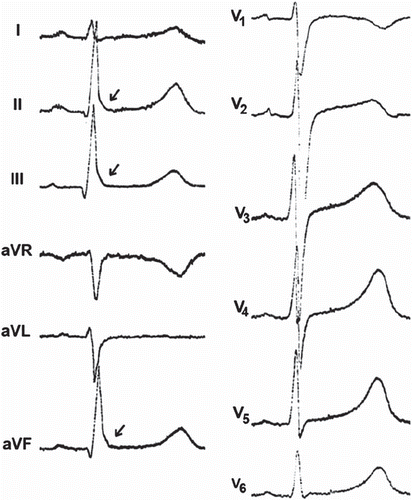
The ECG characteristics of ARVD are dependent on the stage of this progressive genetic disease. According to the recently renewed diagnostic criteria for ARVD, epsilon waves, QRS > 110 ms, inverted T waves in leads V1–3, and left bundle branch block (LBBB) morphology VT with superior axis were viewed as major diagnostic criteria from the ECG characteristics. T wave inversions in two or three consecutive precordial leads, LBBB morphology VT without superior axis, terminal activation delay (from the S wave nadir to the J point >55 ms), and right bundle branch block (RBBB) pattern with T wave inversions in the precordial leads were determined to fulfill minor ECG criteria for ARVD (Citation26–27). The T wave inversions in the leads V1–3 is considered the most common pattern reflecting ARVD () but is less specific than epsilon waves (). In some cases RBBB is present, which causes difficulties in the ECG analysis related to ARVD. Nevertheless, in some patients the diagnosis can be made only by repeated imaging studies during long-term follow-up. In most reports on ARVD, the initial diagnosis of the disease has been made during autopsy in a considerable proportion of subjects.
Figure 4. Arrhythmogenic right ventricular dysplasia. A: ECG on top presents ventricular tachycardia with left bundle branch block QRS morphology, and ECG below taken from the same patient demonstrates T wave inversions in the right precordial leads (V1–3), characteristic of ARVD. Paper speed 25 mm/s, gain 10 mm/mV. B: ECG presents a subtle epsilon wave in leads V1–3 without T wave inversions. Paper speed 25 mm/s, gain 10 mm/mV.
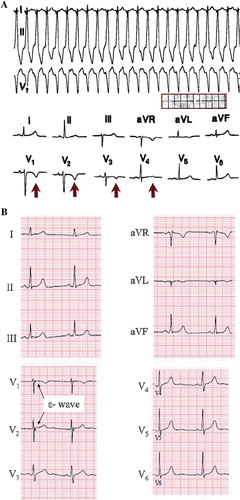
The prevalence of T wave inversions in leads V2–3 has been estimated to be between 1% and 3%, but very little is known of natural history of ARVD ECG findings in the general population (Citation31). For example, T wave inversions or terminal activation delay in the right precordial leads in an otherwise healthy subject are mostly ignored in clinical decision-making due to the lack of information about these ECG changes in the absence of structural changes suggesting ARVD or a family history of premature SCD.
Non-compaction cardiomyopathy
Non-compaction of the LV myocardium is a rare cause of SCD. Echocardiograms or cardiac MRI are required for the diagnosis, since the ECG is normal in up to 25% of cases and ECG manifestations are diverse and non-specific in the remainder (Citation32). Patients with non-compaction cardiomyopathy present with various forms of ST-T and intraventricular conduction abnormalities which are apparent in the precordial leads. Ventricular fibrillation or VT has been rarely described as the first symptom, but later on in symptomatic (heart failure) patients the annual risk can be as high as ∼7% (Citation33,Citation34).
Idiopathic myocardial fibrosis
Infrequent cases of SCD as the first manifestation of underlying disease have been associated with idiopathic myocardial fibrosis. The characteristics of the condition include transmural, interstitial fibrosis and other structural abnormalities. The described ECG characteristics include T wave inversions throughout the precordial leads (Citation35).
ECG manifestations of less common structural cardiac diseases
Wolff–Parkinson–White syndrome
The Wolff–Parkinson–White (WPW) syndrome is caused by the presence of accessory pathway(s) between the atria and the ventricles. They result in impulses bypassing the normal conduction system pattern through the atrio-ventricular node and His-Purkinje system. Accordingly, a portion of ventricular muscle is activated earlier than the normal A-V delay. This early activation, or pre-excitation, generates an early slow waveform of activation at the onset of the QRS complex with a shorter-than-normal PR interval, and the delta wave and the two parallel A-V pathways establish the substrate for re-entry.
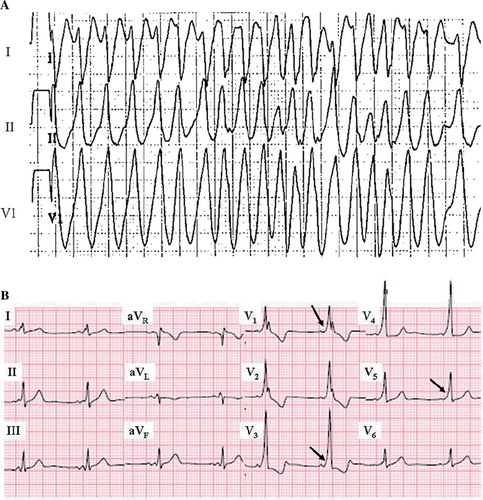
Although most commonly associated with symptomatic but benign A-V re-entrant supraventricular tachycardias, SCD has long been recognized as one of the risks associated with WPW syndrome (Citation36). The consensus regarding the pathophysiology of SCD risk is that rapid conduction to the ventricles may occur through an accessory pathway with a very short refractory period during episodes of atrial fibrillation (), potentially triggering ventricular fibrillation. In some patients, accessory pathways are capable of only retrograde conduction, and no delta wave will be seen (concealed WPW). These patients remain susceptible to re-entrant supraventricular tachycardias, but not to SCD, because the risk associated with atrial fibrillation is not present when antegrade conduction through the accessory pathway is not possible.
Figure 5. Wolff–Parkinson–White syndrome. A: Rapid conduction through an antero-septal accessory pathway during atrial fibrillation in WPW syndrome, mimicking the appearance of ventricular tachycardia. The irregular ventricular response pattern suggests atrial fibrillation. B: Delta waves indicated with arrows. ECG from different patient than in A. Paper speed 25 mm/s, gain 10 mm/mV.
The typical ECG pattern in WPW syndrome is characterized by a short PR interval (<120 ms) and a delta wave in the 12-lead ECG (). Depending on the location of the delta wave and the morphology of the QRS complex, an estimation of the location of the accessory pathway can also be made (Citation37,Citation38).
The accessory pathway has been estimated to be present in 2–4 per 1,000 persons in the general population (Citation39,Citation40). The incidence of symptomatic WPW syndrome is almost 4 in 100,000 persons per year with a bimodal incidence pattern; one peak during infancy and another peak during 20–30 years of age (Citation40). About one-half of the WPW ECG pattern carriers are asymptomatic, and approximately one-fifth present symptoms by middle age. The onset of symptoms after middle age is less common (Citation40), and delta waves may disappear with advancing age (Citation36). The incidence of SCD among previously asymptomatic patients with ECG patterns of WPW syndrome is small at ∼0.17% per year (Citation41). Among asymptomatic children and adolescents, the SCD incidence seems to be slightly higher at ∼0.3% per year (Citation42). Short pre-excited RR intervals during atrial fibrillation (<250 ms) are a sign of increased risk, as well as delta waves suggestive of septal accessory pathways (Citation43). The 12-lead ECG does not disclose the risk of malignant forms due to possible short refractory periods of accessory pathways. Therefore, all patients with antegrade conducting accessory pathway (a delta wave in ECG) should undergo further examinations, i.e. effective refractory period measurement. A stress test can also be used to estimate the refractory period non-invasively. If the delta wave disappears at heart rate 120–130 bpm or less, the likelihood is that the refractory period is long enough to avoid SCD risk.
Coronary artery anomalies
Anatomic variants of coronary arteries include origin from the pulmonary artery, anomalous origin from the wrong sinus of Valsalva in the aortic root, and bridging of a coronary artery by a myocardial band. Anatomically, myocardial bridges and anomalous origins of coronary arteries from the wrong sinus of Valsalva are not uncommon, with incidental findings on cardiac catheterizations of up to 5% for both. However, the majority of these are not clinically important. The incidence of SCD in patients with coronary artery anomalies is reported to be low (Citation44), although it is relatively common as a cause of SCD in athletes.
Origin of a coronary artery from the pulmonary artery is more consistently significant, and may present as myocardial infarction or angina pectoris in infancy or childhood.
The common ECG findings in these entities range from normal in a large number of cases, to non-specific ST segment and T wave changes. The physiology of coronary flow depends upon the duration of diastole. Since 95% of coronary flow occurs during diastole, and an increased heart rate impinges more on diastole than systole, exercise-related increases in heart rate may lead to transient ischemic arrhythmias.
The ECG in uncommon non-structural cardiac disorders
Long QT syndrome
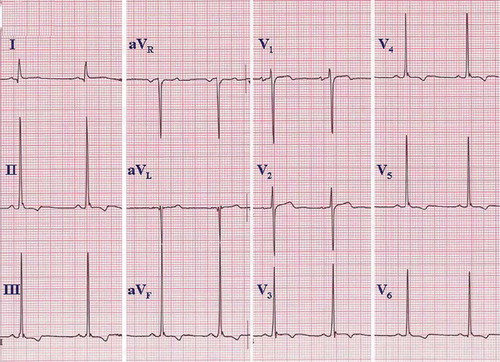
The long QT syndrome (LQTS) has attracted much attention in recent years (Citation45–47) because of advances in knowledge of its genetic patterns. The syndrome is recognized by a prolonged QT interval in the base-line 12-lead ECG, although some carriers of a genetic defect only have lengthened QT intervals induced by triggering events such as hypokalemia or other metabolic, autonomic, or exogenous stimuli. Leads II and V5 have been commonly used for measuring QT, but any lead where the QT seems to be visually longest should be used for measurement (Citation48). Rate correction should always be taken into account, and different equations for these purposes have been generated, among which Bazett's is the most widely used. However, QTc should be interpreted cautiously when the heart rate is less than 50 or greater than 90 bpm. Current limits for prolonged QTc have been set to 440 or 450 ms in men and 460 ms in women (Citation49), based upon population distribution of QTc intervals. Clinically significant QTc prolongation may be greater—in the range of 480–490 ms or more. Considerable QTc overlapping has been described between patients carrying LQTS mutations and the general population, as well as among affected family members who have variable expression of the same mutation (Citation50).
The prevalence of LQTS among neonates has been estimated to be 1:2534. In the same population the prevalence of QTc > 440 ms was 2% (Citation51). In an adult general population the prevalence of QTc > 440 ms in males and > 460 ms in females was 6%, and the relative risk of cardiac mortality in follow-up was 1.3 (Citation52), with excess risk relatively modest when QTc is less than 500 ms (Citation53). The age of manifestation of symptoms is usually in childhood and early adulthood. After 40 years, the incidence of new symptoms is low (Citation53,Citation54). Nevertheless, subjects with prolonged QTc should refrain from using medication/substances that further prolong QTc. In LQT1 and 2, exercise, swimming, and emotional arousal are triggering factors for arrhythmias, and in LQT3 vagal effects facilitate symptoms.
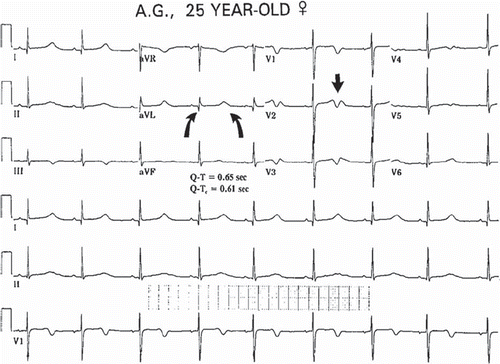
The 12-lead ECG is important both for recognition of prolonged QT intervals and to some degree for risk stratification. In patients with inherited long QT syndrome, serial ECGs over time may show variations in the degree of QTc prolongation (), and one should not rely on a single ECG if there is reason to suspect the syndrome. In addition, the upper limits of normal QT and QTc intervals defined above are based on analyses of population distributions at heart rates between 60 and 100 per minute; but for risk stratification, there is a general relationship between the degree of prolongation of the QTc and risk. While there is a gradient of increasing risk as the QTc increases from the upper limits of normal, the most significant magnitudes of risk of syncope, ventricular tachyarrhythmias, and SCD are associated with a QTc > 500 ms (Citation53). For risk stratification, one should use the longest QTc recognized on sequential ECGs, rather than relying on either the first ECG or an average value.
Figure 6. The ECG in long QT syndrome. QTc of 610 ms, with a T wave morphology suggesting type 2 long QT, is recorded from a 25-year-old female with a history of recurrent syncope and torsade de pointes. Paper speed 25 mm/s, gain 10 mm/mV.
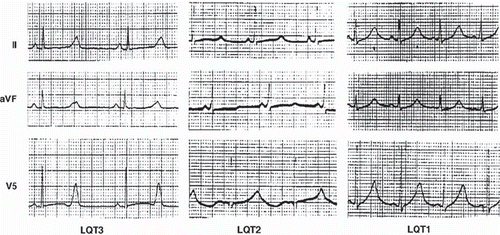
There is a general association between specific patterns of QT prolongation and specific ion current involvement. The type I pattern (LQT1) () is associated with KCNQ1 (slowly activating, delayed rectifier), type II (LQT2) is associated with HERG (rapidly activating delayed rectifier current), and type III (LQT3) is associated with abnormalities in the sodium channel. These are general relationships and do not hold for every case. A recently published clinical test, i.e. the ‘stand-up test’ by Viskin et al., can be helpful in differentiating between mutation carriers and non-carriers in LQTS (Citation55).
Figure 7. ECG manifestations of subtypes of long QT syndrome. Left: Type 3 long QT with the characteristic late onset of the T wave. Center: Type 2 long QT showing a biphasic T wave (i.e. U wave fused to the end of the T wave). Right panel: type 1 long QT with ‘giant’ broad T wave. Paper speed 25 mm/s, gain 10 mm/mV. Reprinted with permission from Moss AJ et al. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929–34. Wolters Kluwer Health. ©
Short QT syndrome
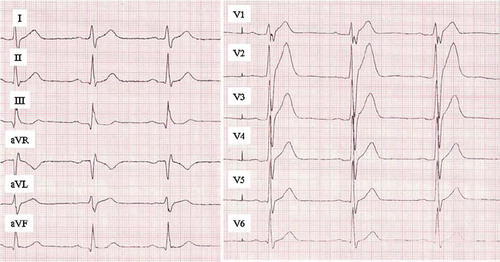
The short QT syndrome (SQTS) is a recently described SCD syndrome which is characterized by a very short (QTc 280–330 ms) QT interval and peaked T waves typically in the right precordial leads V1–3 (). The QT interval should be interpreted cautiously when the heart rate is less than 50 or greater than 90 bpm, and ultimately measurement of the QT should be made from ECG with heart rate as close to 60 bpm as possible. The prevalence of short QT (SQT) interval has been studied in the general population in two large studies (Citation56,Citation57). Anttonen et al. reported a prevalence of 0.4% of QTc under 340 ms, and none of the carriers of the ECG abnormality had died during a ∼40-year follow-up period; similar findings have been described by Kobza et al. Marked differences have been found between subjects with short QT from the general population and symptomatic SQTS patients. The SQTS patients had a clear-cut shorter beginning of repolarization phase measured from J point to T wave peak than subjects of general population SQT, representing a major morphological difference in the 12-lead ECG and evidently also in cardiac electrophysiological properties (Citation58). Recently published diagnostic scores for SQTS include the following ECG characteristics: QTc < 370 ms (1 point), QTc < 350 ms (2 points), QTc < 330 ms (3 points), and J point to T peak interval < 120 ms (1 point). Four points or more equals high probability for SQTS and two points or under a low probability (Citation59). Clinical symptoms, family history, and genotype are also taken into account in the diagnostic chart. Subjects with QTc under 340 ms without a personal or family history of syncope or life-threatening ventricular arrhythmias and with J point to T wave peak measurement over 120 ms should not be alarmed by the phenomenon.
Figure 8. ECG manifestation of short QT syndrome. A short QT interval (QTc 292 ms) with high, peaked T waves in right precordial leads. This patient had ventricular fibrillation at the age of 18 and was successfully resuscitated. Later on the patient received an ICD. Paper speed 50 mm/s, gain 10 mm/mV.
Brugada syndrome
Brugada syndrome is characterized by an atypical right bundle branch block pattern accompanied by ST segment elevation and T wave abnormalities in leads V1–3. The so-called ‘coved’ pattern of the ST-T complex (type 1) is generally accepted as the most specific for Brugada syndrome () (Citation60,Citation61). Types 2 and 3 () are questionable for the diagnosis in the absence of documented arrhythmias, unexplained syncope, and/or drug-induced conversion to a type 1 pattern. The fluctuation between normal ECG and different types of Brugada ECG patterns is common in patients with Brugada syndrome (Citation62), and sometimes ECG recordings from the upper intercostal spaces (second or third) for leads V1–3 might give further diagnostic information (Citation61). The current diagnostic criteria for a type 1 Brugada ECG pattern include a typical coved type ST segment elevation with over 2 mm elevation 40 ms after the J point in two of three right precordial leads at base-line or after drug challenge (Citation61).
Figure 9. ECG manifestations of Brugada syndrome. A: Both ECGs in this panel demonstrate typical type I (coved type) Brugada syndrome ECG pattern; RBBB-like pattern with distinct ST segment elevation and T wave inversion in the right (V1–3) precordial leads. Additional changes on the right side ECG, such as longer QRS duration and prominent R’ wave in aVR, have been associated to poor prognosis. Paper speed 25 mm/s, gain 10 mm/mV. Reprinted with permission from Junttila MJ et al. Differences in 12-lead electrocardiogram between symptomatic and asymptomatic Brugada syndrome patients. J Cardiovasc Electrophysiol. 2008;19:380–3. Wiley-Blackwell Publishing. © B: Brugada syndrome subtypes are shown. Left: Typical type I (coved type) Brugada syndrome ECG pattern. Middle: Type II (saddleback) Brugada ECG pattern. The T waves in the right precordial are upright, differentiating it from type I. Right: Type III Brugada ECG pattern. Similar changes to that of type II, but with minimal (< 0.2 mV) ST segment elevation. Paper speed 50 mm/s, gain 10 mm/mV. Reprinted with permission from Junttila MJ et al. Prevalence and prognosis of subjects with Brugada-type ECG pattern in a young and middle-aged Finnish population. Eur Heart J. 2004;25:874–8. Oxford University Press. ©
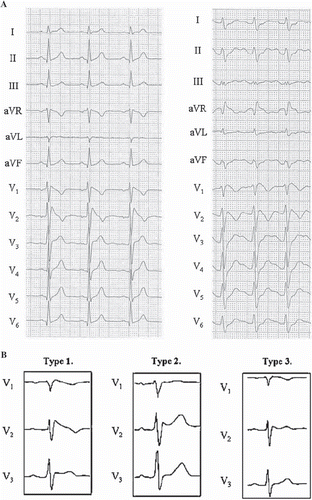
The prevalence of Brugada ECG is modulated by gender and ethnicity. Eighty percent of the cases occur in males, and the syndrome is more common in populations of Southeast-Asian origin than from the Western world. The prevalence of type 1 Brugada ECG among the general population in Japan is ∼0.4% and in Europe from 0% to 0.01% (Citation63–66). The annual incidence of life-threatening arrhythmias among Brugada syndrome patients with aborted cardiac arrest has been estimated to be 7.7%, among patients with syncope 1.9%, and among previously asymptomatic subjects 0.5% (Citation67). Type 2 and 3 Brugada ECGs are far more common also among Western populations, approximately 1 in 200, but they do not seem to carry an elevated risk for SCD (Citation65). Signs of increased risk among type 1 ECG carriers include prolonged QRS (> 100 ms) and markedly elevated J point in right precordial leads () (Citation68). A recent study classified the high risk of ventricular arrhythmias related to medication, anesthesia, or fever-induced type 1 Brugada ECG. In the case of induced Brugada ECG, the culprit medication should be discontinued immediately and fever treated swiftly, since ∼50% of these patients generate ventricular arrhythmias imminently (Citation69).
Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is characterized by exercise-induced syncope caused by polymorphic ventricular arrhythmia, and the first manifestation of the condition may occur during early childhood, adolescence, or young adulthood (Citation70). The disease does not demonstrate any ECG abnormalities at base-line, although during exercise the frequency of polymorphic ventricular extrasystoles, concordant with the level of exercise, and ultimately occurrence of polymorphic or bidirectional VT are typical for this condition (Citation71,Citation72). CPVT causes symptoms, including exercise-induced syncope, in up to 80% of the affected patients under 40 years of age (Citation71). The cumulative mortality may be as high as 40% by age 40. SCD, usually occurring during exercise, is commonly the first manifestation of the disease (Citation70–73).
Early repolarization
Early repolarization has been for long considered as a benign normal variant, but in 2008 Haissaguerre et al. presented cases with idiopathic ventricular fibrillation (VF) and a distinct early repolarization pattern in the infero-lateral leads (Citation74). Recently two large community-based studies confirmed the increased mortality related to the early repolarization pattern especially in the inferior leads (Citation52,Citation75). The typical ECG presentation is a deflection in the R wave down-slope (slurred) or an upward deflection after the deflection in the R down-slope (notched) () in two contiguous infero-lateral leads.
Figure 10. ECG manifestation of inferior early repolarization. Slurred early repolarization pattern in the inferior leads, i.e. deflection from the descending part of the R wave with a smooth transition to ST segment.
The prevalence of infero-lateral early repolarization pattern in the middle-aged population is estimated to be between 4% and 14%. The amplitude of inferior early repolarization over 0.2 mV conveys a 3-fold risk for cardiac mortality and a 4-fold risk of arrhythmic death, i.e. SCD, and the event is presented mostly in ages 40–50 years (Citation52,Citation75). There seems to be two different phenotypes associated with early repolarization ECG pattern. The pattern associated with primary electrical disorder described by Haissaguerre evidently conveys a high risk for SCD, but the prevalence for this disorder is low. The more prevalent phenotype, seen in general population studies, carries a somewhat moderate risk which seems to increase and manifest in SCD in association with other cardiovascular events such as acute coronary events. More studies are needed to elucidate the mechanisms, prevention, and treatment of subjects carrying the early repolarization ECG marker.
ECG manifestation of common acquired cardiac diseases
Coronary artery disease
Coronary artery disease (CAD) accounts for 75%–80% of all SCDs, based upon clinical and post-mortem studies (Citation24). However, as a chronic disease with a long clinical latency and a relatively low absolute risk in an individual within the general population, prediction of SCD due to CAD is clinically and epidemiologically challenging. In addition, SCD is the first clinical manifestation of the underlying disease in approximately 30%–50% of CAD-associated SCDs (Citation2), and one-third of all CAD SCD victims have myocardial infarction (MI) scars even without a clinical history of MI (Citation24).
Identification of ECG markers, specific for SCD risk due to CAD, is difficult. Among the large proportion of patients in whom SCD is the first manifestation of disease, a prior ECG is likely to be normal. Among patients with evidence of CAD, but without a history of MI or documented angina pectoris, the base-line ECG is often normal. At high heart rates, however, typical ST depression or ST-T wave abnormalities may occur in areas of transient ischemia.
The prevalence of miscellaneous ST segment or T wave abnormality has been estimated to be 0.4%–1.4% in Caucasian populations and 0.8%–6.4% in black populations, and the CAD event risk related to this finding is ∼2-fold (Citation76). In a study from the Framingham cohort, ST or T wave abnormality was associated with 8% annual SCD rate in men and 2.8% in women (Citation77). Additionally ECG findings that could be related to cardiovascular diseases such as intraventricular conduction delay, left bundle branch block, moderately prolonged QT, and higher resting heart rates have been associated with higher risk of cardiac mortality and SCD (Citation77–81). Also persons who present LV hypertrophy (LVH) according to the Sokolow–Lyon (V1 S + V6 or V5 R > 35 mm) or Cornell (V3 S + aVL R > 28 for men and 20 mm for women) voltage criteria have a higher risk for SCD (Citation82).
Myocarditis
Myocarditis has been generally thought of as a common cause of SCD among young people, with an estimated 1 per 1,000 deaths prevalence in the age-group under 45 (Citation83). The ECG findings in acute myocarditis are dynamic and in many ways similar to acute MI. The first presentation is a subtle ST elevation as in acute MI, but in myocarditis the changes can be seen globally rather than in a specific coronary vascular area. Additionally PR interval depression in the standard leads can be present. These presentations are followed by T wave inversions within hours or days and full-blown T wave inversions within weeks after the acute phase.
Conclusions
The standard 12-lead ECG can give important information about the risk of SCD in asymptomatic subjects. The early recognition of these ECG abnormalities resides with generalists, hospitalists, and various non-cardiovascular specialists. Correct interpretation of the ECG findings may lead to early recognition of risk and subsequent prevention of SCD.
Acknowledgements
This work was supported by fondation Leducq, Paris, France (MJJ and RJM), Sigrid Juselius Foundation, Helsinki, Finland (HVH), and the Florida Heart Research Foundation, Miami, Florida (RJM). The corresponding author (MJJ) had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104:2158–63.
- Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473–82.
- Myerburg RJ, Vetter VL. Electrocardiograms should be included in preparticipation screening of athletes. Circulation. 2007;116:2616–26; discussion 2626.
- Brock R. Functional obstruction of the left ventricle; acquired aortic subvalvar stenosis. Guys Hosp Rep. 1957;106:221–38.
- Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8.
- Hauer RN, Aliot E, Block M, Capucci A, Luderitz B, Santini M, . Indications for implantable cardioverter defibrillator (ICD) therapy. Study Group on Guidelines on ICDs of the Working Group on Arrhythmias and the Working Group on Cardiac Pacing of the European Society of Cardiology. Eur Heart J. 2001;22:1074–81.
- Codd MB, Sugrue DD, Gersh BJ, Melton LJ 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation. 1989;80:564–72.
- Hada Y, Sakamoto T, Amano K, Yamaguchi T, Takenaka K, Takahashi H, . Prevalence of hypertrophic cardiomyopathy in a population of adult Japanese workers as detected by echocardiographic screening. Am J Cardiol. 1987;59:183–4.
- Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, . The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348: 1647–55.
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–9.
- Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, . The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348:1639–46.
- Ridjab D, Koch M, Zabel M, Schultheiss HP, Morguet AJ. Cardiac arrest and ventricular tachycardia in Japanese-type apical hypertrophic cardiomyopathy. Cardiology. 2007;107: 81–6.
- Eriksson MJ, Sonnenberg B, Woo A, Rakowski P, Parker TG, Wigle ED, . Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;39: 638–45.
- Fananapazir L, Tracy CM, Leon MB, Winkler JB, Cannon RO 3rd, Bonow RO, . Electrophysiologic abnormalities in patients with hypertrophic cardiomyopathy. A consecutive analysis in 155 patients. Circulation. 1989;80:1259–68.
- Lemery R, Kleinebenne A, Nihoyannopoulos P, Aber V, Alfonso F, McKenna WJ. Q waves in hypertrophic cardiomyopathy in relation to the distribution and severity of right and left ventricular hypertrophy. J Am Coll Cardiol. 1990;16:368–74.
- Maron BJ, Wolfson JK, Ciro E, Spirito P. Relation of electrocardiographic abnormalities and patterns of left ventricular hypertrophy identified by 2-dimensional echocardiography in patients with hypertrophic cardiomyopathy. Am J Cardiol. 1983;51:189–94.
- Savage DD, Seides SF, Clark CE, Henry WL, Maron BJ, Robinson FC, . Electrocardiographic findings in patients with obstructive and nonobstructive hypertrophic cardiomyopathy. Circulation. 1978;58(3 Pt 1):402–8.
- Krikler DM, Davies MJ, Rowland E, Goodwin JF, Evans RC, Shaw DB. Sudden death in hypertrophic cardiomyopathy: associated accessory atrioventricular pathways. Br Heart J. 1980;43:245–51.
- Corrado D, Pelliccia A, Heidbuchel H, Sharma S, Link M, Basso C, . Recommendations for interpretation of 12-lead electrocardiogram in the athlete. Eur Heart J. 2010;31:243–59.
- Wolpert C, Veltmann C, Schimpf R, Antzelevitch C, Gussak I, Borggrefe M. Is a narrow and tall QRS complex an ECG marker for sudden death? Heart Rhythm. 2008;5:1339–45.
- Cleland JG, Chattopadhyay S, Khand A, Houghton T, Kaye GC. Prevalence and incidence of arrhythmias and sudden death in heart failure. Heart Fail Rev. 2002;7:229–42.
- van Tintelen JP, Hofstra RM, Katerberg H, Rossenbacker T, Wiesfeld AC, du Marchie Sarvaas GJ, . Working Group on Inherited Cardiac Disorders, line 27/50, Interuniversity Cardiology Institute of The Netherlands. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J. 2007;154:1130–9.
- Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010;375:752–62.
- Adabag AS, Peterson G, Apple FS, Titus J, King R, Luepker RV. Etiology of sudden death in the community: results of anatomical, metabolic, and genetic evaluation. Am Heart J. 2010;159:33–9.
- Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, . Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65: 384–98.
- McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, . Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–8.
- Cox MG, van der Smagt JJ, Wilde AA, Wiesfeld AC, Atsma DE, Nelen MR, . New ECG criteria in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Arrhythm Electrophysiol. 2009;2:524–30.
- Norman MW, McKenna WJ. Arrhythmogenic right ventricular cardiomyopathy: perspectives on disease. Z Kardiol. 1999;88:550–4.
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–33.
- Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart. 2000;83:588–95.
- Marcus FI. Prevalence of T-wave inversion beyond V1 in young normal individuals and usefulness for the diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Am J Cardiol. 2005;95:1070–1.
- Steffel J, Kobza R, Namdar M, Wolber T, Brunckhorst C, Luscher TF, . Electrophysiological findings in patients with isolated left ventricular non-compaction. Europace. 2009;11:1193–200.
- Murphy RT, Thaman R, Blanes JG, Ward D, Sevdalis E, Papra E, . Natural history and familial characteristics of isolated left ventricular non-compaction. Eur Heart J. 2005;26:187–92.
- Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36:493–500.
- Hookana E, Junttila MJ, Sarkioja T, Sormunen R, Niemela M, Raatikainen MJ, . Cardiac arrest and left ventricular fibrosis in a Finnish family with the lamin A/C mutation. J Cardiovasc Electrophysiol. 2008;19:743–7.
- Klein GJ, Yee R, Sharma AD. Longitudinal assessment of asymptomatic patients with the Wolff-Parkinson-White Pattern. N Engl J Med. 1989;320:1229–33.
- Milstein S, Sharma AD, Guiraudon GM, Klein GJ. An algorithm for the electrocardiographic localization of accessory pathways in the Wolff-Parkinson-White syndrome. Pacing Clin Electrophysiol. 1987;10(3 Pt 1):555–63.
- Arruda MS, McClelland JH, Wang X, Beckman KJ, Widman LE, Gonzalez MD, . Development and validation of an ECG algorithm for identifying accessory pathway ablation site in Wolff-Parkinson-White syndrome. J Cardiovasc Electrophysiol. 1998;9:2–12.
- Averill KH, Fosmoe RJ, Lamb LE. Electrocardiographic findings in 67,375 asymptomatic subjects. IV. Wolff-Parkinson-White syndrome. Am J Cardiol. 1960;6:108–29.
- Munger TM, Packer DL, Hammill SC, Feldman BJ, Bailey KR, Ballard DJ, . A population study of the natural history of Wolff-Parkinson-White syndrome in Olmsted County, Minnesota, 1953–1989. Circulation. 1993;87:866–73.
- Santinelli V, Radinovic A, Manguso F, Vicedomini G, Ciconte G, Gulletta S, . Asymptomatic ventricular preexcitation: a long-term prospective follow-up study of 293 adult patients. Circ Arrhythm Electrophysiol. 2009;2:102–7.
- Santinelli V, Radinovic A, Manguso F, Vicedomini G, Gulletta S, Paglino G, . The natural history of asymptomatic ventricular pre-excitation a long-term prospective follow-up study of 184 asymptomatic children. J Am Coll Cardiol. 2009;53:275–80.
- Haghjoo M, Kharazi A, Fazelifar AF, Alizadeh A, Emkanjoo Z, Sadr-Ameli MA. Electrocardiographic and electrophysiologic characteristics of anteroseptal, midseptal, and posteroseptal accessory pathways. Heart Rhythm. 2007;4: 1411–9.
- Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593–601.
- Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59–68.
- Romano C, Gemme G, Pongiglione R. Rare cardiac arrythmias of the pediatric age. II. Syncopal attacks due to paroxysmal ventricular fibrillation. (Presentation of 1st Case in Italian Pediatric Literature). Clin Pediatr (Bologna). 1963;45:656–83.
- Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103–6.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88:782–4.
- Rautaharju PM, Surawicz B, Gettes LS, Bailey JJ, Childers R, Deal BJ, . AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. J Am Coll Cardiol. 2009;53:982–91.
- Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, . Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960–5.
- Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, . Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7.
- Tikkanen JT, Anttonen O, Junttila MJ, Aro AL, Kerola T, Rissanen HA, . Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med. 2009;361:2529–37.
- Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, . Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–74.
- Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358:169–76.
- Viskin S, Postema PG, Bhuiyan ZA, Rosso R, Kalman JM, Vohra JK, . The response of the QT interval to the brief tachycardia provoked by standing: a bedside test for diagnosing long QT syndrome. J Am Coll Cardiol. 2010;55:1955–61.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation. 2007;116:714–20.
- Kobza R, Roos M, Niggli B, Abacherli R, Lupi GA, Frey F, . Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm. 2009;6:652–7.
- Anttonen O, Junttila MJ, Maury P, Schimpf R, Wolpert C, Borggrefe M, . Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm. 2009;6:267–71.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol. 2011;57:802–12.
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–6.
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, . Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–70.
- Veltmann C, Schimpf R, Echternach C, Eckardt L, Kuschyk J, Streitner F, . A prospective study on spontaneous fluctuations between diagnostic and non-diagnostic ECGs in Brugada syndrome: implications for correct phenotyping and risk stratification. Eur Heart J. 2006;27:2544–52.
- Miyasaka Y, Tsuji H, Yamada K, Tokunaga S, Saito D, Imuro Y, . Prevalence and mortality of the Brugada-type electrocardiogram in one city in Japan. J Am Coll Cardiol. 2001;38:771–4.
- Matsuo K, Akahoshi M, Nakashima E, Suyama A, Seto S, Hayano M, . The prevalence, incidence and prognostic value of the Brugada-type electrocardiogram: a population-based study of four decades. J Am Coll Cardiol. 2001;38: 765–70.
- Junttila MJ, Raatikainen MJ, Karjalainen J, Kauma H, Kesaniemi YA, Huikuri HV. Prevalence and prognosis of subjects with Brugada-type ECG pattern in a young and middle-aged Finnish population. Eur Heart J. 2004;25: 874–8.
- Hermida JS, Lemoine JL, Aoun FB, Jarry G, Rey JL, Quiret JC. Prevalence of the brugada syndrome in an apparently healthy population. Am J Cardiol. 2000;86:91–4.
- Probst V, Veltmann C, Eckardt L, Meregalli PG, Gaita F, Tan HL, . Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation. 2010;121: 635–43.
- Junttila MJ, Brugada P, Hong K, Lizotte E, DE Zutter M, Sarkozy A, . Differences in 12-lead electrocardiogram between symptomatic and asymptomatic Brugada syndrome patients. J Cardiovasc Electrophysiol. 2008;19: 380–3.
- Junttila MJ, Gonzalez M, Lizotte E, Benito B, Vernooy K, Sarkozy A, . Induced Brugada-type electrocardiogram, a sign for imminent malignant arrhythmias. Circulation. 2008;117:1890–3.
- Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–9.
- Mohamed U, Napolitano C, Priori SG. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2007;18:791–7.
- Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, . Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89:66–70.
- Postma AV, Denjoy I, Kamblock J, Alders M, Lupoglazoff JM, Vaksmann G, . Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet. 2005;42: 863–70.
- Haissaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L, . Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016–23.
- Sinner MF, Reinhard W, Muller M, Beckmann BM, Martens E, Perz S, . Association of early repolarization pattern on ECG with risk of cardiac and all-cause mortality: a population-based prospective cohort study (MONICA/KORA). PLoS Med. 2010;7:e1000314.
- Machado DB, Crow RS, Boland LL, Hannan PJ, Taylor HA Jr, Folsom AR. Electrocardiographic findings and incident coronary heart disease among participants in the Atherosclerosis Risk in Communities (ARIC) study. Am J Cardiol. 2006;97:1176–81.
- Kreger BE, Cupples LA, Kannel WB. The electrocardiogram in prediction of sudden death: Framingham Study experience. Am Heart J. 1987;113(2 Pt 1):377–82.
- Eriksson P, Wilhelmsen L, Rosengren A. Bundle-branch block in middle-aged men: risk of complications and death over 28 years. The Primary Prevention Study in Goteborg, Sweden. Eur Heart J. 2005;26:2300–6.
- Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QTc prolongation measured by standard 12-lead electrocardiography is an independent risk factor for sudden death due to cardiac arrest. Circulation. 1991;83:1888–94.
- Miller WL, Ballman KV, Hodge DO, Rodeheffer RJ, Hammill SC. Risk factor implications of incidentally discovered uncomplicated bundle branch block. Mayo Clin Proc. 2005;80:1585–90.
- Straus SM, Kors JA, De Bruin ML, van der Hooft CS, Hofman A, Heeringa J, . Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J Am Coll Cardiol. 2006;47:362–7.
- Sullivan JM, Vander Zwaag RV, el-Zeky F, Ramanathan KB, Mirvis DM. Left ventricular hypertrophy: effect on survival. J Am Coll Cardiol. 1993;22:508–13.
- Kyto V, Saraste A, Voipio-Pulkki LM, Saukko P. Incidence of fatal myocarditis: a population-based study in Finland. Am J Epidemiol. 2007;165:570–4.