Abstract
Systemic autoinflammatory diseases are a group of inherited disorders of the innate immune system characterized by seemingly unprovoked inflammation recurring at variable intervals and involving skin, serosal membranes, joints, and gastrointestinal apparatus, with reactive amyloidosis as a possible severe long-term complication. Recent advances in genetics and molecular biology have improved our understanding of the pathogenesis of these diseases, including familial Mediterranean fever, mevalonate kinase deficiency syndrome, tumor necrosis factor receptor-associated periodic syndrome, cryopyrin-associated periodic syndromes, and hereditary pyogenic and granulomatous disorders: the vast majority of these conditions are related to the activation of the interleukin-1 pathway, which results in (or from?) a common unifying pathogenetic mechanism. Their diagnostic identification derives from the combination of clinical data, evaluation of acute phase reactants, clinical efficacy in response to specific drugs, and recognition of specific mutations in the relevant genes, although genetic tests may be unconstructive in some cases. This review will discuss clinical and laboratory clues useful for a diagnostic approach to systemic autoinflammatory diseases.
| Abbreviations | ||
| BS | = | Blau syndrome |
| CAPS | = | cryopyrin-associated periodic syndromes |
| CINCAs | = | chronic infantile neurological cutaneous and articular syndrome |
| DIRA | = | deficiency of interleukin-1 receptor antagonist |
| FCAS | = | familial cold urticaria syndrome |
| FMF | = | familial Mediterranean fever |
| HIDS | = | hyper-gammaglobulinemia D syndrome |
| IL | = | interleukin |
| MK | = | mevalonate kinase |
| MKD | = | mevalonate kinase deficiency syndrome |
| MS | = | Majeed syndrome |
| MWS | = | Muckle–Wells syndrome |
| NLRP12-ad | = | NLRP12-associated autoinflammatory disorder |
| PAPAs | = | pyogenic sterile arthritis, pyoderma gangrenosum, and acne syndrome |
| PFAPA | = | periodic fever, adenitis, pharyngitis, and aphthosis |
| SAA | = | serum amyloid-A |
| SAID | = | systemic autoinflammatory disease |
| TNFRSF1A | = | 55 kDa receptor of TNF-α |
| TNF-α | = | tumor necrosis factor-α |
| TRAPS | = | tumor necrosis factor receptor-associated periodic syndrome |
Key messages
The genes associated with systemic autoinflammatory diseases (SAIDs) have been identified in recent years and, with the exception of hyper-gammaglobulinemia D syndrome, all encode for proteins involved in the inflammatory response and in the processes of cellular apoptosis.
The activation or deregulation of cytokine production is directly implicated in the pathogenesis of SAIDs, and the central role of interleukin-1 has been confirmed in the last decade thanks to advances in techniques used to identify human disease-causing genes, innovations in genomic analysis, and progress in effective targeted biologic therapy.
The diagnostic identification of SAIDs derives from the combination of clinical data, evaluation of acute-phase reactants, clinical efficacy in response to specific drugs, and genetic testing.
Introduction
Systemic autoinflammatory diseases (SAIDs) are a heterogeneous group of genetically determined illnesses that involve the innate immune system and are by definition characterized by the apparently inexplicable recurrence of acute inflammatory episodes affecting the skin, serosal membranes, joints, gastroenteric tube, central nervous system, etc. The term ‘autoinflammatory’ describes the spontaneous appearance of inflammation in the absence of auto-reactive T lymphocytes or specific auto-antibodies, which in point of fact play a pathogenetic role in the autoimmune diseases (Citation1). All SAIDs are caused by lack of regulation in the production of proinflammatory cytokines, such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α, which leads to a pathological delay in inflammatory response deactivation (Citation2). SAIDs can be categorized as hereditary monogenic disorders—the subject of this description—and multifactorial polygenic disorders such as Behçet's disease, adult Still's disease, systemic-onset juvenile idiopathic arthritis, and PFAPA (periodic fever, adenitis, pharyngitis, and aphthosis) syndrome (Citation3).
Classification of the systemic autoinflammatory diseases
The hereditary monogenic SAIDs, characterized by a common clinical phenotype marked by recurrent episodes of fever and varying signs of systemic inflammation, include familial Mediterranean fever (FMF), mevalonate kinase deficiency syndrome (MKD), also known as hyper-gammaglobulinemia D syndrome (HIDS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), the family of cryopyrin-associated periodic syndromes (CAPS), which in turn include familial cold urticaria syndrome (FCAS), Muckle–Wells syndrome (MWS), and CINCA (chronic infantile neurological cutaneous and articular) syndrome (CINCAs), NLRP12-associated autoinflammatory disorder (NLRP12-ad), hereditary pyogenic disorders including PAPA (pyogenic sterile arthritis, pyoderma gangrenosum, and acne) syndrome (PAPAs), Majeed syndrome (MS), and IL-1 receptor antagonist deficiency (DIRA), and finally a granulomatous disorder with familiar presentation, called Blau syndrome (BS) (). Some of these (namely FMF, HIDS, MS, and DIRA) are transmitted by autosomal recessive inheritance, while the others (TRAPS, FCAS, MWS, CINCAs, NLRP12-ad, PAPAs, and BS) are autosomal-dominant. The genes associated with these diseases have been identified in recent years and, with the exception of HIDS, all encode for proteins involved in the inflammasome, a multi-protein complex regulating the innate immunity and activating the processing and secretion of proinflammatory cytokines (Citation4).
Table I. Descriptive summary of the systemic autoinflammatory diseases.
Prominent clinical details of the systemic autoinflammatory diseases
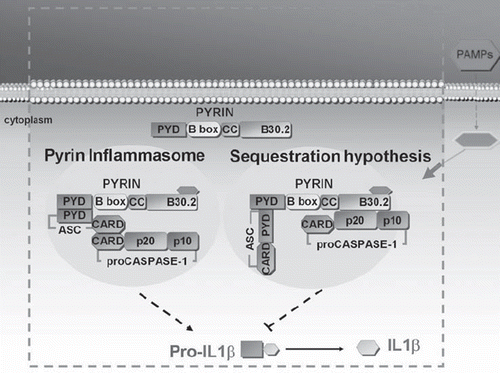
FMF is the most common autoinflammatory disease in the world, with a particularly high incidence throughout the Mediterranean basin, especially among Sephardic (non-Ashkenazi) Jews, Turks, Armenians, and Arabs. The gene responsible for FMF, denominated MEFV (from MEditerranean FeVer), identified in 1997 on chromosome 16, is made up of 10 exons and encodes for a 781-amino acid protein called pyrin (or ‘marenostrin’, from the ancient Latin name for the Mediterranean sea, Mare nostrum) (Citation5,Citation6). Pyrin () is believed to orchestrate IL-1β processing and release in various types of cells, including neutrophil and eosinophil granulocytes, monocytes, dendritic cells, and fibroblasts, through a down-regulation of the inflammasome function (Citation7–9). Diagnosis of FMF is based on the use of Tel Hashomer criteria (recurrent episodes of fever associated with serositis, presence of amyloidosis, and favorable response to colchicine) (Citation10). Although the disease typically appears in childhood, diagnosis after age 20 is not rare, even in the presence of a clinical phenotype virtually identical to that noted in childhood cases (Citation11), i.e. brief recurrent episodes of fever, serositis manifesting with chest and/or abdominal pain, arthralgia and/or arthritis of the hip, knee, or ankle, and erysipelas-like erythema, frequently on the lower limbs. Fever episodes begin suddenly, last 1–4 days, tend to resolve spontaneously, and alternate with periods of perfect health. The most worrisome complication of undiagnosed/untreated FMF is AA-amyloidosis, which can involve all of the organs, but frequently impacts the kidneys, gut, spleen, and liver (Citation12). Possible muscular involvement has also been described, ranging from minor forms of myalgia to protracted febrile myalgias, even with no increase in muscular enzymes, except in very rare cases in which such an increase reflects the depositing of amyloid fibrils in the skeletal musculature or iatrogenic damage linked to colchicine treatment (Citation13,Citation14). Most patients with amyloidosis and renal involvement develop a nephrotic syndrome and may face chronic renal insufficiency (Citation15).
Figure 1. Proposed mechanisms of pyrin action in FMF. Pyrin inflammasome hypothesis: Pyrin can assemble the ‘inflammasome’ complex through pyrin–pyrin domain interaction with ASC, resulting in activation of caspase-1. Sequestration hypothesis: Pyrin has an inhibitory effect on caspase-1-mediated activation of IL-1β by competitive binding of ASC, as well as caspase-1. (PAMPs = pathogen-associated molecular patterns; PYD = pyrin domain; B-box = B-box zinc finger; CC = coiled-coiled region; ASC = apoptosis-associated speck-like protein containing a CARD; CARD = caspase recruitment domain; IL = interleukin).
MKD, better known as HIDS, or ‘Dutch fever’ as it was first described in 1984 in the Netherlands (Citation16), is caused by mutations in the MVK gene located on chromosome 12 that encodes for mevalonate kinase (MK)—the second enzyme in the metabolic pathway of cholesterol—which lead to a reduction of enzymatic activity, but not its complete abolition (Citation17–19): this enzyme normally catalyzes the phosphorylation of mevalonic acid into 5-phosphomevalonate and supplies numerous bioactive molecules, such as sterols with isoprenilic groups (Citation20). However, the molecular mechanism at the root of the inflammatory phenotype has not yet been identified, and the relationship between MK deficiency and inflammation remains enigmatic. In 75% of cases, the disease initially manifests itself within the first year of life and in all cases within the first 5 years; it usually persists throughout the patient's entire life, but attacks tend to diminish over time in intensity and frequency (Citation21). From the clinical point of view, attacks start with high fever, often accompanied by chills, lasting 4–7 days and generally recurring every 3–4 weeks; these fever episodes may be preceded by headache, weakness, or irritability and are accompanied by arthralgia, lymphadenopathy, vomiting, diarrhea, abdominal pain, splenomegaly, and mouth ulcers. Cutaneous involvement is very frequent, in the form of various types of rashes, from macular and papular to urticarial and nodular (Citation22).
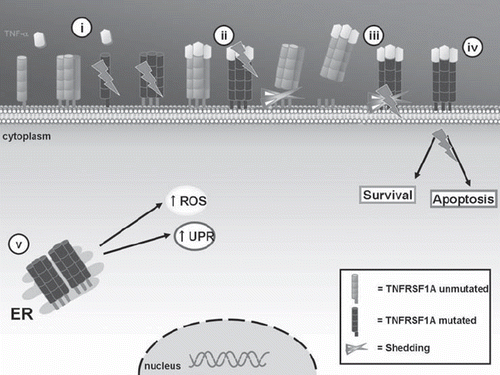
TRAPS, first described in 1982 in a family of both Irish and Scottish ancestry and initially called ‘Hibernian fever’ in reference to the ancient Latin name for Ireland, ‘Hibernia’, is the most common autosomal-dominant autoinflammatory pathology (Citation23). It is caused by mutations in the TNFRSF1A gene, which encodes for the 55 kDa receptor of TNF-α (TNFRSF1A or TNFR p55) (), a member of the family of TNF receptors involved in the processes of T cell activation and B cell homeostasis (Citation24–26). The average age at which the disease makes its appearance is around 3, but it may sometimes arise in late adolescence. Attacks last 20 days on average and recur at varying intervals, generally longer than those seen in other SAIDs (Citation27), initiating with muscle cramps or myalgia that migrates in a centrifugal pattern, followed by fever associated with cutaneous, joint, abdominal, and ocular manifestations (Citation28). The most common cutaneous manifestation is centrifugal migratory erythematous rash, which overlies the area affected by myalgia; this type of lesion is painful and warm to the touch. Arthralgias are common, and in rare cases arthritis may be present, mainly affecting single joints. Abdominal pain is another typical sign of TRAPS and is an indication of inflammation of the peritoneal cavity and abdominal wall muscles. Finally, eye involvement is also quite characteristic and can manifest itself in the form of conjunctivitis, periorbital edema, and uveitis (Citation29).
Figure 2. Mechanisms of TNFRSF1A action in the pathogenesis of TRAPS. The TNF receptor (TNFRSF1A) participates in the regulation of inflammatory responses through TNF-α binding. TRAPS-associated TNFRSF1A mutations may present alterations in: i) receptor trafficking to the cell surface; ii) TNF-α binding; iii) activation-induced shedding of the receptor; iv) TNF-α induced activation of transcription factors or apoptosis; and v) accumulation of aberrant receptors in the ER with increased formation of ROS and activation in UPR. (TNF = tumor necrosis factor; ER = endoplasmic reticulum; ROS = reactive oxygen species; UPR = unfolded protein response).
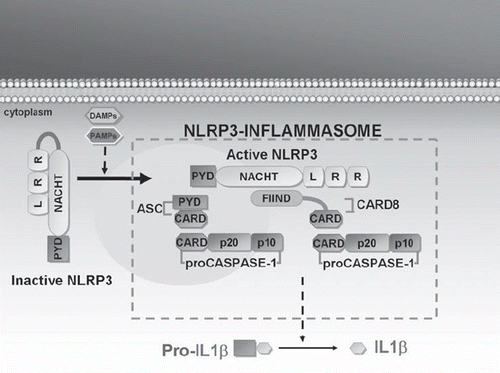
CAPS include three phenotypical expressions of the same autosomal-dominant pathology: FCAS, MWS, and CINCAs, respectively, from the least to most serious. Between 1999 and 2001 these conditions were linked with mutations of the NLRP3 gene located on chromosome 1 (Citation30), which encodes for a protein called ‘cryopyrin’, one of the intracellular sensors that participate in host defense mechanisms and induction of the inflammatory response () (Citation31). In particular, FCAS is characterized by fever episodes associated with cutaneous rashes and arthralgias, most often triggered by exposure to cold (Citation32). MWS is characterized by an analogous group of symptoms associated with migrating urticaria-like lesions, ocular abnormalities, progressive neuro-sensorial deafness, and risk of amyloidosis (Citation33). The clinical triad of skin rash, chronic meningeal involvement, and arthropathy, usually involving the knees, distinguishes the CINCAs. A non-itching, urticaria-like rash is persistently present in 75% of cases from birth, and skin biopsy usually reveals a moderate neutrophil infiltration of derma. Central nervous system involvement manifests itself in the first years of life with seizures, ventriculomegaly, headache, and in some cases mental retardation. Eye involvement may lead to progressively worsening reduction of visual acuity and is associated with chronic anterior uveitis in half of cases (Citation34). The major musculoskeletal deformities are caused by a premature and aberrant ossification of the knee-caps, with serious anomalies involving the epiphysis and hypertrophy of growth plates; shortening of the hands and feet and nail clubbing can often be observed as well (Citation35). NLRP12-ad is a rare genetic disease with close similarity with FCAS, characterized by recurrent bouts of fever lasting several days, joint symptoms, and skin rash triggered by exposure to cold temperatures, requiring genetic differentiation from CAPS (Citation36).
Figure 3. Mechanism of NLRP3 action in the pathogenesis of CAPS. Cryopyrin (or NLRP3) plays a key role in the inflammatory response by regulating IL-1β secretion. At rest, NLRP3 is inactive. Upon NLRP3 activation by means of agonist recognition, as with PAMPs (such as muramyl dipeptides, lipopolysaccharide, peptidoglycan, bacterial or viral RNA) and DAMPs (such as uric acid, ATP, or ultraviolet B radiations), NLRP3 interacts with several proteins (such as ASC and CARD8) to form the NLRP3–inflammasome: this multimolecular complex leads to caspase-1 activation, which in turn catalyzes the processing of IL precursor to IL-1β. (IL = interleukin; PAMPs = pathogen-associated molecular patterns; DAMPs = damage-associated molecular pattern molecules; PYD = pyrin domain; NACHT = nucleotide-binding oligomerization domain; LRR = leucine-rich repeat; ASC = apoptosis-associated speck-like protein containing a CARD; CARD = caspase recruitment domain; CARD8 = caspase recruitment domain family, member 8; FIIND = function to find domain).
Hereditary pyogenic disorders pertain to the SAID group and are characterized by the recurrence of multiple inflammatory manifestations, mainly linked to the multi-site formation of a granulocytic exudate. They include PAPAs, characterized by erosive pyogenic arthritis, pyoderma gangrenosum, cutaneous abscesses, and nodule-cystic acne (Citation37); MS, characterized by recurrent precociously arising multifocal osteomyelitis, dyserythropoietic anemia, and neutrophilic dermatosis (Citation38); and DIRA, recently described as a neonatal autoinflammatory disorder characterized by multifocal osteomyelitis and pustular rash, caused by unopposed IL-1 activity and uncontrolled inflammatory responses involving bone and skin (Citation39).
Among the granulomatous disorders, BS can be considered an autoinflammatory syndrome of the joints, eyes, and skin, manifesting through the association of non-erosive granulomatous polyarthritis, severe granulomatous uveitis, and chronic panniculitis with juvenile onset (Citation40).
Genetic diagnosis of the systemic autoinflammatory diseases
The clear Mendelian inheritance of SAIDs combined with improved genetic mapping methods has allowed for the identification of the underlying genes since 1997. To date, more than 180 mutations of the MEFV gene have been identified, the most frequent of which affect exons 2, 3, 5, and 10; mutations described in exons 1, 7, and 9 are rarer. Although there have been reports of symptomatic heterozygous patients, genetic tests are considered positive when they reveal two mutations, one for each allele (Citation41). Over the years, various studies have been carried out with the goal of identifying a genotype–phenotype correlation, and the results obtained indicate that the most frequent mutations in homozygous subjects, such as M694V, M680I, and M694I, are associated with a more marked clinical severity than forms linked to compound heterozygosity (Citation42).
Most of the MVK gene mutations identified to date are of the missense type, and most of the patients are heterozygous for two different mutations (Citation43): the prevalent mutation is V377I, observed in approximately 80% of patients and associated with a modest reduction in the stability and catalytic activity of the enzyme MK (Citation44).
More than 60 mutations of the TNFRSF1A gene have been described, about 60% of which are associated with TRAPS, consisting mainly of point mutations that lead to the replacement of a single amino acid in the external section of the mature protein (Citation45). For the NLRP3 gene, approximately 100 different mutations have been described so far, nearly all present on exon 3, which encodes the NACHT domain of cryopyrin; two exceptions are mutations located on exons 4 and 5, which encode for LRR (leucine-rich repeat) domain: a common model proposes that CAPS mutations result in a gain-of-function phenotype leading to a constitutive hyperproduction of IL-1 (Citation46). NLRP12-ad (NLRP12 stands for NACHT, LRR, and PYD domains containing protein 12, also named ‘monarch-1’) is related to NLRP12 mutations, which have been identified very recently, underlining the importance of screening for the NLRP12 gene in patients presenting with unexplained recurrent fevers and symptoms similar to CAPS (Citation47).
PAPAs is transmitted as an autosomal-dominant trait and is linked to the PSTPIP1 gene located on chromosome 15; MS is linked to the LPIN2 gene and has been described only in Jordan; and DIRA is due to mutations in the IL1RN gene located on chromosome 2q14, which stop the physiological secretion of the IL-1 receptor antagonist (Citation48).
BS is linked to the CARD15/NOD2 gene: the mutations reported involve the NACHT portion and are associated with activation of the NF-κB protein complex (Citation49).
All information regarding mutations described in patients with SAID can be found at the web address: http://fmf.igh.cnrs.fr/infevers (Citation50).
The biohumoral picture of the systemic autoinflammatory diseases
In SAIDs, laboratory tests commonly reveal increases in indicators of inflammation during each acute inflammatory episode; in particular, marked increases are observed for erythro-sedimentation rate and C-reactive protein, as well as fibrinogen and haptoglobin, which characteristically return to normal levels during non-acute intervals. These increases can also be associated with abnormalities in blood cell count, such as neutrophil leukocytosis, thrombocytosis, and hypo- or normochromic anemia, which is typical of chronic inflammatory diseases. Also fairly frequent are findings of polyclonal hyper-gammaglobulinemia due to stimulation of immunoglobulin synthesis by numerous proinflammatory cytokines, such as IL-6 (Citation51).
Characteristic but not specific to MKD are reports of elevated serum IgD and IgA levels, in agreement with what has been observed in other periodic fever disorders, as well as in numerous inflammatory disorders, immunodeficiencies, lymphoproliferative diseases, and even in completely healthy subjects (Citation52). The exact role of IgD in the pathogenesis of MKD is still unclear, but its serum level can substantiate the diagnosis: serum IgD tests carried out during phases of clinical good health with results above 100 IU/mL contribute to the diagnosis but must be repeated in at least two consecutive tests at 1-month intervals (Citation53). More precise for diagnostic purposes is the dosage of urinary mevalonic acid during the acute inflammatory attack, although the gold standard remains the quantification of MK enzyme activity (Citation54).
Acute-phase reactants are often elevated in patients with TRAPS even between fever attacks, although at a lower level than during attacks, but the most determinant laboratory element of the quiescent phase is the finding of low serum levels of the soluble TNF receptor (<1 ng/mL), as the illness is linked to a defective release of the receptor from cell membranes (Citation55).
In CAPS, and especially in CINCAs, a marked rise in the inflammatory parameters and a finding of nearly constant neutrophil leukocytosis are highly indicative (Citation56).
The diagnostic contribution of serum amyloid-A and protein S100A12
Serum amyloid-A (SAA) is an acute-phase protein, synthesized and secreted by the liver upon stimulation by proinflammatory cytokines as IL-1, IL-6, and TNF-α (Citation57). Its amino-terminal fragment may be deposited in various organs in the form of amyloid fibrils, leading to the development of AA-amyloidosis. The measurement of SAA is a useful diagnostic aid: elevated concentrations are associated with a risk of progressive amyloid fibril deposits in various parenchymas (Citation58). It has been determined that a variable but considerable percentage of patients with FMF, TRAPS, and CAPS may develop renal amyloidosis: the most common clinical manifestation is peripheral edema caused by proteinuria, which may even reach the nephrotic range, the usual outcome of which is chronic renal insufficiency. In addition to the possibility of renal damage, there is also the risk of autonomous nervous system involvement—which leads to alterations in bowel habits and orthostatic hypotension—while heart involvement, with the potential development of restrictive cardiomyopathy, is rather unexpected (Citation59). The ultimate goal of therapy in SAIDs is control of the inflammatory state, which must be accompanied by a reduction in SAA concentration to below 10 mg/L (Citation60). In studies on the treatment of CAPS, SAA has been shown to be a useful parameter in the evaluation of clinical activity in response to therapy, as well as for response to treatment with TNF inhibitors in TRAPS (Citation61–63).
Like that of SAA in inflammatory processes of various origins, the role of the protein S100A12 (or calgranulin C) has also been clarified (Citation64): it is a calcium-binding protein expressed and secreted by neutrophil granulocytes which, through the NF-κB pathway, activates the inflammatory response in the endothelial cells and leukocytes of patients with CAPS. It has been demonstrated that determination of serum S100A12 levels is closely correlated not only with the disease activity, but also with the clinical efficacy of therapy, providing a reliable new marker for future utilization (Citation65).
The cytokine pattern in the systemic autoinflammatory diseases
Cytokines are an important group of signal molecules that regulate numerous aspects of intercellular communication: cytokine signaling is fundamental for the proper functioning of the immune system, including its response to pathogens and extraneous cells, as well as self tolerance. The activation or deregulation of cytokine production is directly implicated in the pathogenesis of SAIDs, and the central role of IL-1 has been confirmed in the last decade thanks to advances in the techniques used to identify human disease-causing genes, innovations in genomic analysis, and progress in effective targeted biologic therapy. In particular, the activation of the IL-1β pathway appears to be the principal mechanism in most SAIDs (Citation66). For example, in FMF, the mutated form of pyrin is capable of activating the caspase-1, which leads to secretion of IL-1β and triggering of the inflammatory response; there are also increases in IL-6 and IL-10 in FMF (Citation67–69). In HIDS IL-1β, TNF-α, and IL-6 levels may all be elevated (Citation70). A characteristic of TRAPS is increased levels of IL-6, TNF-α, IL-8, and IFN-γ (Citation71,Citation72), while in CAPS every clinical manifestation correlates with serum levels of IL-1β and, to a lower degree, IL-18 (Citation73). It has been demonstrated that cultured monocytes from patients with FCAS release significant amounts of IL-1 when exposed to mild hypothermia (32°C), which in turn induces autocrine secretion of IL-6 and TNF-α (Citation74). A similar cold-induced response has been observed in bone-marrow-derived dendritic cells isolated from mice with a common FCAS mutation (Citation75).
Concluding remarks
The aggregate of these recent gains in knowledge have allowed physicians to establish targeted therapies with ‘anti-cytokine’ activity directed against those target molecules responsible for the inflammatory manifestations of SAIDs and thus to revolutionize the clinical pictures and outcomes of many patients. Although IL-1 is not the only effector cytokine driving the inflammatory process in all SAIDs, therapeutic approaches targeting the IL-1 pathway have shown dramatic responses especially in CAPS, as proven by several double-blind placebo-controlled studies, but other biologic agents have also shown mid-term clinical efficacy, as in the case of etanercept in patients with TRAPS (Citation76,Citation77). As new research continues to unravel the innate immune system's arsenal of sensory proteins, and the family of SAIDs expands, it is likely that the many pathways comprising IL-1 function will be explained, and new clinical uses for IL-1 inhibitors will be recognized.
Acknowledgements
L. Cantarini and D. Rigante contributed equally to this paper.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Touitou I, Koné-Paut I. Autoinflammatory diseases. Best Pract Res Clin Rheumatol. 2008;22:811–29.
- Joost PH, van Der Meer JW. The inflammasome: a linebacker of innate defense. N Engl J Med. 2006;355:730–2.
- Rigante D. Autoinflammatory diseases in children. J Pediatr Sci. 2010;4:e65.
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621–68.
- The French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31.
- Yepiskoposyan L, Harutyunyan A. Population genetics of familial Mediterranean fever: a review. Eur J Hum Genet. 2007;15:911–6.
- Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, . Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006;13:236–49.
- Chae JJ, Wood G, Richard K, Jaffe H, Colburn NT, Masters SL, . The familial Mediterranean fever protein, pyrin, is cleaved by caspase-1 and activates NF-kappaB through its N-terminal fragment. Blood. 2008;112:1794–803.
- Centola M, Wood G, Frucht DM, Galon J, Aringer M, Farrell C, . The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223–31.
- Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, . Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40:1879–85.
- Sayarlioglu M, Cefle A, Inanc M, Kamali S, Dalkilic E, Gul A, . Characteristics of patients with adult-onset familial Mediterranean fever in Turkey: analysis of 401 cases. Int J Clin Pract. 2005;59:202–5.
- Touitou I, Sarkisian T, Medlej-Hashim M, Tunca M, Livneh A, Cattan D, . Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum. 2007;56:1706–12.
- Cantarini L, Volpi N, Galeazzi M, Giani T, Fanti F, Lucherini OM, . Colchicine myopathy and neuromyopathy: two cases with different characteristics. J Clin Rheumatol. 2010;16:229–32.
- Cantarini L, Volpi N, Lucherini OM, Giannini F, Galeazzi M. A case of amyloid myopathy in a patient with familial Mediterranean fever. Clin Exp Rheumatol. 2009;27(2 Suppl 53):S106–7.
- Rigante D, La Torraca I, Ansuini V, Compagnone A, Sallì A, Stabile A. The multi-face expression of familial Mediterranean fever. Eur Rev Med Pharmacol Sci. 2006;10:163–71.
- van der Meer JM, Vossen JM, Radl J, van Nieuwkoop JA, Meyer CJ, Lobatto S, . Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet. 1984;1:1087–90.
- Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, . Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nat Genet. 1999;22:178–81.
- Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, . Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet. 1999;22:175–7.
- Houten SM, Wanders RJ, Waterham HR. Biochemical and genetic aspects of mevalonate kinase and its deficiency. Biochim Biophys Acta. 2000;1529:19–32.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30.
- Frenkel J, Houten SM, Waterham HR, Wanders RJ, Rijkers GT, Duran M, . Clinical and molecular variability in childhood periodic fever with hyperimmunoglobulinaemia D. Rheumatology (Oxford). 2001;40:579–84.
- Frenkel J, Houten SM, Waterham HR, Wanders RJ, Rijkers GT, Kimpen JL, . Mevalonate kinase deficiency and Dutch type periodic fever. Clin Exp Rheumatol. 2000;18:525–32.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial hibernian fever. Q J Med. 1982;51: 469–80.
- Hull KM, Drewe E, Aksentijevich I, Singh HK, Wong K, McDermott EM, . The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore). 2002;81: 349–68.
- McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, . Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–44.
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65.
- Kimberley FC, Lobito AA, Siegel RM, Screaton GR. Falling into TRAPS—receptor misfolding in the TNF receptor 1-associated periodic fever syndrome. Arthritis Res Ther. 2007;9:217.
- Dodé C, André M, Bienvenu T, Hausfater P, Pêcheux C, Bienvenu J, .; French Hereditary Recurrent Inflammatory Disorder Study Group. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2002;46:2181–8.
- Aksentijevich I, Galon J, Soares M, Mansfield E, Hull K, Oh HH, . The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet. 2001;69:301–14.
- Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, . Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203.
- Fritz JH. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7:1250–7.
- Hoffman HM, Wanderer AA, Bride DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001;108:615–20.
- Muckle TJ, Wells M. Urticaria, deafness and amyloidosis: a new heredo-familial syndrome. Q J Med. 1962;31:235–48.
- Lepore L, Paloni G, Caorsi R, Alessio M, Rigante D, Ruperto N, . Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with anakinra. J Pediatr. 2010;157:310–5.
- Ebrahimi-Fakhari D, Wahlster L, Mackensen F, Blank N. Clinical manifestations and longterm followup of a patient with CINCA/NOMID syndrome. J Rheumatol. 2010;37: 2196–7.
- Jéru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, . Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A. 2008;105:1614–9.
- Tallon B, Corkill M. Peculiarities of PAPA syndrome. Rheumatology. 2006;45:1140–3.
- Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, . Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet. 2005;42:551–7.
- Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, . An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426–37.
- Okafuji I, Nishikomori R, Kanazawa N, Kambe N, Fujisawa A, Yamazaki S, . Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis. Arthritis Rheum. 2009;60:242–50.
- Touitou I. The spectrum of familial Mediterranean fever (FMF) mutations. Eur J Hum Genet. 2001;9:473–83.
- Gershoni-Baruch R, Brik R, Shinawi M, Livneh A. The differential contribution of MEFV mutant alleles to the clinical profile of familial Mediterranean fever. Eur J Hum Genet. 2002;10:145–9.
- Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, . The Infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat. 2008;29:803–8.
- Simon A, Mariman EC, van der Meer JWM, Drenth JP. A founder effect in the hyperimmunoglobulinemia D and periodic fever syndrome. Am J Med. 2003;114:148–52.
- Rebelo SL, Bainbridge SE, Amel-Kashipaz MR, Radford PM, Powell RJ, Todd I, . Modeling of tumor necrosis factor receptor superfamily 1A mutants associated with tumor necrosis factor receptor-associated periodic syndrome indicates misfolding consistent with abnormal function. Arthritis Rheum. 2006;54:2674–87.
- Aksentijevich I, D Putnam C, Remmers EF, Mueller JL, Le J, Kolodner RD, . The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56:1273–85.
- Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, . Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum. 2011;63:830–9.
- Ferguson PJ, El-Shanti HI. Autoinflammatory bone disorders. Curr Opin Rheumatol. 2007;19:492–8.
- Villanueva-Mendoza C, Arellanes-García L, Cubas-Lorenzo V, Jimenez-Martinez MC, Flores-Suárez LF, Zenteno JC. Familial case of Blau syndrome associated with a CARD15/NOD2 mutation. Ophthalmic Genet. 2010;31:155–8.
- Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, . Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat. 2004;24:194–8.
- Manukyan G, Ghazaryan K, Ktsoyan Z, Khachatryan Z, Kelly D, Tatyan M, . Comparative analysis of cytokine profiles in autoinflammatory and autoimmune conditions. Cytokine. 2010;50:146–51.
- Steichen O, van der Hilst J, Simon A, Cuisset L, Grateau G. A clinical criterion to exclude the hyperimmunoglobulin D syndrome (mild mevalonate kinase deficiency) in patients with recurrent fever. J Rheumatol. 2009;36:1677–81.
- Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis. 2006;1:13.
- Simon A, Cuisset L, Vincent MF, van der Velde-Visser SD, Delpech M, van der Meer JW, . Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-IgD and periodic fever syndrome: its application as a diagnostic tool. Ann Intern Med. 2001;135:338–43.
- Aganna E, Hammond L, Hawkins PN, Aldea A, McKee SA, van Amstel HK, . Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum. 2003;48:2632–44.
- Lachmann HJ, Lowe P, Felix SD, Rordorf C, Leslie K, Madhoo S, . In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J Exp Med. 2009;206:1029–36.
- Rocken C, Shakespeare A. Pathology, diagnosis and pathogenesis of AA amyloidosis. Virchows Arch. 2002;440:111–22.
- Grateau G. Clinical and genetic aspects of the hereditary periodic fever syndromes. Rheumatology. 2004;43:410–5.
- Lachmann HJ, Gillmore JD. Renal amyloidosis. Br J Hosp Med (Lond). 2010;71:83–6.
- Gillmore JD, Lovat LB, Persey MR, Pepys MB, Hawkins PN. Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet. 2001;358:24–9.
- Aganna E, Hawkins PN, Ozen S, Pettersson T, Bybee A, McKee SA, . Allelic variants in genes associated with hereditary periodic fever syndromes as susceptibility factors for reactive systemic AA amyloidosis. Genes Immun. 2004;5:289–93.
- Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, . Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355:581–92.
- Cantarini L, Rigante D, Lucherini OM, Cimaz R, Laghi Pasini F, Baldari CT, . Role of etanercept in the treatment of tumor necrosis factor receptor-associated periodic syndrome: personal experience and review of the literature. Int J Immunopathol Pharmacol. 2010;23:701–7.
- Pietzsch J, Hoppmann S. Human S100A12: a novel key player in inflammation? Amino Acids. 2009;36:381–9.
- Wittkowski H, Frosch M, Wulffraat N, Goldbach-Mansky R, Kallinich T, Kuemmerle-Deschner J, . S100A12 is a novel molecular marker differentiating systemic-onset juvenile idiopathic arthritis from other causes of fever of unknown origin. Arthritis Rheum. 2008;58:3924–31.
- Simon A, van der Meer JW. Pathogenesis of familial periodic fever syndromes or hereditary autoinflammatory syndromes. Am J Physiol Regul Integr Comp Physiol. 2007;292:R86–98.
- Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, . The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A. 2006;103:9982–7.
- Direskeneli H, Ozdogan H, Korkmaz C, Akoglu T, Yazici H. Serum soluble intercellular adhesion molecule 1 and interleukin 8 levels in familial Mediterranean fever. J Rheumatol. 1999;26:1983–6.
- Köklü S, Oztürk MA, Balci M, Yüksel O, Ertenli I, Kiraz S. Interferon-gamma levels in familial Mediterranean fever. Joint Bone Spine. 2005;72:38–40.
- Drenth JP, van Deuren M, van der Ven-Jongekrijg J, Schalkwijk CG, van der Meer JW. Cytokine activation during attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Blood. 1995;85:3586–93.
- Nowlan ML, Drewe E, Bulsara H, Esposito N, Robins RA, Tighe PJ, . Systemic cytokine levels and the effects of etanercept in TNF receptor-associated periodic syndrome (TRAPS) involving a C33Y mutation in TNFRSF1A. Rheumatology (Oxford). 2006;45:31–7.
- Baykal Y, Saglam K, Yilmaz MI, Taslipinar A, Akinci SB, Inal A. Serum IL-2r, IL-6, IL-10 and TNF-alpha level in familial Mediterranean fever patients. Clin Rheumatol. 2003;22:99–101.
- Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JC, . IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol. 2005;175:2630–4.
- Rosengren S, Mueller JL, Anderson JP, Niehaus BL, Misaghi A, Anderson S, . Monocytes from familial cold autoinflammatory syndrome patients are activated by mild hypothermia. J Allergy Clin Immunol. 2007;119:991–6.
- Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, . Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–87.
- Lachmann HJ, Koné-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, . Canakinumab in CAPS Study Group. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416–25.
- Gattorno M, Pelagatti MA, Meini A, Obici L, Barcellona R, Federici S, . Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58:1516–20.