Abstract
Characteristic morphological and molecular alterations such as vessel wall thickening and reduction of nitric oxide occur in the aging vasculature leading to the gradual loss of vascular homeostasis. Consequently, the risk of developing acute and chronic cardiovascular diseases increases with age. Current research of the underlying molecular mechanisms of endothelial function demonstrates a duality of reactive oxygen and nitrogen species in contributing to vascular homeostasis or leading to detrimental effects when formed in excess. Furthermore, changes in function and redox status of vascular smooth muscle cells contribute to age-related vascular remodeling. The age-dependent increase in free radical formation causes deterioration of the nitric oxide signaling cascade, alters and activates prostaglandin metabolism, and promotes novel oxidative posttranslational protein modifications that interfere with vascular and cell signaling pathways. As a result, vascular dysfunction manifests. Compensatory mechanisms are initially activated to cope with age-induced oxidative stress, but become futile, which results in irreversible oxidative modifications of biological macromolecules. These findings support the ‘free radical theory of aging’ but also show that reactive oxygen and nitrogen species are essential signaling molecules, regulating vascular homeostasis.
Key messages
Reactive nitrogen and oxygen species (RNOS) are elevated in aging, which cause oxidative stress, undirected oxidative posttranslational modifications of biological macromolecules, and interference with important redox signaling processes.
In aging, the (physiological) modulator role of RNOS, especially peroxynitrite, in platelets on the prostanoid cascade via modulation of prostaglandin endoperoxide synthase is attenuated and causes platelet hyperreactivity.
Enhanced superoxide formation in vascular endothelial cells impairs •NO signaling by reducing free bioavailable nitric oxide and by reduction of prostacyclin via peroxynitrite-mediated inactivation of prostacyclin synthase.
Mitochondria are a major source for elevated ROS in aging.
Introduction
The future demographics of Western societies indicate that the proportion of people older than 65 years of age will dramatically increase within the next few decades (Citation1). The major foreseeable problems associated with this development are the increasing costs for medical care for older people, primarily due to more frequent and longer periods of hospitalization and rehabilitation due to the increase in cardiovascular, cerebrovascular, and neurodegenerative diseases. Individually, the occurrence of most chronic diseases in the elderly will often be accompanied by a critical reduction in the quality of life and the need for reliance on outside assistance. Therefore, the major objective of aging research should not be the discovery of the ‘fountain of eternal youth’, but rather improving the quality of the life. Scientifically, this aim can be achieved by a better understanding of molecular processes involved in the development of age-dependent diseases, which could ultimately result in earlier diagnostic procedures and preventive measures. Cardiovascular diseases and their acute and chronic manifestations such as myocardial infarction, stroke, and peripheral arterial occlusive disease increase in frequency with age and are further aggravated by the onset of other risk factors (Citation2), such as adiposity, hyperlipidemia, diabetes, or hypertension. Pathophysiologically, vascular aging is mainly characterized by endothelial dysfunction, which is considered a precursor of arteriosclerosis. The aging vasculature displays typical morphological and molecular alterations leading to increased vascular stiffness, reduced compliance, and the loss of vascular homeostasis.
Current research focuses on the regulation of vascular tone and vessel remodeling mediated by free radicals, particularly the role of superoxide (•O2−) and nitric oxide (•NO). In the past, free radicals were regarded as mitochondrial-derived harmful by-products of oxidative phosphorylation, but since the discovery of vascular NADPH oxidases it is obvious that reactive nitrogen and oxygen species (RNOS) play a pivotal role in cell signaling. Besides the classic •NO function of activating soluble guanylyl cyclase and increasing the second messenger cyclic guanosine monophosphate (cGMP), •NO can lead to S-nitros(yl)ation, tyrosine nitration, and glutathiolation of important enzymes involved in vascular signaling via secondary species such as peroxynitrite (Citation3–6). These processes and novel signaling cascades are part of the emerging field termed redox regulation or redox signaling. Therefore, enhanced age-dependent free radical formation will not only cause non-specific oxidation of biological macromolecules but will also interfere with vascular redox regulation. Current knowledge about vascular redox signaling, sources of free radicals, posttranslational oxidative modifications of proteins, oxidative defense systems, and compensatory mechanisms will be discussed with particular regard to cardiovascular aging and development of cardiovascular disease.
Functions and mediators of the endothelium
The endothelium has an important role as a modulator of vascular tone, structure, barrier, and function (Citation7). This multimodal regulation is controlled by the endothelium-derived autacoids •NO (formerly referred to as endothelium-derived relaxing factor (EDRF)), prostacyclin (PGI2), and endothelium-derived hyperpolarizing factor (EDHF). Under physiological conditions, these mediators cause vasodilatation and ensure vascular homeostasis. However, with increasing age, the modulatory role of the endothelium cannot be preserved. An imbalance between vasoprotection and vascular damage occurs ().
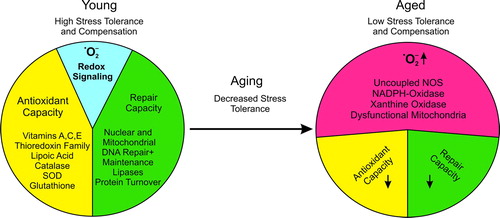
Figure 1. General aspects of RNOS in aging. Young individuals have a higher stress tolerance due to a high antioxidant potential, and efficient repair mechanisms restore homeostatic conditions in response to damage. Under these physiological conditions redox regulation can normally proceed in special compartments but differs in a cell type-specific manner. Prostaglandin endoperoxide H2 synthase (PGHS, COX) is an excellent example of a highly redox-controlled enzyme. In aging the stress tolerance to RNOS decreases. The molecular basis for the loss of balance between damage and repair is the enhanced RNOS generation which must be compensated by antioxidant systems. These exhibit a reduced efficiency in repair with increasing age. As a consequence, oxidatively altered macromolecules accumulate and burden physiological cellular processes. This attenuates redox regulation and also interferes with intercellular communication.
Vanhoutte and co-workers have introduced the definition of a second set of endothelium-derived mediators, which cause vasoconstriction (Citation8). This group of mediators includes superoxide anion (•O2−), prostaglandin endoperoxide H2 (PGH2), and endothelin (ET) (Citation8). As exemplified by the paradoxical reaction in response to acetylcholine, the endothelium may also mediate constriction, which can be reversed by denudation of the endothelium. Certainly, the molecular definition of endothelial dysfunction, i.e. imbalance of endothelium-derived mediators in favor of constriction, needs to be refined.
Under normal conditions, the vascular endothelium is characterized by a basal thromboresistant and vasodilatory phenotype. Following stimulation of a quiescent endothelium, an imbalance in this equilibrium between vasodilatory and constrictive mediators can be observed as demonstrated in numerous reports.
Endothelial cell activation is a transient process covering events occurring in the range of minutes to a few hours and describes the involvement of the endothelial immune response. The endothelium is highly integrated in an orchestrated series of immunological events starting with the trapping of circulating leukocytes, transmigration across the endothelial barrier, the removal of invaded pathogens by immune cells, and, indeed, resolution of inflammation (Citation9).
As a prerequisite for this process, endothelial cells modify their resting properties towards a pro-adhesive and permeable state by temporarily reducing the production of homeostatic autacoids and, instead, cause activation and expression of cellular adhesion molecules such as vascular cell adhesion molecule (VCAM), intercellular adhesion molecule (ICAM), and E-selectin (Citation10). Thus, cells of the immune system can adhere to the endothelium, infiltrate the tissue, and are then guided by chemotaxis to the sites of inflammation, which is termed diapedesis. Endothelial cell activation becomes important in several vascular disorders such as the development of atherosclerosis and can occur in conjunction with endothelial dysfunction. Endothelial cell activation is a reversible physiological reaction to inflammation or injury, but chronic exposure to RNOS, as in the aging process, will lead to irreversible changes of endothelial function (Citation11).
Endothelial stunning has been observed upon a brief exposure to lipopolysaccharide or pro-inflammatory cytokines in healthy individuals, which can lead to an impaired endothelium-dependent relaxation that can last days to weeks (Citation12,Citation13). These events are associated with a decreased ability of the endothelium to maintain its anti-aggregatory and vasodilatory properties and are often accompanied by a decrease in constitutively expressed NO synthase-3 (NOS-3) and prostaglandin endoperoxide H2 synthase-1 (PGHS-1; COX-1). These events are typically observed after moderate systemic bacteremia and increase the risk for thromboembolic events.
Endothelial dysfunction, however, is characterized by a pronounced and persistent stimulation of the endothelium that occurs in situations such as sepsis. It is usually accompanied by an excessive formation of superoxide anion that causes severe vasoconstriction and platelet aggregation (Citation14,Citation15). Persistent stimulation, e.g. by lipopolysaccharide (LPS), can ultimately lead to a complete detachment of endothelial cells, an observation also known as the Shwartzman reaction (Citation16). As a compensatory mechanism, the underlying smooth muscle can help regulate these dysfunctional events in the endothelium. Resting vascular smooth muscle cells (VSMC) express prostacyclin (PGI2) synthase abundantly but are devoid of significant PGHS activity. Upon stimulation by pro-inflammatory cytokines or LPS, VSMCs express inducible PGHS-2 and become a rich source of PGI2. In case the dysfunction occurs locally only, the VSMC-derived PGI2 partially compensates for the impaired endothelial function. However, systemic inflammation in humans is mostly responsible for the large amounts of PGI2 that cause pronounced vasodilation in progressive stages of septic conditions.
General aspects of vascular aging
Vascular aging already starts in young adults by slow and progressive vascular remodeling (Citation17,Citation18). Early signs of declining endothelial function manifest before the fourth decade of life (Citation17). Morphological changes involve intima–media thickening and vascular hypertrophy/hyperplasia accompanied by reorganization of the extracellular matrix (Citation18,Citation19). Vascular wall thickening in otherwise ‘healthy’ (i.e. non-atherosclerotic) regions is mainly caused by proliferation of medial VSMC. Wall thickness increases in a linear manner up to 3-fold (Citation20), accompanied by lumen size enlargement. Unlike aging, intimal thickening in atherosclerosis involves the invasion and de-differentiation of macrophages to lipid-laden foam cells as well as recruitment of VSMC and neointima formation. This process causes severe luminal reduction, and unstable plaques may promote thrombus formation and arterial occlusive disease. Vascular stiffness is based on augmented elastin synthesis, replaced by deposition of collagens and glycosaminoglycans, ground substance, and partial calcification (Citation18). Free radicals can worsen these processes by oxidizing and cross-linking matrix constituents. Furthermore, RNOS can promote VSMC motility and proliferation and therefore accelerate vascular hypertrophy, stiffness, and cellular senescence. Hypertension in the elderly is also, at least in part, related to impaired endothelial function and reduced vessel wall elasticity (Citation21).
Age-associated endothelial dysfunction is generally defined by reduced bioavailability of free •NO. Various molecular explanations have emerged so far to explain this alteration, but the most prominent one is the inactivation of •NO by •O2−. From this interaction a more reactive RNOS species peroxynitrite can arise, which, in turn, can influence other enzymes involved in the maintenance of vascular homeostasis such as prostacyclin (PGI2) synthase (Citation22) or sarco/endoplasmic reticulum calcium ATPase (SERCA) (Citation23). This illustrates that RNOS may play a pivotal role in cardiovascular aging and the development of cardiovascular disease, both by direct oxidative damage and by interfering with redox signaling cascades in the vasculature.
Models and theories of aging
Lifestyle, diseases, genetic predispositions, and environmental factors modulate the aging process in a multimodal fashion and further complicate basic research into the underlying mechanisms of aging. Several theories of aging have emerged over the past decades, and molecular evidence for each of these hypotheses has been provided. A summary is given in . According to experimental data, it is obvious that these theories and hypotheses are oversimplifications that are interconnected with each other. The most relevant of these theories, which also best explain cardiovascular aging, are those of replicative senescence, chronic oxidative stress (free radical theory of aging), and mitochondrial dysfunction.
Table I. Theories of aging.
Vascular senescence
Hayflick and Moorhead discovered that primary human fibroblasts have a limited number of cell division cycles and a finite lifespan upon continuous subculture. The ‘Hayflick limit’ is commonly known as replicative or intrinsic senescence (Citation24). The molecular basis of this limit in replicative capacity of cells are telomeres, specialized tandem G-rich repeats that extend until the chromosome end. These structures were first discovered in the 1930s by McClintock and Mueller, and later found by Watson and Olovnikov to serve as primers for DNA replication and chromosomal stability. Blackburn (Citation25) and others revealed that telomeres were maintained and elongated by telomerase or the so-called alternative lengthening of telomeres (ALT) pathway (Citation26,Citation27) and that telomere shortening was important in aging. This important concept of aging was confirmed in telomerase deletion animals (Citation28–30) that demonstrated reduced tissue regeneration and impairment of bone marrow or skin-derived stem cells. Furthermore, many of the known premature aging syndromes in humans including Werner syndrome (Citation31) or dyskeratosis congenita (Citation32) can be attributed to enhanced telomere attrition. In recent years, it has become evident that various tumors (Citation33) as well as primary cell lines (Citation34) maintain telomeres by either activating telomerase or the ALT pathway, thus avoiding replicative senescence and conferring infinite cell proliferation and immortality. This also illustrates that cancer and aging are linked to genomic instability. This topic is outside of this review, but the interested reader is referred to other excellent reviews (Citation35).
In early life, cell senescence is considered to ensure reproduction (germ line) by preventing tumor formation and preserving tissue integrity (somatic cells). However, in later life, senescent cells clearly compromise the function of tissues and may accelerate the process of age-related diseases such as atherosclerosis.
General characteristics of these cells are morphological changes such as increased cell size, polymorphic nuclei, cell flattening (fried egg appearance), vacuolization, increased lysosomal mass, and accumulation of a transcriptionally inactive heterochromatic structure, senescence-associated heterochromatic foci (SAHF). Histological identification is achieved by visualization of the senescence-associated β-galactosidase (SA-β-Gal) staining (Citation36). The molecular basis of the ‘programmed lifespan’ is still obscure (Citation37) but seems to be primarily related to telomere attrition, which causes chromosomal and genetic instability (termed state of ‘cell crisis’) as well as expression of cell cycle inhibitors such as p53, retinoblastoma protein (Rb), or Ras (Citation37,Citation38).
Besides the exhibition of these typical markers of senescence, endothelial cells in culture rapidly cease to produce homeostatic mediators such as PGI2 or •NO (Citation39,Citation40). In conjunction with increased expression of plasminogen activator inhibitor (PAI)-1 and cell adhesion molecules this indicates endothelial cell dysfunction. In vivo, senescent endothelial cells have mainly been identified at atherosclerosis-prone regions such as branching sites (Citation41,Citation42). Impaired healing/repair of vascular lesions and impaired angiogenesis may be some of the consequences (Citation43) of senescent cells. This concept of aging is highly contentious since most results were obtained in artificial cell culture experiments.
In contrast, an ‘acute form’ of senescence, which is unrelated to extensive proliferation or telomere damage, can be induced by a number of sub-lethal stressors. This has been termed as ‘stress-induced premature senescence’ (SIPS) (Citation44) and ‘stress or aberrant signaling-induced senescence’ (STASIS) (Citation45). In the vasculature, pro-atherogenic and pro-inflammatory factors such as oxidized low-density lipoprotein (LDL) (Citation46), pro-inflammatory cytokines such as tumor necrosis factor (TNF)-alpha (response to injury hypothesis) (Citation46), RNOS (Citation47), p38-mitogen-activated protein kinase (MAPK) signaling cascade (Citation48), hyperglycemia (Citation42), and advanced glycation end-products (AGE) (Citation42,Citation49) may all lead to activation of the endothelium and ultimately cause senescence. Telomere shortening and inhibition of the endogenous telomerase activity are often accompanying phenomena (Citation50,Citation51). With respect to RNOS, addition of •NO donors or antioxidants can delay endothelial senescence and telomere erosion (Citation52,Citation53).
Recent evidence suggests that senescence also plays a role in media calcification (Citation54). The increased expression of osteoblastic genes suggests an osteoblastic transition of VSMCs, partially through runt-related transcriptional factor-2 (RUNX-2), initiating osteoblastic differentiation dependent mechanisms (Citation54). Interestingly, statin and Rho-kinase inhibitors prevent calcification of senescent VSMCs (Citation54).
Free radical theory of aging
In 1956, Harman described for the first time the free radical theory of aging: ‘the reaction of active free radicals, normally produced in the organism, with cellular constituents’ (Citation55,Citation56). By this time, radicals were regarded as unstable molecules that had biological effects. With the discovery of superoxide dismutase (SOD) by Fridovich and McCord (Citation57) and various other antioxidant systems (Citation58), this hypothesis gained substantial scientific footing, and a new research area on oxidative stress phenomena emerged.
In general, RNOS can irreversibly oxidize biological macromolecules, which leads to cumulative impairment of cell function. This phenomenon is observed naturally with aging and in diseases, such as atherosclerosis or Alzheimer's disease, that have a high prevalence in the elderly. Similar to oxidative stress-related diseases, in various animal models and in humans, an accumulation of oxidized proteins, lipids, and DNA has been confirmed for aging. Typical examples include protein-bound nitrotyrosine (Citation5,Citation59,Citation60), protein-carbonyls (Citation61), lipofuscin (Citation62,Citation63), malondialdehyde (Citation64), ceroid bodies (Citation65), or 8-oxo-deoxy-guanosine (Citation66). These modifications typically serve as molecular footprints of RNOS generation and increase in quantity with age in the cardiovascular system. In particular, nitrotyrosine and dityrosine are important markers, which correlate well with age-dependent endothelial dysfunction. This is of particular interest since nitrotyrosine is regarded as a marker of peroxynitrite (PN), a highly reactive RNOS that is formed from the reaction of •O2− with •NO. Besides causing oxidative damage, this reaction also reduces the bioavailability of •NO, which is a widely accepted cause of endothelial dysfunction and a hallmark of cardiovascular aging. The details on the •NO/•O2− antagonism will be explained in a later section on •NO, •O2−, and RNOS.
Elevated levels of other oxidative markers including protein-carbonyls, oxidatively damaged DNA, or protein lipid adducts that affect tissue function can be abundantly detected in the aging myocardium and vasculature. The role of RNOS is further supported by mouse models in which important antioxidant systems were ablated. For example ALDH-2−/− (aldehyde dehydrogenase) mice, which have an impaired capacity to remove reactive aldehydes including 4-hydroxynoneal, develop severe endothelial dysfunction at medium age (Citation67). In addition, mice with specific cardiac over-expression of catalase directed to either the peroxisomes or to the mitochondria appear to have increased survival rates and improved cardiac contractility with aging (Citation68). These data suggest that free radicals may play a crucial role in the aging process.
However, reports on dietary supplementation with antioxidants or on over-expression of antioxidant enzymes interfering with aging are inconsistent. The low or absent efficacy of orally administered antioxidants (e.g. vitamin C) may be due to the inability of these compounds to reach the compartments of increased RNOS formation in sufficiently high concentration. Genetic manipulation of SOD in yeast or Drosophila melanogaster supported the role of free radicals and extended lifespan, although this failed in a mouse model (Citation69). To evaluate Harman's oxidative stress theory, Muller and co-workers reviewed results grouped by selected model organisms (Citation58). The authors conclude that, unlike data in Drosophila melanogaster, studies in mice clearly support a role of oxidative stress in age-related diseases but not in determining lifespan. Although an increased longevity in a number of genetically modified mouse models has been demonstrated, the mechanisms by which these genetic modifications regulate aging largely remain unclear, and systematic validation would be highly desirable (Citation70).
Lastly, although radicals can cause oxidative stress and destroy biological macromolecules, at low concentrations they serve as important messenger molecules. Consequently, enhanced radical formation and manipulations in the antioxidant system will interfere with the more recently identified redox signaling pathways as well.
Mitochondrial theory of aging
In the 1970s, Harman extended the free radical theory of aging (Citation56) on mitochondria as the ‘seed of senescence’ (Citation71). In most cell types, mitochondria are the principal source of endogenous oxidants. It can be assumed that 0.1% of oxygen consumed is converted into RNOS. Aging-dependent impairment of tissue function parallels with dysfunctional mitochondria and enhanced mitochondrial free radical generation. Research has mainly focused on mitochondria-derived oxidative stress phenomena such as nuclear (genomic) DNA mutations as shown in Caenorhabditis elegans (Citation72). The organelle was ascribed the role of a ‘cellular power plant’, which generates free radicals by misdirection of electrons from the respiratory chain to molecular oxygen and hence into ROS production (Citation73,Citation74). Autoxidation of components of the respiratory chain will lead to the formation of •O2−, which is the one-electron reduction product of oxygen. Most, but not all, oxidative damage is mediated by one-electron oxidation processes. Novel findings suggest that mitochondria are organized in a flexible network that is controlled by fission and fusion. Within this network, heterogeneous mitochondrial populations seem to exist. These were characterized by genetic variability (Citation75), differences in the membrane potential (Citation76,Citation77), calcium sensitivity for permeability transition (Citation78–80), or by histology (in the heart: interfibrillary or subsarcolemmal mitochondria) (Citation81,Citation82). This network is extremely sensitive to metabolic changes such as high glucose and can respond to an increase in oxygen radical formation (Citation83). However, the physiological consequences remain unclear and need further elucidation.
Typical age-mediated alterations, involving mitochondrial physiology and RNOS generation, are reduction of ATP synthesis by inhibiting enzymes of the citric acid cycle, creatine kinase, or respiratory chain components (Citation84–86), mutation of mitochondrial DNA (mtDNA) (Citation87,Citation88), and an increase in apoptosis by oxidative activation of the permeability transition pore (Citation89).
Several animal models provide experimental evidence for the mitochondrial theory of aging and disease, such as mitochondrial MnSOD−/− and MnSOD−/+ animals (Citation90). Major mitochondrial alterations include reduced antioxidant capacity, increased mtDNA damage, and reduced activities in enzymes of the respiratory chain and citric acid cycle leading to enhanced apoptosis. Newborn MnSOD−/− mice suffer from severe oxidative stress and die within a few days after birth (Citation90). The heart and central nervous system, tissues of high oxygen consumption, are adversely affected. Heterozygous MnSOD−/+ animals have a normal lifespan but show enhanced age-dependent endothelial dysfunction and arteriosclerosis (Citation67,Citation69,Citation91–94).
The biogenesis of mitochondrial respiratory chain complexes requires concerted contribution from physically separated genomes: the nuclear DNA and the maternally inherited mtDNA. This ‘genome in the genome’ only codes for respiratory chain components and is more susceptible to oxidative stress than nuclear DNA due to the proximity to respiratory chain components, the lack of histones (Citation95), and the lack of an efficient DNA repair system (mt polymerases lack specificity for base excision repair) (Citation96). Therefore, mtDNA is considered as the ‘seed of senescence’. Age-associated mitochondrial dysfunction implies increased generation of ROS (Citation97), which initiates a vicious cycle of mtDNA damage leading to a dysfunctional respiratory chain accompanied by •O2− generation and promotes further mtDNA mutations and damage (Citation98,Citation99). MtDNA deletions associated with an increased induction of oxidative phosphorylation in nuclear genes coding for mitochondrial respiratory complexes accumulate with age and were found to be significantly higher in human hearts with coronary artery disease than in hearts of age-matched controls (Citation100). Recent investigations show that the mutation rate of mtDNA is enhanced in transgenic animals carrying a proof-reading domain-deficient mitochondrial polymerase γ. This is accompanied by typical signs of premature aging. The phenotype of these animals is characterized by kyphosis (curvature of the spine), cardiac hypertrophy, and osteoporosis, all of which corresponds to enhanced RNOS formation and apoptosis (Citation101,Citation102). A recent study showed that transgenic mice expressing cardiac-targeted mutated human Polγ developed severe cardiomyopathy similar to observations in Polγ transgenic animals (Citation103). However, the exact signaling pathways determining the molecular basis for the link between lifelong damage to mtDNA, RNOS, and development of vascular disease are not yet fully understood (Citation104,Citation105).
Recently, the classic mitochondrial theory of aging has been challenged with experimental results suggesting that oxidative damage may occur without severe mtDNA mutation (Citation106) and, conversely, that mtDNA mutagenesis may have causes other than RNOS (Citation58).
A recent groundbreaking publication by Sahin and co-workers (Citation107) suggested a concept that unifies the mitochondrial theory of aging, the free radical theory of aging, and replicative senescence. According to this hypothesis, telomere dysfunction triggers a chronic activation of p53 that leads to a suppression of PPAR-γ coactivator 1α and-β (PGC-1α/PGC-1β) expression, which normally is involved in the regulation of gene expression required for mitochondrial biogenesis. Hence, replicative senescence would automatically lead to a decline in the mitochondrial maintenance and repair machinery and cause an accumulation of damaged mitochondria. This would allow for the clonal expansion of mitochondria containing mutated DNA. Subsequently, greater mtROS would be formed that, in a vicious cycle, would lead to more mitochondrial, nuclear, and, in particular, to telomeric DNA damage.
Taken together, RNOS and cellular oxidative damage appear to be associated with age-related disease, although a causal relationship between oxidative stress and lifespan in humans and in mammalian animal models remains to be definitively proven. In any case, the oxidative stress theories of aging are mutually interdependent, and interactions are described by other theories such as replicative senescence, telomere shortening, and ‘extensions’ such as mitochondrial DNA damage.
•NO, •O2−, and RNOS
RNOS sources in vascular aging
Nitric oxide—the classic paradigm
•NO was the first gaseous free radical discovered to have a physiological signaling function and is one of the most important endothelium-derived mediators in the cardiovascular system. •NO, which can easily cross cell membranes by diffusion, can mediate a plethora of effects via cGMP-dependent and independent mechanisms such as smooth muscle relaxation (Citation108), leukocyte repulsion (Citation109), inhibition of platelet activation and thrombus formation (Citation110), or suppression of vascular remodeling by inhibition of smooth muscle cell de-differentiation and growth (Citation111).
Classically, •NO activates soluble guanylyl cyclase (sGC) in SMC via nitrosylation (reaction of •NO with the heme moiety and not with sulfhydryls) thus increasing intracellular cGMP levels. This second messenger activates protein kinase G (PKG) which reduces [Ca2 + ]i by re-uptake into the sarcoplasmic reticulum (Citation112), activation of Ras homolog gene family (Rho)/Rho kinases (Citation113,Citation114), or myosin light chain phosphatase (dephosphorylation of myosin light chains leads to smooth muscle relaxation) (Citation113).
The major •NO source in the endothelium is NOS-3. However, depending on the tissue, other NOS isoforms may have significant impact on vascular function, particularly microcirculation () (Citation115–118).
Table II. Major cellular NO sources and regulation.
Nerve endings in the corpus cavernosum contain NOS-1 (Citation119), which becomes activated by nerve excitation leading to vasorelaxation and induction of penile erection by filling the lacunae-like structures with blood. Similarly, the skeletal muscle splice variant nNOSμ (spliced and truncated variant of neuronal NO synthase) becomes concomitantly activated by Ca2 + -triggered muscle constriction ensuring adequate oxygen supply and blood circulation. In inflammation, NOS-2 is induced in many cell types (e.g. atherosclerotic lesions), generating high levels of •NO. NOS-3 is the most important isoform in the vasculature to maintain homeostasis. NO synthesis by NOS-3 is not only triggered by [Ca2 + ]i levels but can be modulated by phosphorylation as an immediate adaptation to stimuli such as shear stress.
In contrast to the classic cGMP-mediated effects, •NO can directly modulate enzymatic activities via more reactive intermediate nitrogen species, reversibly or irreversibly modifying protein-bound sulfhydryls or tyrosine residues. This requires the participation of •O2−, which generates a whole new set of RNOS and opens an amazing new insight into vascular redox signaling (). In this context, •O2− can be regarded as an •NO antagonist, representing a ‘Yin–Yang system’ similar to the effects exerted by prostanoids and thromboxane in the vasculature.
Figure 2. The vascular •NO signaling cascade and the antagonizing •O2−. Agonist stimulation of endothelial cells, including angiotensin II, acetylcholine, serotonin, etc., or shear stress causes activation of endothelial NO synthase by a transient increase in intracellular calcium or phosphorylation. •NO as a freely diffusible molecule can easily cross cell membranes and activate soluble guanylyl cyclase in smooth muscle cells, increasing intracellular cGMP levels. Protein kinase G is activated and can cause a decrease in intracellular calcium [Ca2 + ]i and mediates the dephosphorylation of myosin light chain leading to vasorelaxation. Superoxide can react with and reduce free •NO, impairing the •NO-cGMP signaling pathway. Inactivation of soluble guanylyl cyclase by superoxide has been reported, too. Various superoxide sources in the endothelium and vascular smooth muscle cells have been identified, including mitochondria, uncoupled NO synthase, xanthine oxidase, NADPH oxidases, and epoxygenase. Depending on the model and pathology, different sources of •O2− can be activated. (Ag = agonist; [Ca2 + ]i = intracellular calcium; NOS-3 = endothelial NO synthase; sGC = soluble guanylyl cyclase; •NO = nitric oxide; cGMP = cyclic guanosine monophosphate; Rec = receptor; •O2− = superoxide anion)
![Figure 2. The vascular •NO signaling cascade and the antagonizing •O2−. Agonist stimulation of endothelial cells, including angiotensin II, acetylcholine, serotonin, etc., or shear stress causes activation of endothelial NO synthase by a transient increase in intracellular calcium or phosphorylation. •NO as a freely diffusible molecule can easily cross cell membranes and activate soluble guanylyl cyclase in smooth muscle cells, increasing intracellular cGMP levels. Protein kinase G is activated and can cause a decrease in intracellular calcium [Ca2 + ]i and mediates the dephosphorylation of myosin light chain leading to vasorelaxation. Superoxide can react with and reduce free •NO, impairing the •NO-cGMP signaling pathway. Inactivation of soluble guanylyl cyclase by superoxide has been reported, too. Various superoxide sources in the endothelium and vascular smooth muscle cells have been identified, including mitochondria, uncoupled NO synthase, xanthine oxidase, NADPH oxidases, and epoxygenase. Depending on the model and pathology, different sources of •O2− can be activated. (Ag = agonist; [Ca2 + ]i = intracellular calcium; NOS-3 = endothelial NO synthase; sGC = soluble guanylyl cyclase; •NO = nitric oxide; cGMP = cyclic guanosine monophosphate; Rec = receptor; •O2− = superoxide anion)](/cms/asset/0353c7af-ae72-4238-9ed6-364f03e10686/iann_a_645498_f0002_b.jpg)
The versatile chemistry allows •NO to act as a signaling molecule and permits potent antioxidant properties (Citation120). •NO at physiological concentrations effectively prevents Fenton chemistry through ligand–metal interactions. The formation of a metal nitrosyl complex limits the addition of peroxides to biological metal co-ordination sites and thus inhibits the formation of more reactive species including hydroxyl radical, peroxo- or oxo-metal complexes (Citation120). •NO also acts through radical–radical interaction and terminates radical chains. An example is the efficient abatement of lipid peroxidation by •NO (Citation121) and the scavenging of reactive intermediates formed during decomposition of peroxynitrite (Citation122). Such mechanisms can regulate cyclo- oxygenase-mediated vascular tone (Citation123), platelet aggregation (Citation124), or contribute to the prevention of atherogenesis (Citation125,Citation126).
Superoxide anion—a nitric oxide antagonist and mediator of redox regulation
•O2− may have reducing rather than oxidizing properties, as exemplified by cytochrome c (Citation57). Target enzymes that are directly affected by •O2− include protein kinase C (Citation127), calcineurin (Citation128), and aconitase (Citation129). This likely occurs by metal catalysis under physiological •O2− concentrations (10−9–10−12 M). Its physiological/pathophysiological role in the vasculature mainly originates from the almost diffusion-controlled reaction with •NO, which exceeds the dismutation rate by SODs. At very high concentrations of •O2−, oxidative stress occurs via Fenton chemistry resulting in the formation of the highly toxic and, for biological macromolecules, deleterious hydroxyl radical (•OH).
Several vascular •O2− sources exist (), being activated under particular physiological and pathophysiological situations. Sources differ in their mode of activation and capacity to release •O2−. The most intensively studied enzymatic family members are subunits of the NADPH oxidase. Besides the classic well characterized phagocyte (NADPH) oxidase (gp91phox = glycoprotein 91 phagocyte oxidase or NOX-2), six more members have emerged, among which NOX-1 and NOX-4 are expressed in vascular smooth muscle cells (Citation130).
Table III. Potential sources of superoxide in the vasculature.
At membranes, an active NOX complex comprises the catalytic integral membrane and cytosolic regulatory proteins. This complex generates •O2− or H2O2. In contrast to phagocytes, the levels of superoxide formed in smooth muscle and other types of cells are rather low. The non-phagocytic NADPH oxidase family requires a different set of cytosolic subunits (NOX-1) or is constitutively active (NOX-4). Increased gp91phox expression in heart (Citation131) and aortic endothelium (Citation20) was reported in aged rats. NADPH oxidase contributes at least in part to increased superoxide radical formation in aging (Citation132).
Another source of •O2− production is an uncoupled NOS-3, which has recently gained more recognition with respect to cardiovascular disease and cardiovascular aging. It is now widely accepted that reduced levels of the co-factors tetrahydrobiopterin (BH4) and/or L-arginine are a common cause for uncoupling. In particular, BH4 is very sensitive to oxidation, and its levels may be reduced under conditions of oxidative stress (Citation133). Beside the lack of co-factors, the involvement of heat shock protein (HSP) 90, disruption of the Zn-finger (Citation117), which changes dimer–monomer distribution (Citation134), phosphorylation (Citation135,Citation136), or protein S-glutathionylation (Citation137) are alternative explanations for uncoupling of NOS-3. Meanwhile, NOS-3 uncoupling has been accepted to occur in conditions such as diabetes mellitus (Citation138,Citation139), hypertension, and hypercholesterolemia (Citation135), as well as in chronic smokers or patients treated with nitroglycerin-containing drugs (Citation117). Its role in vascular aging, however, has not yet been definitively proven.
Xanthine oxidase is the classic •O2− source, which becomes activated during ischemia/reperfusion. Under physiological conditions the enzyme exists in the dehydrogenase form, catalyzing the final step of purine degradation in man and primates, by converting xanthine via hypoxanthine into uric acid. Conversion to the oxidase can occur either by proteolytic cleavage or oxidation of sulfhydryl groups and disruption of Fe-sulphur clusters. Therefore, xanthine oxidase may also represent an enzyme which is capable of modulating, enhancing, and amplifying radical formation via reversible oxidation of sulfhydryl groups in conjunction with other radical sources. Allopurinol or oxypurinol has successfully been used therapeutically to inhibit xanthine oxidase and lower plasma uric acid levels. Interestingly, uric acid as a potent scavenger for peroxynitrite may inhibit platelet aggregation and restore endothelial function, and it is believed to prolong lifespan in man and Drosophila melanogaster (Citation140,Citation141).
Apart from metabolism by the prostaglandin endoperoxide and the lipoxygenase pathways, arachidonic acid can be metabolized via the epoxygenase pathway as well (Citation142). Cytochrome P450 (CYP) epoxygenases contribute to the generation of oxygen-derived free radicals in the vascular wall (Citation143).
Mitochondria represent the most potent and yet most obscure source of superoxide formation. It is still not clear whether ‘redox cross-talk’ between cytosolic RNOS sources and the mitochondria exist and, if so, through which molecular mechanisms it is controlled (Citation144–147). Nevertheless, uncoupling proteins, the permeability transition pore, and a putative anion channel have been suggested to modulate mitochondrial radical formation as well as posttranslational modifications of complex I and II (Citation148).
The free radical signaling network—the non-classic nitric oxide pathways
Initially, •NO was assumed to nitros(yl)ate sulfhydryls directly, but chemically an intermediate •NO-derived S-nitrosating species is required. The parallel release of low amounts of •O2−, and of •NO at concentrations that are 2–3 times higher, causes peroxynitrite formation as a short-lived intermediate (Citation149) which is rapidly converted to an S-nitrosating species like N2O3 as in-vitro experiments demonstrated (Citation150). The maximal generation of peroxynitrite alone occurs when •NO and •O2− are in equimolar ratios. The occurrence of N2O3 as a physiological nitrosating species is not fully elucidated yet, and other secondary RNOS or heme-catalyzed mechanisms may play a more important role (Citation151,Citation152).
In general, redox-based cysteine thiol modifications can be seen as the basis of redox signaling but also as a protective mechanism to prevent essential cysteines from being irreversibly oxidized to sulphinic or sulphonic acid during interim stress situations. Such modifications include S-nitrosation, S-glutathiolation, or intramolecular disulphide formation (Citation153).
S-nitrosation of essential catalytic cysteines has been shown to alter enzyme activity, such as inhibition of glyceraldehyde dehydrogenase (Citation154), NADP + isocitrate dehydrogenase (Citation155), caspase-3 (Citation156,Citation157), mitochondrial complex I (Citation89,Citation158), and mitochondrial aldehyde dehydrogenase (Citation159,Citation160).
The prostanoid pathway
Prostanoids play a vital role in numerous different cell types and tissues throughout the body. These mediators are involved in the maintenance of steady-state conditions but can also become highly up-regulated under acute and chronic pathophysiological conditions such as sepsis, diabetes, atherosclerosis, or aging. The formation of prostanoids involves a cascade of three enzymes (), starting with the liberation of membrane-bound arachidonic acid by Ca2 + -activated phospholipase A2 (PLA2). Fluid shear stress is also a potent activator of PLA2 via phosphorylation. Then, arachidonic acid is converted in a two-step process into PGH2 by the rate-limiting enzyme, prostaglandin endoperoxide H2 synthase (PGHS), also referred to as cyclo-oxygenase (COX). In a final step, PGH2 is converted into different prostanoids by terminal synthases and isomerases such as PGI2 synthase or thromboxane A2 (TxA2) synthase. The most important prostanoids in the vascular system are PGI2 and PGH2, which are endothelium-derived autacoids, and TxA2, which is solely platelet-derived. PGI2 acts synergistically with •NO and mediates its anti-aggregatory, anti-adhesive, anti-proliferative, and anti-thrombotic effects via the G-protein coupled prostacyclin receptor (IP-receptor), leading to an increase in cyclic adenosine monophosphate (cAMP). TxA2 and PGH2 have opposing properties, but both act on the TxA2/PGH2 receptor (TP receptor) to increase intracellular [Ca2 + ]i levels. Peroxynitrite formation, which is increased in an age-dependent manner, may nitrate and inactivate PGI2 synthase, causing vasoconstriction, platelet aggregation, and thrombus formation (Citation161).
Figure 3. The Yin–Yang system of prostanoids. Agonist stimulation of endothelial cells causes an increase in intracellular calcium levels [Ca2 + ]i, which releases arachidonic acid via activation of phospholipase A2 from membrane phospholipids. Alternatively, phospholipase A2 is regulated by fluid or cyclic shear stress through phosphorylation. Arachidonic acid is converted by prostaglandin endoperoxide H2 synthase (cyclo-oxygenase) into the intermediate prostaglandin endoperoxide H2, which is further processed into prostacyclin by prostacyclin synthase. Prostacyclin mediates vasorelaxation by acting on the prostacyclin receptor, activating adenylyl cyclase, and elevating cyclic AMP, resulting in an increase in protein kinase A activity. Furthermore, prostacyclin inhibits platelet aggregation and leukocyte adhesion to the endothelium. Under conditions that cause inhibition of prostacyclin synthase, PGH2 levels increase and PGH2 exerts thromboxane-like effects via stimulation of the thromboxane A2/PGH2 receptor. Thromboxane A2 stimulates platelet aggregation and enhances this process in an autocrine loop or promotes leukocyte adhesion. Activation of the thromboxane A2/PGH2 receptor leads to activation of phospholipase C and an increase in intracellular calcium levels. (AA = arachidonic acid; AC = adenylyl cyclase; Ag = agonist; [Ca2 + ]i = intracellular calcium; G = G-protein; ONOO− = peroxynitrite; •O2− = superoxide; PLA2 = phospholipase A2; •NO = nitric oxide; PGHS-1 = prostaglandin endoperoxide H2 synthase; PGH2 = prostaglandin endoperoxide H2; PGIS = prostacyclin synthase; PLC = phospholipase C; PKA = protein kinase A; Rec = receptor; TxA2 = thromboxane A2; TxAS = thromboxane synthase)
![Figure 3. The Yin–Yang system of prostanoids. Agonist stimulation of endothelial cells causes an increase in intracellular calcium levels [Ca2 + ]i, which releases arachidonic acid via activation of phospholipase A2 from membrane phospholipids. Alternatively, phospholipase A2 is regulated by fluid or cyclic shear stress through phosphorylation. Arachidonic acid is converted by prostaglandin endoperoxide H2 synthase (cyclo-oxygenase) into the intermediate prostaglandin endoperoxide H2, which is further processed into prostacyclin by prostacyclin synthase. Prostacyclin mediates vasorelaxation by acting on the prostacyclin receptor, activating adenylyl cyclase, and elevating cyclic AMP, resulting in an increase in protein kinase A activity. Furthermore, prostacyclin inhibits platelet aggregation and leukocyte adhesion to the endothelium. Under conditions that cause inhibition of prostacyclin synthase, PGH2 levels increase and PGH2 exerts thromboxane-like effects via stimulation of the thromboxane A2/PGH2 receptor. Thromboxane A2 stimulates platelet aggregation and enhances this process in an autocrine loop or promotes leukocyte adhesion. Activation of the thromboxane A2/PGH2 receptor leads to activation of phospholipase C and an increase in intracellular calcium levels. (AA = arachidonic acid; AC = adenylyl cyclase; Ag = agonist; [Ca2 + ]i = intracellular calcium; G = G-protein; ONOO− = peroxynitrite; •O2− = superoxide; PLA2 = phospholipase A2; •NO = nitric oxide; PGHS-1 = prostaglandin endoperoxide H2 synthase; PGH2 = prostaglandin endoperoxide H2; PGIS = prostacyclin synthase; PLC = phospholipase C; PKA = protein kinase A; Rec = receptor; TxA2 = thromboxane A2; TxAS = thromboxane synthase)](/cms/asset/6f0509ab-6a04-4aef-b2cc-e32afb486d3f/iann_a_645498_f0003_b.jpg)
Nitration of PGI2 synthase—a change in PGH2 metabolic pathways
Steric inhibition of PGI2 synthase occurs via peroxynitrite- mediated nitration of Tyr430 close to the active site, thus limiting the accessibility of the substrate. Physiologic concentrations of peroxynitrite (IC50 = 50–200 nM) can already inactivate the enzyme (Citation162,Citation163), catalyzed via its heme-thiolate structure and the close proximity of the Tyr residue to the heme. Such low concentrations of peroxynitrite may be formed under numerous conditions such as diabetes, inflammation, atherosclerosis, ischemia/reperfusion injury, and aging. An increase in •O2− formation can be regarded as one of the major initial causes of an imbalanced prostanoid formation in favor of PGH2.
Under homeostatic conditions and upon stimulation with vasoactive agonists, TxA2 and PGI2 are released in balanced amounts, and PGH2 represents an intermediate metabolite at negligible concentrations only, since PGH2 is quickly converted by terminal synthases. However, if disturbances in the utilization of PGH2 by terminal synthases occur, e.g. by inhibition of prostacyclin synthase through nitration, PGH2 accumulates and acts on the TxA2 receptor (TP) leading to vasoconstriction. Those pathophysiological events typically occur in the aged vasculature (). A persistent imbalance and TP receptor stimulation causes vascular hypertrophy and proliferation of vascular smooth muscle cells in TP receptor over-expressing or IP-receptor deletion mice (Citation164,Citation165). There is still an on-going debate about the origin of TxA2 in the vasculature, as TxA2 synthase inhibitors, unlike TP receptor antagonists, have no clear beneficial effects on endothelial function. This can be sufficiently explained by an early hypothesis by Gryglewski et al. (Citation166). Dysfunctional or activated endothelium may luminally release PGH2, which can undergo transcellular conversion. PGH2 may then trigger the TP receptors on platelets, thus stimulating platelet-derived TxA2 synthesis, but can also cross the plasma membrane and be converted to TxA2 via platelet TxA2 synthase.
Activation of PGHS by peroxynitrite—the peroxide tone
In recent years, the endogenous regulation of the PGHS catalytic cycle by RNOS has raised controversy. It became quite apparent that •NO, •O2−, and their derivatives have a highly specific impact on PGHS catalysis. The model of the ‘peroxide tone’ describes the transient activation and maintenance of PGHS activity by peroxides. Peroxides convert the resting PGHS into its active form in which a ferric heme together with a neighboring tyrosine residue are converted to a ferryl (FeIV = O, Compound I) species and a tyrosyl radical. The interaction of this tyrosyl radical with the substrate arachidonic acid yields an arachidonyl radical, which interacts with molecular oxygen to form 15-hydroperoxy-prostaglandin-9,11-endoperoxide (PGG2) in a controlled reaction. The second step again utilizes the peroxidase activity of the enzyme to reduce PGG2 to the 15-hydroxy derivative PGH2. Although all peroxides can provide the peroxide tone for the PGHS catalytic cycle, peroxynitrite was shown to be the most potent endogenous species to activate and maintain PGHS catalysis (Citation124).
Interestingly, the peroxide tone for inducible PGHS-2 was reported about 10-fold lower (2 nM) than that for the constitutively expressed PGHS-1 (21 nM). This implies that PGHS-2, which is usually expressed under the conditions of elevated •NO and •O2− formation, is already activated after its expression.
The extent of activation, however, is highly dependent on the cell type, the balanced formation of •NO and •O2− to form peroxynitrite, and on cellular antioxidant capacity. In the case of PGHS activity our group has proven that a 1:1 ratio stimulates the enzyme maximally, although it is still unclear how the cell controls the concerted generation of equimolar concentrations of •NO and •O2− (Citation167). An excess of either radical will reduce PGHS activity by enhancing the decomposition of peroxynitrite without inducing posttranslational modifications of the enzyme. Uric acid and vitamin E, in particular γ-tocopherol, both of which are potent scavengers of peroxynitrite, can also lower the peroxide tone and PGHS activity.
The constitutively expressed PGHS-1 can be found in a wide variety of cell types and tissues, such as vascular endothelium or platelets. The considerably higher peroxide requirement for the activation of PGHS-1 could be considered as an effective regulatory mechanism to prevent uncontrolled prostanoid synthesis under resting conditions. Growing evidence is emerging that the concept of peroxynitrite as an activator of prostanoid synthesis is crucially involved in the increased formation of prostanoids observed in cardiovascular aging () (Citation168).
In the light of recent findings, the role of lipoxygenases both as a source of lipid peroxides and as a potential leak of RNOS has become important. Seiler and co-workers identified a signaling pathway linking oxidative stress, GPx4 (glutathione peroxidase-4), lipoxygenase-derived lipid peroxidation, and apoptosis-inducing factor-mediated cell death (Citation169). GPx4 inactivation was found to cause lipid peroxidation and cell death, mediated by 12/15 lipoxygenase. The identification of a redox-regulated pro-apoptotic signaling cascade may also turn out to be a promising therapeutic target in the future.
Free radicals and prostanoids in vascular aging
Aging is a multifactorially controlled process, and divergence in molecular findings might partly be due to the use of different models. Studies of cardiovascular aging in humans are often hampered by the fact that other risk factors (diabetes, hypertension, hypercholesterolemia) are also present. As an example, age-dependent alterations in kidney physiology may be caused by up-regulation of the renin–angiotensin system witnessed in hypertension. Angiotensin II (Ang II) is a very potent vasoactive mediator causing cardiovascular remodeling and hypertension. On a molecular basis, Ang II activates and increases the expression of vascular NADPH oxidases (NOX-1, -2, and -4 in endothelial cells, smooth muscle cells, and fibroblasts). Under these conditions NADPH oxidases represent the major •O2− source and contribute to reduction of free •NO. Persistent up-regulation of the renin–Ang II system eventually leads to endothelial dysfunction and triggers the mitogenic response in smooth muscle cells via the MAPK cascade leading to remodeling of the vasculature. Under the premise of ‘action and reaction’, the cell always reacts with compensation on the age-dependent ‘gain’ or ‘loss of function’ in physiological processes. The cellular capacity to compensate for these alterations by alternative pathways might add new knowledge to the understanding of the aging process.
Superoxide sources in aging and effects on •NO signaling
In agreement with the free radical hypothesis of aging, it is widely accepted that RNOS and in particular •O2− formation increase with age. Several posttranslational oxidative modifications of bio-macromolecules have been described and provide substantial evidence of on-going altered radical formation in the aged vasculature, but some of the assumed enzymatic •O2− sources still remain controversial ().
We and others have demonstrated that vascular •O2− formation mainly occurs in adventitial fibroblasts and endothelial cells (Citation131,Citation170,Citation171). Denudation of the endothelial cell layer decreases the detectable amount of •O2− in aged vessels compared with young controls (Citation170). In contrast, smooth muscle cells only contribute a minor amount, which might be due to a higher antioxidant capacity. The highest increase in peroxynitrite-derived nitrotyrosine formation can be found in mitochondria (4–5-fold increase), which points to the mitochondrial electron transport chain as the major age-dependent •O2− source. Uncoupled NOS-3 in the endothelium also seems to be a likely candidate, since NOS-3 is over-expressed in aged tissues (Citation170). The enzyme was proven to be active by confirmation of the conversion of L-arginine to L-citrulline, although the release of •NO was reduced in these vessels. However, other investigators have found a decrease in NOS-3 expression accounting for the reduced •NO bioavailability (Citation51,Citation172). In our opinion, mitochondrial •O2− formation will scavenge free •NO, forming peroxynitrite, as proven by enhanced protein nitration, and might lead to oxidation of the NOS co-factor BH4. This could then, to some extent, uncouple NOS-3, further amplifying and aggravating the •O2− formation in the endothelium. In order to maintain vascular homeostasis, the endothelium reacts with an increase in NOS-3 expression partially to compensate the loss of •NO, eventually leading to a vicious cycle, and thereby representing a futile mechanism to prevent endothelial dysfunction.
There are several reports indicating that vascular NADPH oxidases are major contributors to enhanced •O2− formation (Citation19,Citation171). These data often should be interpreted with caution in those cases in which lucigenin-enhanced chemiluminescence together with the addition of NADPH as substrate had been used. This is because the non-specific autoxidation of NADPH by membrane-localized flavoproteins may result in measurement of artifacts. However, the suitability of lucigenin below 50 μM as a highly specific probe for •O2− was demonstrated, by comparison, with high-performance liquid chromatography (HPLC)-based hydroxyethidium measurements and electron paramagnetic resonance (EPR) measurements (Citation67,Citation144,Citation173).
Immunohistochemical localizations are mostly restricted to one subunit of the NADPH oxidase complex, e.g. p22phox, demonstrating a marginal alteration only (Citation171). In contrast, in aged hypertensive animals a significant increase can be observed, related to the well known age-dependent increase in the activation of the renin–Ang II system (Citation18,Citation19). Using quantitative real-time PCR, no significant transcriptional alterations induced by aging could be detected, neither in the mouse (NOX-1, NOX-2, NOX-4, p47phox, p67phox; unpublished data) nor in the rat model (protein kinase C translocation, p47phox, p67phox) (Citation174). However, the possibility of an enhanced activation during the aging process will need further investigation.
The proteasome, which is responsible for the removal of oxidatively damaged proteins accumulating during cellular aging, is itself affected by oxidative stress. Consequently, its activity declines in an age-related manner (Citation175,Citation176), although, in contrast to the cytosolic proteasome the nuclear proteasome is relatively resistant to this age-dependent decline (Citation177). These changes, however, are still as of yet unconfirmed in the VSMC proteasome (Citation178).
Peroxide tone in aging and its impact on the prostanoid signaling cascade
Most studies of vascular disease and aging demonstrate that a decline in endothelium-derived bioavailable •NO, an increase in vascular oxidants and PGHS-2 expression, along with an elevation and shift in the prostanoid spectrum lead to vasomotor dysfunction. Endothelium-derived constriction factors (EDCF), which result from PGHS metabolism and include PGH2, PGF2α, or TxA2, appear to be central in mediating age-dependent vasoconstriction, vascular stiffness, and hypertrophy. The identity of EDCF is controversial, but the effects are blocked by TxA2/PGH2 receptor antagonists such as SQ-29548 (Citation179) and terutroban (Citation180), or removal of the endothelium (Citation180). This shift in the prostanoid pattern, which seems to be mainly mediated by the endothelium, can be explained by PGHS-2 expression and the inhibition of prostacyclin synthase through peroxynitrite-mediated nitration. In addition, the elevated levels of RNOS and the lower peroxide tone requirement to activate PGHS-2 would explain the increase in prostanoid levels—in particular, that of PGE2, and EDCF. PGE2 is a pro-inflammatory mediator that has been linked to a variety of age-associated diseases such as immune senescence, arthritis, and cardiovascular disease including atherosclerosis (Citation181). This concept of the peroxide tone was proven in macrophages derived from young and old rats (Citation182). Stimulated macrophages from mature rats have a significantly higher release of PGE2 as compared with younger animals. An up-regulation of PGE2 is believed to cause dysregulation of the immune function (Citation183), up-regulation of cellular adhesion molecules on the endothelium such as P-selectin (Citation184), smooth muscle cell proliferation (hypertrophy and vascular stiffness), and sustained cell senescence (Citation185).
Furthermore, Vanhoutte and co-workers (Citation180) noticed that PGHS-2 expression increased with age in the endothelium of rat femoral arteries, which leads to the release of endothelium-dependent vasoconstricting prostaglandins and activation of the TxA2/PGH2 receptor. In parallel, catalase activity in the endothelium increased, and formation of reactive oxygen species was demonstrated as well. These findings further suggest that an age-dependent imbalance of vasoconstrictive mediators acutely affects vasotone but also results in remodeling and stiffening of the vessel wall by promoting hypertrophy (Citation186). Therefore, PGHS inhibitors, including aspirin, may not only lower the risk of vascular occlusion, but could also improve vascular stiffness.
In addition, other PGHS-derived and lipid peroxidation products including 4-hydroxynoneal, malondialdehyde, or the PGD2-derived 15d-PGJ2 are increased by RNOS and can also promote ROS generation in mitochondria of the endothelium (Citation187). These products have been shown to increase in inflammation, atherosclerosis, and aging and are known to form protein adducts by reacting with histidine or cysteine residues. At low concentrations, such electrophilic compounds can regulate protein expression and activity. However, continuous exposure and high abundance may lead to non-specific and irreversible protein modifications that accumulate as lipid-modified protein aggregates within the cell. A reason for the accumulation is that heavily oxidized and lipidated proteins cannot be further processed and degraded by the ubiquitin/proteasome system.
A prime example for the regulatory role of electrophilic compounds or RNOS is the Keap1/Nrf2 complex that controls the antioxidant- or electrophile-response element (ARE) (Citation188). Various important genes for type II detoxification of RNOS and electrophiles are under control of this element, including SODs and glutathione metabolism (Citation189). Reactive electrophiles such as 15d-PGJ2 or RNOS react with a critical cysteine residue on Keap1, leading to the dissociation of Nrf2 and to ubiquitination and degradation of Keap1 (Citation190). Free Nrf2 translocates to the nucleus, heterodimerizes with Maf protein, and activates the AREs. This example illustrates that at low concentrations the RNOS- and PGHS-derived electrophilic 15d-PGJ2 acts as a signaling molecule that leads to the induction of cytoprotective genes. These examples suggest that the aging process may be counterbalanced by the induction of PGHS-2 to release more PGI2 from the endothelium and smooth muscle cells. Increased, RNOS that support the peroxide tone for PGHS and the formation of electrophilic side products initially induce cytoprotective mechanisms. However, these cascades become futile with age, and the over-activation may lead to hyperoxidized proteins by RNOS or electrophilic lipid oxidation products. This in turn leads to progressive deterioration of multiple signaling cascades including Nrf2 (Citation191,Citation192), AP-1, NF-kB (Citation193,Citation194), FoxO (forkhead box O) (Citation178) and may be the cause of aging. Nevertheless, these examples also underline the challenges for aging research to decipher what is cause and what is consequence.
Age-dependent compensatory mechanisms
Platelets are a major source of pro-aggregatory TxA2 in the vascular system. The aging process is associated with an increased risk of thrombus formation which can ultimately lead to capillary plugging and infarction. Platelets from elderly people are more sensitive to various stimuli (Citation195). Furthermore, this increased susceptibility to stimulation is accompanied by an increased basal formation of TxA2 in aged individuals. We recently demonstrated peroxynitrite-mediated activation of PGHS-1 and subsequent TxA2 formation and aggregation in human platelets. Sources of free radicals in platelets include NADPH oxidase, mitochondria (Citation196), xanthine oxidase (Citation197), and uncoupled NOS-3. It could be shown that an age-dependent increase in •O2− formation in platelets leads to an increased formation of peroxynitrite (Schildknecht et al., unpublished). From this interaction, two consequences arise: initially, a certain portion of anti-aggregatory •NO, which is released in small quantities under basal conditions and which can be up-regulated following stimulation, is trapped to form peroxynitrite. Secondly, the formed peroxynitrite provides the peroxide tone for PGHS-1 in platelets, thus supporting TxA2 formation and ultimately aggregation. Interestingly, it was also observed that platelets of aged individuals are more sensitive to PGI2 (Bachschmid et al., unpublished). This may represent a compensatory mechanism to counteract the increased platelet aggregability in aging (). This concept is further supported by findings of increased 6-keto-PGF1α levels in the plasma of elderly people (Citation198), presumably originating from the vascular endothelium by a likewise elevated peroxide tone. Since endothelial PGI2 synthase was shown to become nitrated and inactivated in an age-dependent manner, this event may represent a turning-point from counter-regulation to a state accompanied by a high risk of thrombotic events.
Figure 4. Free radical-mediated platelet hyperreactivity in aging. Platelets use peroxynitrite at low concentrations (< 21 nM) in order to provide the peroxide tone for modulating basal prostaglandin endoperoxide H2 synthase (cyclo-oxygenase-1) activity after agonist-mediated platelet activation. NADPH oxidase and endothelial NO synthase concertedly release superoxide anion and •NO to form peroxynitrite. Cyclo-oxygenase-1 provides the substrate prostaglandin endoperoxide H2 for thromboxane synthase, which is converted into thromboxane A2. This potent eicosanoid further activates platelets in an autocrine loop or causes the recruitment of other platelets to the thrombus. However, aging leads to an elevated superoxide formation via uncoupling of NO synthase, conversion of xanthine dehydrogenase into an oxidase, activation of NADPH oxidase, or by electron leakage in the mitochondrial electron transport chain. This saturates the platelet peroxide tone, resulting in loss of the modulation of COX-1 activity, which subsequently leads to hyperreactivity of platelets. Therefore, platelet aggregation readily occurs at lower doses of agonists and proceeds faster. (Ag = agonist; Rec = receptor; PLA2 = phospholipase A2; PGHS-1 = prostaglandin endoperoxide H2 synthase; AA = arachidonic acid; PGH2 = prostaglandin endoperoxide H2; TxAS = thromboxane synthase; ONOO− = peroxynitrite; TxA2 = thromboxane A2; [Ca2 + ]i = intracellular calcium; •NO = nitric oxide; •O2− = superoxide)
![Figure 4. Free radical-mediated platelet hyperreactivity in aging. Platelets use peroxynitrite at low concentrations (< 21 nM) in order to provide the peroxide tone for modulating basal prostaglandin endoperoxide H2 synthase (cyclo-oxygenase-1) activity after agonist-mediated platelet activation. NADPH oxidase and endothelial NO synthase concertedly release superoxide anion and •NO to form peroxynitrite. Cyclo-oxygenase-1 provides the substrate prostaglandin endoperoxide H2 for thromboxane synthase, which is converted into thromboxane A2. This potent eicosanoid further activates platelets in an autocrine loop or causes the recruitment of other platelets to the thrombus. However, aging leads to an elevated superoxide formation via uncoupling of NO synthase, conversion of xanthine dehydrogenase into an oxidase, activation of NADPH oxidase, or by electron leakage in the mitochondrial electron transport chain. This saturates the platelet peroxide tone, resulting in loss of the modulation of COX-1 activity, which subsequently leads to hyperreactivity of platelets. Therefore, platelet aggregation readily occurs at lower doses of agonists and proceeds faster. (Ag = agonist; Rec = receptor; PLA2 = phospholipase A2; PGHS-1 = prostaglandin endoperoxide H2 synthase; AA = arachidonic acid; PGH2 = prostaglandin endoperoxide H2; TxAS = thromboxane synthase; ONOO− = peroxynitrite; TxA2 = thromboxane A2; [Ca2 + ]i = intracellular calcium; •NO = nitric oxide; •O2− = superoxide)](/cms/asset/6356a6ff-cf80-4706-83ff-7ac12af05858/iann_a_645498_f0004_b.jpg)
Vitamins and the role of antioxidant enzymes in vascular aging
Based upon the oxidative stress hypothesis of vascular aging, it would seem particularly attractive to prevent the production of RNOS and their subsequent reactions with naturally occurring antioxidants, i.e. vitamins, and/or other antioxidative substances. Prevention of oxidative stress depends on a fully functional endogenous system, so that a loss of endogenous antioxidants and antioxidant enzymes, even without a change in RNOS formation, would increase oxidative stress and demand for dietary antioxidant supplementation. The beneficial effects of dietary antioxidants to extend lifespan are, however, highly controversial. For example the biological antioxidant vitamin E (tocopherol), which is an efficient scavenger of lipid-derived radicals and an electron source for the reduction of peroxynitrite, is reduced in rat serum (Citation199). In contrast, a dramatic increase of α-tocopherol, its most biologically active form, was found in the aortic vessel wall of aged rats fed a normal diet (Citation199). This appears to be a compensatory mechanism for age-associated oxidative stress and underscores that additional vitamin E supplementation may be obsolete since the cell is already utilizing the antioxidant to its limit.
Nevertheless, various studies could demonstrate that vitamin supplementation improves age-related diseases. In an aging model of rat macrophages, vitamin E reduced PGHS-2 activity by lowering the peroxide tone, which in turn reduced immune senescence and dysfunction (Citation182) caused by PGE2. Meydani and co-workers further developed this concept and showed in a human trial that vitamin E supplementation improved the T cell-mediated immune response in elderly over 65. Similarly, vitamin E attenuated the expression of adhesion molecules in endothelial cells, which participate in the innate immune response and slowed down the progression of atherosclerosis (Citation200,Citation201). Vitamin E also reduced the expression of genes leading to cardiac hypertrophy (Citation202) in old mice.
SODs are the main scavengers of •O2− and best studied antioxidant system in the vasculature. Three different isoenzymes exist: manganese SOD (MnSOD) is the major antioxidant enzyme in mitochondria of all mammals (see below). EC-SOD is the main scavenger of •O2− in the extracellular space (Citation203). Cu,ZnSOD, normally localized in the cytosol, was recently shown to relocate to mitochondria in an age-dependent manner (Citation204), thus becoming part of a compensatory defense against compromised mitochondrial function. The importance of these enzymes, in particular Cu,ZnSOD and MnSOD, for vascular homeostasis was demonstrated by gene ablation in mice, which significantly impaired endothelium-dependent relaxation (Citation205,Citation206). Various strategies in the vasculature exist that target the restoration of endothelium-dependent •NO formation by scavenging the antagonizing superoxide or preventing NOS-3 from age-dependent uncoupling by restoration of BH4 levels with sepiapterin (Citation207). SOD- mimetics (Citation208), peroxynitrite decomposition catalysts (Citation209), and over-expression of EC-SOD improved vascular relaxation in old mice or rats (Citation210).
Interestingly, the long-lived white-footed house mouse (Peromyscus leucopus) has unchanged levels of Cu,ZnSOD and MnSOD expression levels compared to the typical mouse (Mus musculus) but increased levels of glutathione peroxidase-1, catalase, and heme oxygenase that are associated with reduction of hydrogen peroxide (Citation211).
The most complex antioxidant and detoxification systems in the cell are glutathione and glutathione utilizing proteins including peroxiredoxins, thioredoxin, glutaredoxins, glutathione peroxidases, and glutathione-S transferases. Effects on these systems by aging are far more complex, vary among the tissues, and are less studied in the cardiovascular system. Thioredoxin (Trx), which is a multifunctional enzyme that has a chaperone function, modulates the signaling function of thiol-controlled transcription factors, including NF-kB and AP-1, and has an antioxidant function by reducing intra- and intermolecular protein disulfides. Over-expression of cytosolic Trx in mice (Citation212) extended median and maximum lifespans, increased stress resistance, and enhanced telomerase activity. It has also been observed that N-acetylcysteine, which can replete cellular glutathione levels, prevented hydrogen peroxide-induced telomere shortening and endothelial cell senescence (Citation52). From the study of Trx transgenics it is not clear whether Trx enhanced the lifespan by its antioxidant function or by influencing important signaling cascades.
Another approach in aging research is to eliminate highly reactive radical species such as the •OH, which is derived from Fenton chemistry via free iron and other transition metals. One such approach utilized cardiac specific metallothionin transgenic mice (Citation213, Citation214). This low-molecular-weight enzyme is a chelator for free metal ions such as iron and copper, and almost 30% of its amino acid residues are comprised of cysteines. This potent thiol antioxidant may have contributed and alleviated the aging-induced cardiac contractile defects leading to a 4-month lifespan extension in the transgenic mouse model.
Mitochondria: the seed of senescence?
Mitochondrial antioxidants
Cardiovascular diseases are associated with a chronic increase in mitochondrial ROS formation. Under conditions such as aging, an increased amount of electrons is misdirected from the respiratory chain into ROS production. An age-associated decline of enzymatic function normally responsible for the detoxification of ROS is one of the most important reasons. This has been shown for aconitase (Citation215) and MnSOD (Citation170). Mice lacking MnSOD exhibit increased mitochondria-derived oxidative stress and decreased mitochondrial function (Citation94). Nitration of an essential tyrosine residue in MnSOD, which causes inactivation of the enzyme, may lead to subsequent peroxynitrite formation and has been associated with age-related endothelial dysfunction (Citation67,Citation170). Genetic inactivation of MnSOD results in premature death, while treatment with the SOD mimetic manganese-tetrakis-benzoic acid porphyrin (MnTBAP) dramatically prolongs survival of MnSOD−/− mice (Citation90). Conversely, MnSOD over-expression increased yeast chronological lifespan (Citation216), and a synthetic SOD/catalase mimetic extended lifespan in C. elegans (Citation208). Furthermore, over-expression of catalase, another enzyme crucially involved in the antioxidative defense, enhanced protection of mitochondria from ROS and led to an extended lifespan in mice (Citation68). Interestingly, compared with catalase targeted to peroxisomes and to the nucleus, catalase targeted to mitochondria maximally increases lifespan (Citation68,Citation217), thus strongly supporting the role of ROS derived from mitochondria as a pivotal limiting factor of longevity in rodents. Even in cardiac specific transgenic mice, catalase over-expression still extended lifespan and attenuated or even nullified various aging-induced myocyte defects (Citation218). For other mitochondrial antioxidant enzymes little information on the vasculature is available. We could recently demonstrate that ALDH-2−/− (aldehyde dehydrogenase) mice develop severe endothelial dysfunction at medium age (Citation67). This enzyme removes reactive aldehydes in the mitochondria and was shown to activate organic nitrites that lead to vasorelaxation. Moreover, the enzyme contains various reactive cysteines, which also may exert peroxidase activity and antioxidant activity.
Based upon a better understanding of the role of mitochondria in the aging process, specifically designed therapies to delay aging may have a promising future.
Mitochondrial DNA and network dynamics
In the past, it was believed that mitochondrial DNA (mtDNA) must have a much higher mutation rate due to its plasmid-like appearance, the absence of efficient DNA repair systems, and histones. Recent research, however, demonstrates that mtDNA is organized in protein-DNA complexes in which 2–8 DNA molecules form a nucleoid. The mitochondrial transcription factor A (TFAM) plays a dual role of controlling DNA transcription and replication as well as a histone-like protein. Moreover, nucleoids are tethered to the inner mitochondrial membrane in close proximity to the respiratory chain, which suggests that mtDNA and the respiratory chain build a functional cluster or super-complexes (Citation219,Citation220). We have recently shown that MnSOD is an integral component of the nucleoids in VSMCs, where it can protect mtDNA from RNOS. In addition, MnSOD is acetylated (Citation221), which might modulate its association with mtDNA within the nucleoids as well as activity. One of the most active deacetylases in the mitochondria is sirtuin 3, which has been shown to deacetylate MnSOD and regulate its activity (Citation222,Citation223). A polymorphism of Sirt3 was demonstrated in humans to correlate with centenarians (Citation224).
Since mtDNA solely encodes components of the respiratory chain (components of all complexes except for complex II, which is exclusively encoded by nuclear genes) a deterioration of the mtDNA would result in a dysfunctional respiratory chain, which could be detected by the cell machinery for degradation. In fact, vascular mitochondria are organized into dynamic networks that undergo constant fission and fusion. This network structure is important during cell division, since mitochondria as well as mtDNA can be distributed to the daughter cell, which is also known as mtDNA segregation. However, single particle-like mitochondria with spherical to tubular appearance can be formed. These mitochondria have variable membrane potentials, which is an indicator for the integrity of respiratory chain complexes and consequently of the mtDNA. It has been observed that such mitochondria are removed by a specialized lysosomal pathway of autophagy, known as mitophagy (Citation225,Citation226). Some researchers believe that this pathway is important for maintaining functional mtDNA and mitochondria, thus preventing aging. Indeed, we have observed that the network in umbilical vein endothelial cells is more dynamic than in aortic endothelial cells (unpublished observation) (Citation227). Also, RNOS, hyperglycemia, and other xenobiotics that may oxidatively damage the DNA were shown to disrupt the mitochondrial network. This probably enables the cell immediately to identify and remove damaged mitochondria and mtDNA. Such processes are of particular importance for low-dividing cells such as endothelial cells or postmitotic tissues such as the myocardium (Citation100). In these tissues, mtDNA lesions such as major deletions or point mutations have been observed with aging, whereas somatic cells that undergo frequent cell divisions seem to maintain and remove damaged mtDNA molecules during the cell cycle.
Metabolism, mitochondria, and sirtuins
Caloric restriction is a simple and universal intervention in the aging process, which has shown effectiveness from yeast up to mammals (Citation228). In the past decade, sirtuins, a group of class III NAD + -dependent deacetylases and mono-ADP-ribosylases, have gained interest as master regulators in metabolism, aging (Citation229,Citation230), and mitochondrial function. Sirt1, currently the most studied isoform, can directly influence telomere length and reduces senescence (Citation231) but has also a significant impact on metabolism. In this context, the French paradox, which is the consumption of a fat-rich diet in combination with red wine, has surprisingly few adverse consequences in the development of obesity, metabolic syndrome, and aging. Resveratrol, a polyphenol that was isolated from red wine, appeared to be the major active compound in mediating beneficial effects analogous to those induced by caloric restriction. Sinclair and co-workers demonstrated that sirtuins (Citation232,Citation233), in particular Sirt1, also known as Sir2, are the pharmacological targets of resveratrol and executers of caloric restriction in mammals. Currently, this concept has been questioned, since newer Sirt1 activators as well as resveratrol may not be direct activators of Sirt1 after all (Citation234). Nevertheless, improvements in type II diabetes, atherosclerosis (Citation235), angiogenesis (Citation236), and activation of NOS-3 (Citation237,Citation238) due to resveratrol-dependent up-regulation of Sirt1 and compensation of Sirt1 deletion in endothelial cells (Citation239,Citation240) have been demonstrated. Additional targets of resveratrol include Nrf2, which up-regulates mitochondrial genes such as NAD(P)H:quinone oxidoreductase-1 and MnSOD or other antioxidant enzymes including γ-glutamylcysteine synthetase and heme oxygenase (Citation241), all of which can counteract the aging process. As already mentioned, resveratrol was shown to activate NOS-3, probably via Sirt1, and promoted mitochondrial biogenesis leading to improved metabolism in aging and caloric restriction-like effects. Besides resveratrol, over-expression of Sirt1 showed beneficial effects in disease and aging by up-regulation of mitochondrial genes via FoxO1 and PGC-1α. This leads to an increase in the cellular mitochondrial mass as well as induction of antioxidant enzymes such as MnSOD that protects against oxidative damage and promotes molecular activation (Citation242,Citation243) of caloric restriction.
Summary
Our mechanistic model of vascular aging combines concepts from different theories, especially the oxidative stress hypothesis and the mitochondrial theory of aging, with a more extended and modernized view based on ideas partly yet to be proven.
Enhanced formation of RNOS is a central regulator of vascular aging which is mainly characterized by endothelial dysfunction. The sources of increased •O2− production involve the endothelial NADPH oxidase system, mitochondrial respiratory chain components, and NOS-3. Age-associated up-regulation of NOS-3 may lead to either a scavenger mechanism of •O2− or as a source of •O2− itself, especially in the absence of co-factors. The mechanisms leading to •O2− generation in the mitochondria, the cytoplasm, and at the cell membrane may well occur in parallel without a special order or sequence, and, in the end, it is irrelevant which is the first to trigger vascular dysfunction in aging. The link between increased •O2− production and decreased •NO bioavailability to form peroxynitrite is stringent in view of the chemistry, kinetics, and diffusion properties of both free radicals. The most significant generation of peroxynitrite occurs within endothelial mitochondria. Nitration and inactivation of key enzymes of antioxidant defense means that counterbalancing mechanisms cannot be preserved with increasing age. However, it has also to be pointed out that peroxynitrite is an important signaling molecule as exemplified by the fact that PGH2 synthase activity is maintained by peroxides.
Increased generation of •O2− and peroxynitrite entails increased nuclear and mitochondrial DNA damage. Oxidative DNA lesions promote mutation and dysfunction of respiratory chain components, thus leading to yet more RNOS generation and hence vascular dysfunction. We have recently shown that MnSOD is an integral component of the nucleoids in VSMCs, where it can protect mtDNA from RNOS. MnSOD can be acetylated which may modulate its association with mtDNA within the nucleoids. Mitochondria harbor three isoforms of sirtuins, master switches in metabolism which may, by deacetylation, increase MnSOD association with mtDNA. Age-associated increased production of peroxynitrite has been shown to inactivate Sirt1, suggesting that sirtuins may age-dependently lose their ability to modulate, by acetylation/deacetylation, key enzymes involved in antioxidative defense.
The dysregulation of various cellular signaling pathways in conjunction with oxidative stress induces cellular senescence. In particular for the endothelium, senescent cells may have a negative impact on neighboring cells, furthering endothelial dysfunction. Cellular senescence can be induced by stress or by shortening of indispensable telomeres on nuclear chromosomes.
Studying the molecular basis of the link between DNA damage and vascular dysfunction will help better to understand and ultimately to treat vascular aging.
Declaration of interest: This work was supported by NIH grants PO1 HL 068758, R37 HL104017, K08 HL71563, NIH pre-doctoral training grant HL007969-06A1, as well as by NHLBI, National Institutes of Health, Department of Health and Human Services under Contract No. HHSN268201000031C. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the awarding offices. M. M. Bachschmid and S. Schildknecht contributed equally to the writing of the manuscript.
References
- Kelly DT. Paul Dudley White International Lecture. Our future society. A global challenge. Circulation. 1997;95:2459–64.
- Burke GL, Evans GW, Riley WA, Sharrett AR, Howard G, Barnes RW, . Arterial wall thickness is associated with prevalent cardiovascular disease in middle-aged adults. The Atherosclerosis Risk in Communities (ARIC) Study. Stroke. 1995;26:386–91.
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66.
- Cohen RA, Adachi T. Nitric-oxide-induced vasodilatation: regulation by physiologic s-glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends Cardiovasc Med. 2006;16:109–14.
- Xu S, Ying J, Jiang B, Guo W, Adachi T, Sharov V, . Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol. 2006;290:H2220–7.
- Bachschmid M, Schildknecht S, Ullrich V. Redox regulation of vascular prostanoid synthesis by the nitric oxide-superoxide system. Biochem Biophys Res Commun. 2005;338:536–42.
- Lüscher TF, Vanhoutte PM. The endothelium: modulator of card?ovascular function. Boca Raton, FL, USA:CRC Press; 1991.
- Vanhoutte PM. Ageing and endothelial dysfunction. Eur Heart J Supplements. 2002;49:A8–17.
- Bachschmid M, Thurau S, Zou MH, Ullrich V. Endothelial cell activation by endotoxin involves superoxide/NO-mediated nitration of prostacyclin synthase and thromboxane receptor stimulation. FASEB J. 2003;17:914–6.
- Prescott SM, McIntyre TM, Zimmerman GA. Events at the vascular wall: the molecular basis of inflammation. J Investig Med. 2001;49:104–11.
- van der Loo B, Schildknecht S, Zee R, Bachschmid MM. Signalling processes in endothelial ageing in relation to chronic oxidative stress and their potential therapeutic implications in humans. Exp Physiol. 2009;94:305–10.
- Bhagat K, Moss R, Collier J, Vallance P. Endothelial “stunning” following a brief exposure to endotoxin: a mechanism to link infection and infarction? Cardiovascular research. 1996;32:822–9.
- Vallance P, Collier J, Bhagat K. Infection, inflammation, and infarction: does acute endothelial dysfunction provide a link? Lancet. 1997;349:1391–2.
- Frein D, Schildknecht S, Bachschmid M, Ullrich V. Redox regulation: a new challenge for pharmacology. Biochem Pharmacol. 2005;70:811–23.
- Virdis A, Ghiadoni L, Giannarelli C, Taddei S. Endothelial dysfunction and vascular disease in later life. Maturitas. 2010;67:20–4.
- Gaynor E, Bouvier C, Spaet TH. Vascular lesions: possible pathogenetic basis of the generalized Shwartzman reaction. Science. 1970;170:986–8.
- Gerhard M, Roddy MA, Creager SJ, Creager MA. Aging progressively impairs endothelium-dependent vasodilation in forearm resistance vessels of humans. Hypertension. 1996;27:849–53.
- Wang M, Takagi G, Asai K, Resuello RG, Natividad FF, Vatner DE, . Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41:1308–16.
- Basso N, Paglia N, Stella I, de Cavanagh EM, Ferder L, del Rosario Lores Arnaiz M, . Protective effect of the inhibition of the renin-angiotensin system on aging. Regul Pept. 2005;128:247–52.
- Oudot A, Martin C, Busseuil D, Vergely C, Demaison L, Rochette L. NADPH oxidases are in part responsible for increased cardiovascular superoxide production during aging. Free Radic Biol Med. 2006;40:2214–22.
- Gaballa MA, Jacob CT, Raya TE, Liu J, Simon B, Goldman S. Large artery remodeling during aging: biaxial passive and active stiffness. Hypertension. 1998;32:437–43.
- Zou M, Martin C, Ullrich V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol Chem. 1997;378:707–13.
- Viner RI, Ferrington DA, Huhmer AF, Bigelow DJ, Schoneich C. Accumulation of nitrotyrosine on the SERCA2a isoform of SR Ca-ATPase of rat skeletal muscle during aging: a peroxynitrite-mediated process? FEBS Lett. 1996;379:286–90.
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621.
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–13.
- Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell. 1993;73:347–60.
- Dunham MA, Neumann AA, Fasching CL, Reddel RR. Telomere maintenance by recombination in human cells. Nat Genet. 2000;26:447–50.
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, . Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34.
- Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee HW, Blasco MA. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J. 1999;18:2950–60.
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–74.
- Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet. 2008;124:369–77.
- Mason PJ, Wilson DB, Bessler M. Dyskeratosis congenita—a disease of dysfunctional telomere maintenance. Curr Mol Med. 2005;5:159–70.
- Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 1997; 3:1271–4.
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, . Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–52.
- Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–74.
- van der Loo B, Fenton MJ, Erusalimsky JD. Cytochemical detection of a senescence-associated beta-galactosidase in endothelial and smooth muscle cells from human and rabbit blood vessels. Exp Cell Res. 1998;241:309–15.
- Sherr CJ, DePinho RA. Cellular senescence: mitotic clock or culture shock? Cell. 2000;102:407–10.
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602.
- Sato I, Morita I, Kaji K, Ikeda M, Nagao M, Murota S. Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cell. Biochem Biophys Res Commun. 1993;195:1070–6.
- Nakajima M, Hashimoto M, Wang F, Yamanaga K, Nakamura N, Uchida T, . Aging decreases the production of PGI2 in rat aortic endothelial cells. Exp Gerontol. 1997;3?:685–93.
- Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol. 2005;40:634–42.
- Chen J, Goligorsky MS. Premature senescence of endothelial cells: Methusaleh's dilemma. Am J Physiol Heart Circ Physiol. 2006;290:H1729–39.
- Rivard A, Fabre JE, Silver M, Chen D, Murohara T, Kearney M, . Age-dependent impairment of angiogenesis. Circulation. 1999;99:111–20.
- Toussaint O, Medrano EE, von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol. 2000;35:927–45.
- Drayton S, Peters G. Immortalisation and transformation revisited. Curr Opin Genet Dev. 2002;12:98–104.
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9.
- Colavitti R, Finkel T. Reactive oxygen species as mediators of cellular senescence. IUBMB Life. 2005;57:277–81.
- Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8:131–44.
- Chen J, Brodsky SV, Goligorsky DM, Hampel DJ, Li H, Gross SS, . Glycated collagen I induces premature senescence-like phenotypic changes in endothelial cells. Circ Res. 2002;90:1290–8.
- Vasa M, Breitschopf K, Zeiher AM, Dimmeler S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res. 2000; 87:540–2.
- Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res. 2001;89:793–8.
- Haendeler J, Hoffmann J, Diehl JF, Vasa M, Spyridopoulos I, Zeiher AM, . Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ Res. 2004;94:768–75.
- Scalera F, Borlak J, Beckmann B, Martens-Lobenhoffer J, Thum T, T ger M, Bode-B ger SM. Endogenous nitric oxide synthesis inhibitor asymmetric dimethyl L-arginine accelerates endothelial cell senescence. Arterioscler Thromb Vasc Biol. 2004;24:1816–22.
- Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, . Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol. 2009;297:H1673–84.
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300.
- Harman D. Free radical theory of aging: an update: increasing the functional life span. Ann N Y Acad Sci. 2006;1067:10–21.
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244:6049–55.
- Muller FL, Lustgarten MS, Ja?g Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503.
- Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci USA. 2004;101:4003–8.
- Klumpp G, Schildknecht S, Nastainczyk W, Ullrich V, Bachschmid M. Prostacyclin in the cardiovascular system: new aspects and open questions. Pharmacol Rep. 2005;57 Suppl:120–6.
- Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–4.
- Brunk UT, Jones CB, Sohal RS. A novel hypothesis of lipofuscinogenesis and cellular aging based on interactions between oxidative stress and autophagocytosis. Mutat Res. 1992;275:395–403.
- Sohal RS, Marzabadi MR, Galaris D, Brunk UT. Effect of ambient oxygen concentration on lipofuscin accumulation in cultured rat heart myocytes—a novel in vitro model of lipofuscinogenesis. Free Radic Biol Med. 1989;6:23–30.
- Gil P, Farinas F, Casado A, Lopez-Fernandez E. Malondialdehyde: a possible marker of ageing. Gerontology. 2002;48:209–14.
- Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–30.
- Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14:312–8.
- Wenzel P, Schuhmacher S, Kienhofer J, Muller J, Hortmann M, Oelze M, . Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–9.
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, . Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11.
- Landis GN, Tower J. Superoxide dismutase evolution and life span regulation. Mech Ageing Dev. 2005;126:365–79.
- Ladiges W, Van Remmen H, Strong R, Ikeno Y, Treuting P, Rabinovitch P, . Lifespan extension in genetically modified mice. Aging Cell. 2009;8:346–52.
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–7.
- Hartman P, Ponder R, Lo HH, Ishii N. Mitochondrial oxidative stress can lead to nuclear hypermutability. Mech Ageing Dev. 2004;125:417–20.
- Miwa S, Brand MD. The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim Biophys Acta. 2005;1709:214–9.
- Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–7.
- Jacobs HT, Lehtinen SK, Spelbrink JN. No sex please, we're mitochondria: a hypothesis on the somatic unit of inheritance of mammalian mtDNA. Bioessays. 2000;22:564–72.
- O’Rourke B. Pathophysiological and protective roles of mitochondrial ion channels. J Physiol. 2000;529 Pt 1:23–36.
- Romashko DN, Marban E, O’Rourke B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proc Natl Acad Sci USA. 1998;95:1618–23.
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, . The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–96.
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–53.
- Duchen MR, Leyssens A, Crompton M. Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J Cell Biol. 1998;142:975–88.
- Riva A, Tandler B, Lesnefsky EJ, Conti G, Loffredo F, Vazquez E, . Structure of cristae in cardiac mitochondria of aged rat. Mech Ageing Dev. 2006;127:917–21.
- Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–9.
- Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–8.
- Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J Biol Chem. 1994; 269:29409–15.
- Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys. 1994; 308:89–95.
- Stachowiak O, Dolder M, Wallimann T, Richter C. Mitochondrial creatine kinase is a prime target of peroxynitrite-induced modification and inactivation. J Biol Chem. 1998;273:16694–9.
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2:324–9.
- Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci USA. 1988; 85:6465–7.
- Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–34.
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, . A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–63.
- Ohashi M, Runge MS, Faraci FM, Heistad DD.?MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2331–6.
- Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. 2007; 27:1941–6.
- Strassburger M, Bloch W, Sulyok S, Schuller J, Keist AF, Schmidt A, . Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med. 2005;38:1458–70.
- Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+ /-) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci USA. 2001;98:2278–83.
- Wei YH. Oxidative stress and mitochondrial DNA mutations in human aging. Proc Soc Exp Biol Med. 1998;217:53–63.
- Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108.
- van der Loo B, Lüscher TF. Endothelium-mediated signaling and vascular aging. In: Hagen, T, editor. Mechanisms of cardiovascular aging. Advances in Cell Aging and Gerontology. Vol. 11. Amsterdam: Elsevier Science; 2002. p. 127–44.
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47.
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95.
- Corral-Debrinski M, Shoffner JM, Lott MT, Wallace DC. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat Res. 1992;275:169–80.
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, . Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23.
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, . Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–4.
- Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SS, Green EC, . Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab Invest. 2007;87:326–35.
- Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, . Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–9.
- Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res. 2006;99:924–32.
- Bonawitz ND, Shadel GS. Rethinking the mitochondrial theory of aging: the role of mitochondrial gene expression in lifespan determination. Cell Cycle. 2007;6:1574–8.
- Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, . Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–65.
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–6.
- Korbut R, Trabka-Janik E, Gryglewski RJ. Cytoprotection of human polymorphonuclear leukocytes by stimulators of adenylate and guanylate cyclases. Eur J Pharmacol. 1989;165:171–2.
- Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;2:1057–8.
- Marks DS, Vita JA, Folts JD, Keaney JF Jr, Welch GN, Loscalzo J. Inhibition of neointimal proliferation in rabbits after vascular injury by a single treatment with a protein adduct of nitric oxide. J Clin Invest. 1995;96:2630–8.
- Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35:430–42.
- Pfitzer G. Invited review: regulation of myosin phosphorylation in smooth muscle. J Appl Physiol. 2001;91:497–503.
- Loirand G, Guilluy C, Pacaud P. Regulation of Rho proteins by phosphorylation in the cardiovascular system. Trends Cardiovasc Med. 2006;16:199–204.
- Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem. 2004;279:36167–70.
- Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–94.
- Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–14.
- Raman CS, Martasek P, Masters BSS. Structural themes determining function in nitric oxide synthases. San Diego:Academic Press; 2000.
- Dashwood MR, Crump A, Shi-Wen X, Loesch A. Identification of neuronal nitric oxide synthase (nNOS) in human penis: a potential role of reduced neuronally-derived nitric oxide in erectile dysfunction. Curr Pharm Biotechnol. 2011 Apr 20. [Epub ahead of print]
- Wink DA, Miranda KM, Espey MG, Pluta RM, Hewett SJ, Colton C, . Mechanisms of the antioxidant effects of nitric oxide. Antioxid Redox Signal. 2001;3:203–13.
- Hummel SG, Fischer AJ, Martin SM, Schafer FQ, Buettner GR. Nitric oxide as a cellular antioxidant: a little goes a long way. Free Radic Biol Med. 2006;40:501–6.
- Wink DA, Cook JA, Kim SY, Vodovotz Y, Pacelli R, Krishna MC, . Superoxide modulates the oxidation and nitrosation of thiols by nitric oxide-derived reactive intermediates. Chemical aspects involved in the balance between oxidative and nitrosative stress. J Biol Chem. 1997; 272:11147–51.
- Schildknecht S, Bachschmid M, Ullrich V. Peroxynitrite provides the peroxide tone for PGHS-2-dependent prostacyclin synthesis in vascular smooth muscle cells. FASEB J. 2005;19:1169–71.
- Schildknecht S, van der Loo B, Weber K, Tiefenthaler K, Daiber A, Bachschmid MM. Endogenous peroxynitrite modulates PGHS-1- dependent thromboxane A2 formation and aggregation in human platelets. Free Radic Biol Med. 2008;45:512–20.
- Hogg N, Struck A, Goss SP, Santanam N, Joseph J, Parthasarathy S, . Inhibition of macrophage-dependent low density lipoprotein oxidation by nitric-oxide donors. J Lipid Res. 1995;36:1756–62.
- Hogg N, Kalyanaraman B, Joseph J, Struck A, Parthasarathy S. Inhibition of low-density lipoprotein oxidation by nitric oxide. Potential role in atherogenesis. FEBS Lett. 1993;334:170–4.
- Knapp LT, Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J Biol Chem. 2000;275:24136–45.
- Ullrich V, Namgaladze D, Frein D. Superoxide as inhibitor of calcineurin and mediator of redox regulation. Toxicol Lett. 2003;139:107–10.
- Gardner PR, Raineri I, Epstein LB, White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995;270:13399–405.
- Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–83.
- Adler A, Messina E, Sherman B, Wang Z, Huang H, Linke A, . NAD(P)H oxidase-generated superoxide anion accounts for reduced control of myocardial O2 consumption by NO in old Fischer 344 rats. Am J Physiol Heart Circ Physiol. 2003;285:H1015–22.
- Krause KH. Aging: a revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp Gerontol. 2007;42:256–62.
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, . Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003; 111:1201–9.
- Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109:817–26.
- Fleming I, Mohamed A, Galle J, Turchanowa L, Brandes RP, Fisslthaler B, . Oxidized low-density lipoprotein increases superoxide production by endothelial nitric oxide synthase by inhibiting PKCalpha. Cardiovasc Res. 2005;65:897–906.
- Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, . Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–65.
- Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, . S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–8.
- Zou MH, Cohen R, Ullrich V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium. 2004;11:89–97.
- Hink U, Li H, Mo?lnau H, Oelze M, Matheis E, Hartmann M, . Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–22.
- Alderman M, Aiyer KJ. Uric acid: role in cardiovascular disease and effects of losartan. Curr Med Res Opin. 2004;20:369–79.
- Massie HR, Shumway ME, Whitney SJ. Uric acid content of Drosophila decreases with aging. Exp Gerontol. 1991;26:609–14.
- Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–62.
- Fleming I, Michaelis UR, Bredenkotter D, Fisslthaler B, Dehghani F, Brandes RP, . Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88:44–51.
- Wenzel P, Mollnau H, Oelze M, Schulz E, Wickramanayake JM, Muller J, . First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid Redox Signal. 2008;10:1435–47.
- Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906.
- Wosniak J Jr, Santos CX, Kowaltowski AJ, Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal. 2009;11:1265–78.
- Lee SB, Bae IH, Bae YS, Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J biol chem. 2006;281:36228–35.
- Burwell LS, Brookes PS. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:579–99.
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–4.
- Daiber A, Schildknecht S, Muller J, Kamuf J, Bachschmid MM, Ullrich V. Chemical model systems for cellular nitros(yl)ation reactions. Free Radic Biol Med. 2009;47:458–67.
- Jourd'heuil D, Lancaster JR Jr, Fukuto J, Roberts DD, Miranda KM, Mayer B, . The bell-shaped curve for peroxynitrite-mediated oxidation and nitration of NO/O2-* is alive and well. J Biol Chem. 2010;285:le15; author reply le16.
- Bosworth CA, Toledo JC Jr, Zmijewski JW, Li Q, Lancaster JR Jr. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc Natl Acad Sci USA. 2009;106:4671–6.
- Pimentel D, Haeussler DJ, Matsui R, Burgoyne J, Cohen RA, Bachschmid M. Regulation of cell physiology and pathology by protein S-glutathionylation. Lessons learned from the cardiovascular system. Antioxid Redox Signal. 2011 Oct 19. [Epub ahead of print] PubMed PMID: 22010840.
- Mohr S, Stamler JS, Brune B. Mechanism of covalent m?dification of glyceraldehyde-3-phosphate dehydrogenase at its active site thiol by nitric oxide, peroxynitrite and related nitrosating agents. FEBS Lett. 1994;348:223–7.
- Lee JH, Yang ES, Park JW. Inactivation of NADP + -dependent isocitrate dehydrogenase by peroxynitrite. Implications for cytotoxicity and alcohol-induced liver injury. J Biol Chem. 2003;278:51360–71.
- Kim PK, Kwon YG, Chung HT, Kim YM. Regulation of caspases by nitric oxide. Ann N Y Acad Sci. 2002;962:42–52.
- Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun. 2001;282:1075–9.
- Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–6.
- Moon KH, Kim BJ, Song BJ. Inhibition of mitochondrial aldehyde dehydrogenase by nitric oxide-mediated S-nitrosylation. FEBS Lett. 2005;579:6115–20.
- Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, . Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–9.
- Zou MH, Bachschmid M. Hypoxia-reoxygenation triggers coronary vasospasm in isolated bovine coronary arteries via tyrosine nitration of prostacyclin synthase. J Exp Med. 1999;190:135–9.
- Kozak AJ, Liu F, Funovics P, Jacoby A, Kubant R, Malinski T. Role of peroxynitrite in the process of vascular tone regulation by nitric oxide and prostanoids—a nanotechnological approach. Prostaglandins Leukot Essent Fatty Acids. 2005;72:105–13.
- Zou MH, Ullrich V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett. 1996;382:101–4.
- Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, . Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–41.
- Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim HS, . Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab. 2005;2:201–7.
- Gryglewski RJ, Dembinska-Kiec A, Korbut R. A possible role of thromboxane A2 (TXA2) and prostacyclin (PGI2) in circulation. Acta Biol Med Ger. 1978;37:715–23.
- Espey MG, Thomas DD, Miranda KM, Wink DA. Focusing of nitric oxide mediated nitrosation and oxidative nitrosylation as a consequence of reaction with superoxide. Proc Natl Acad Sci USA. 2002;99:11127–32.
- Im JW, Kim HK, Kim ND, Choi JS, Yu BP, Yang HS, . Activation of cyclooxygenases by H2O2 and t-butylhydroperoxide in aged rat lung. Biotechnol Lett. 2004;26:1665–9.
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, . Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lip?xygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48.
- van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, . Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–44.
- Hamilton CA, Brosnan MJ, McIntyre M, Graham D, Dominiczak AF. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension. 2001;37:529–34.
- Yoon HJ, Cho SW, Ahn BW, Yang SY. Alterations in the activity and expression of endothelial NO synthase in aged human endothelial cells. Mech Ageing Dev. 2010;131:119–23.
- Wenzel P, Schulz E, Oelze M, Muller J, Schuhmacher S, Alhamdani MS, . AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radic Biol Med. 2008;45:619–26.
- Bachschmid M, van der Loo B, Schuler K, Labugger R, Thurau S, Eto M, . Oxidative stress-associated vascular aging is independent of the protein kinase C/NAD(P)H oxidase pathway. Arch Gerontol Geriatr. 2004;38:181–90.
- Breusing N, Arndt J, Voss P, Bresgen N, Wiswedel I, Gardemann A, . Inverse correlation of protein oxidation and proteasome activity in liver and lung. Mech Ageing Dev. 2009;130:748–53.
- Breusing N, Grune T. Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Biol Chem. 2008;389:203–9.
- Merker K, Ullrich O, Schmidt H, Sitte N, Grune T. Stability of the nuclear protein turnover during cellular senescence of human fibroblasts. FASEB J. 2003;17:1963–5.
- Li M, Chiu JF, Kelsen A, Lu SC, Fukagawa NK. Identification and characterization of an Nrf2-mediated ARE upstream of the rat glutamate cysteine ligase catalytic subunit gene (GCLC). J Cell Biochem. 2009; 107:944–54.
- Stewart KG, Zhang Y, Davidge ST. Aging increases PGHS-2-dependent vasoconstriction in rat mesenteric arteries. Hypertension. 2000;35:1242–7.
- Shi Y, Man RY, Vanhoutte PM. Two isoforms of cyclooxygenase contribute to augmented endothelium-dependent contractions in femoral arteries of 1-year-old rats. Acta Pharmacol Sin. 2008;29:185–92.
- Rolland PH, Jouve R, Pellegrin E, Mercier C, Serradimigni A. Alteration in prostacyclin and prostaglandin E2 production. Correlation with changes in human aortic atherosclerotic disease. Arteriosclerosis. 1984;4:70–8.
- Beharka AA, Wu D, Serafini M, Meydani SN. Mechanism of vitamin E inhibition of cyclooxygenase activity in macrophages from old mice: role of peroxynitrite. Free Radic Biol Med. 2002;32:503–11.
- Goodwin JS, Messner RP. Sensitivity of lymphocytes to prostaglandin E2 increases in subjects over age 70. J Clin Invest. 1979;64:434–9.
- Hailer NP, Oppermann E, Leckel K, Cinatl J, Markus BH, Blaheta RA. Prostaglandin E2 induces expression of P-selectin (CD62P) on cultured human umbilical vein endothelial cells and enhances endothelial binding of CD4-T-cells. Transplantation. 20?0;70:236–40.
- Yang HH, Kim C, Jung B, Kim KS, Kim JR. Involvement of IGF binding protein 5 in prostaglandin E(2)-induced cellular senescence in human fibroblasts. Biogerontology. 2011;12:239–52.
- Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107(3):490–7. Review. PubMed PMID: 12551876.
- Landar A, Zmijewski JW, Dickinson DA, Le Goffe C, Johnson MS, Milne GL, . Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am J Physiol Heart Circ Physiol. 2006;290:H1777–87.
- Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, . Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–82.
- Luo C, Urgard E, Vooder T, Metspalu A. The role of COX-2 and Nrf2/ARE in anti-inflammation and antioxidative stress: Aging and anti-aging. Med Hypotheses. 2011;77:174–8.
- McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox- insensitive Neh6 degron. J Biol Chem. 2004;279:31556–67.
- Ungvari Z, Bailey-Downs L, Gautam T, Sosnowska D, Wang M, Monticone RE, . Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J Gerontol A Biol Sci Med Sci. 2011;66:866–75.
- Ungvari Z, Bailey-Downs L, Sosnowska D, Gautam T, Koncz P, Losonczy G, . Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am J Physiol Heart Circ Physiol. 2011;301:H363–72.
- Cernuda-Morollon E, Pineda-Molina E, Canada FJ, Perez-Sala D. 15-Deoxy-delta 12,14-prostaglandin J2 inhibition of NF-kappaB-DNA binding through covalent modification of the p50 subunit. J Biol Chem. 2001;276:35530–6.
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, . Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–8.
- Kasjanovova D, Adameckova D, Gratzlova J, Hegyi L. Sex-related and age-related differences in platelet function in vitro: influence of hematocrit. Mech Ageing Dev. 1993;71:103–9.
- Genova ML, Pich MM, Bernacchia A, Bianchi C, Biondi A, Bovina C, . The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann N Y Acad Sci. 2004;1011:86–100.
- Wachowicz B, Olas B, Zbikowska HM, Buczynski A. Generation of reactive oxygen species in blood platelets. Platelets. 2002;13:175–82.
- Kim JW, Zou Y, Yoon S, Lee JH, Kim YK, Yu BP, . Vascular aging: molecular modulation of the prostanoid cascade by calorie restriction. J Gerontol A Biol Sci Med Sci. 2004;59:B876–85.
- van der Loo B, Labugger R, Aebischer CP, Skepper JN, Bachschmid M, Spitzer V, . Cardiovascular aging is associated with vitamin E increase. Circulation. 2002;105:1635–8.
- Koga T, Kwan P, Zubik L, Ameho C, Smith D, Meydani M. Vitamin E supplementation suppresses macrophage accumulation and endothelial cell expression of adhesion molecules in the aorta of hypercholesterolemic rabbits. Atherosclerosis. 2004;176:265–72.
- Liu L, Meydani M. Combined vitamin C and E supplementation retards early progression of arteriosclerosis in heart transplant patients. Nutr Rev. 2002;60:368–71.
- Park SK, Page GP, Kim K, Allison DB, Meydani M, Weindruch R, . alpha- and gamma-Tocopherol prevent age-related transcriptional alterations in the heart and brain of mice. J Nutr. 2008;138:1010–8.
- Stralin P, Karlsson K, Johansson BO, Marklund SL. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler Thromb Vasc Biol. 1995;15:2032–6.
- van der Loo B, Bachschmid M, Skepper JN, Labugger R, Schildknecht S, Hahn R, . Age-associated cellular relocation of Sod 1 as a self-defense is a futile mechanism to prevent vascular aging. Biochem Biophys Res Commun. 2006;344:972–80.
- Wenzel P, Schuhmacher S, Kienh fer J, M ller J, Hortmann M, Oelze M, Schulz E, Treiber N, Kawamoto T, Scharffetter-Kochanek K, M nzel T, B rkle A, Bachschmid MM, Daiber A. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–9. Epub 2008 Jul 2. PubMed PMID: 18596060.
- Didion SP, Kinzenbaw DA, Schrader LI, Faraci FM. Heterozygous CuZn superoxide dismutase deficiency produces a vascular phenotype with aging. Hypertension. 2006;48:1072–9.
- Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009; 297:H1829–36.
- Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, . Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289:1567–9.
- Radovits T, Seres L, Gero D, Lin LN, Beller CJ, Chen SH, . The peroxynitrite decomposition catalyst FP15 improves ageing-associated cardiac and vascular dysfunction. Mech Ageing Dev. 2007;128:173–81.
- Brown KA, Chu Y, Lund DD, Heistad DD, Faraci FM. Gene transfer of extracellular superoxide dismutase protects against vascular dysfunction with aging. Am J Physiol Heart Circ Physiol. 2006;290:H2600–5.
- Csiszar A, Labinskyy N, Zhao X, Hu F, Serpillon S, Huang Z, . Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007;6:783–97.
- Mitsui A, Hamuro J, Nakamura H, Kondo N, Hirabayashi Y, Ishizaki-Koizumi S, . Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxid Redox Signal. 2002;4:693–6.
- Yang X, Doser TA, Fang CX, Nunn JM, Janardhanan R, Zhu M, . Metallothionein prolongs survival and antagonizes senescence- associated cardiomyocyte diastolic dysfunction: role of oxidative stress. FASEB J. 2006;20:1024–6.
- Fang CX, Doser TA, Yang X, Sreejayan N, Ren J. Metallothionein antagonizes aging-induced cardiac contractile dysfunction: role of PTP1B, insulin receptor tyrosine phosphorylation and Akt. Aging Cell. 2006;5:177–85.
- Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci USA. 1997;94:11168–72.
- Harris N, Costa V, MacLean M, Mollapour M, Moradas-Ferreira P, Piper PW. Mnsod overexpression extends the yeast chronological (G(0)) life span but acts independently of Sir2p histone deacetylase to shorten the replicative life span of dividing cells. Free Radic Biol Med. 2003;34:1599–606.
- Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, . Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–97.
- Ren J, Li Q, Wu S, Li SY, Babcock SA. Cardiac overexpression of antioxidant catalase attenuates aging-induced cardiomyocyte relaxation dysfunction. Mech Ageing Dev. 2007;128:276–85.
- Schafer E, Seelert H, Reifschneider NH, Krause F, Dencher NA, Vonck J. Architecture of active mammalian respiratory chain supercomplexes. J Biol Chem. 2006;281:15370–5.
- Wang Y, Bogenhagen DF. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J Biol Chem. 2006;281:25791–802.
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, . Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–18.
- Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, . Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12:534–41.
- Kienhofer J, Haussler DJ, Ruckelshausen F, Muessig E, Weber K, Pimentel D, . Association of mitochondrial antioxidant enzymes with mitochondrial DNA as integral nucleoid constituents. FASEB J. 2009; 23:2034–44.
- Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, . A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005; 85:258–63.
- Kowald A, Kirkwood TB. Accumulation of defective mitochondria through delayed degradation of damaged organelles and its possible role in the ageing of post-mitotic and dividing cells. J Theor Biol. 2000;202:145–60.
- Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011;14:1939–51.
- Lugus JJ, Ngoh GA, Bachschmid MM, Walsh K. Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J Mol Cell Cardiol. 2011;51:885–93.
- Masoro EJ. Caloric restriction-induced life extension of rats and mice: a critique of proposed me?hanisms. Biochim Biophys Acta. 2009; 1790:1040–8.
- Baur JA, Chen D, Chini EN, Chua K, Cohen HY, de Cabo R, . Dietary restriction: standing up for sirtuins. Science. 2010;329:1012–3; author reply 13–14.
- Johnson FB, Sinclair DA, Guarente L. Molecular biology of aging. Cell. 1999;96:291–302.
- Palacios JA, Herranz D, De Bonis ML, Velasco S, Serrano M, Blasco MA. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J Cell Biol. 2010;191:1299–313.
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, . Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–42.
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506.
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, . SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285:8340–51.
- Xu S, Jiang B, Hou X, Shi C, Bachschmid MM, Zang M, . High-fat diet increases and the polyphenol, S17834, decreases acetylation of the sirtuin-1-dependent lysine-382 on p53 and apoptotic signaling in atherosclerotic lesion-prone aortic endothelium of normal mice. J Cardiovasc Pharmacol. 2011;58:263–71.
- Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, . SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–58.
- Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, . SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:14855–60.
- Potente M, Dimmeler S. NO targets SIRT1: a novel signaling network in endothelial senescence. Arterioscler Thromb Vasc Biol. 2008;28:1577–9.
- Ungvari Z, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, . Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H1876–81.
- Csiszar A, Labinskyy N, Pinto JT, Ballabh P, Zhang H, Losonczy G, . Resveratrol induces mitochondrial biogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H13–20.
- Ungvari Z, Bagi Z, Feher A, Recchia FA, Sonntag WE, Pearson K, . Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. 2010;299:H18–24.
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, . Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–12.
- Zhong L, Mostoslavsky R. Fine tuning our cellular factories: sirtuins in mitochondrial biology. Cell Metab. 2011;13:621–6.
- Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–81.
- Grollman AP, Moriya M. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 1993;9:246–9.
- Schodel J, Padmapriya P, Marx A, Huang PL, Ertl G, Kuhlencordt PJ. Expression of neuronal nitric oxide synthase splice variants in atherosclerotic plaques of apoE knockout mice. Atherosclerosis. 2009;206:383–9.
- Schwarz PM, Kleinert H, Forstermann U. Potential functional significance of brain-type and muscle-type nitric oxide synthase I expressed in adventitia and media of rat aorta. Arterioscler Thromb Vasc Biol. 1999;19:2584–90.
- Fleming I, Busse R. Signal transduction of eNOS activation. Cardiovasc Res. 1999;43:532–41.
- Nohl H, Staniek K, Sobhian B, Bahrami S, Redl H, Kozlov AV. Mitochondria recycle nitrite back to the bioregulator nitric monoxide. Acta Biochim Pol. 2000;47:913–21.
- Maia LB, Moura JJ. Nitrite reduction by xanthine oxidase family enzymes: a new class of nitrite reductases. J Biol Inorg Chem. 2011;16:443–60.
- Zhang Z, Naughton DP, Blake DR, Benjamin N, Stevens CR, Winyard PG, . Human xanthine oxidase converts nitrite ions into nitric oxide (NO). Biochem Soc Trans. 1997;25:524S.
- Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, . Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol. 2009;5:865–9.
- van Faassen EE, Bahrami S, Feelisch M, Hogg N, Kelm M, Kim-Shapiro DB, . Nitrite as regulator of hypoxic signaling in mammalian physiology. Med Res Rev. 2009;29:683–741.
- Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem. 2008;283:27038–47. Epub 2008 Jul 13. PubMed PMID: 18622039; PubMed Central PMCID: PMC2556006.
- Song Y, Cardounel AJ, Zweier JL, Xia Y. Inhibition of superoxide generation from neuronal nitric oxide synthase by heat shock protein 90: implications in NOS regulation. Biochemistry. 2002;41:10616–22.
- Brandes RP, Schroder K. Differential vascular functions of Nox family NADPH oxidases. Curr Opin Lipidol. 2008;19:513–8.
- Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:653–61.
- Herkert O, Djordjevic T, BelAiba RS, Gorlach A. Insights into the redox control of blood coagulation: role of vascular NADPH oxidase-derived reactive oxygen species in the thrombogenic cycle. Antioxid Redox Signal. 2004;6:765–76.
- George J, Struthers AD. Role of urate, xanthine oxidase and the effects of allopurinol in vascular oxidative stress. Vasc Health Risk Manag. 2009;5:265–72.