Abstract
Inflammation and activation of immunity are central features in the pathogenesis of atherosclerosis, ischemic myocardial injury, and hypertension-induced target-organ damage. The renin-angiotensin-aldosterone system can initiate not only innate but also acquired immunity. The latter process includes formation of activating antibodies directed at the angiotensin (Ang) II receptor. Ang II not only regulates vascular tone and sodium balance, but also activates immune cells and promotes cell infiltration into target organs. Studies showed that macrophages and various T cell subtypes play a pivotal role in target-organ damage and even in the regulation of blood pressure and responses to Ang II. Experimental and clinical evidence shows that adaptive transfer of immune cells, rendering mice deficient for a certain subset of immune cells, or immunosuppressive treatment affects blood pressure and ameliorates target-organ damage. Neural mechanisms interact with and regulate these processes. Understanding the mechanisms could direct us to novel therapies.
Key messages
Hypertensive target-organ damage features inflammation and immunity.
Angiotensin elicits innate and acquired immunity.
Understanding the mechanisms could suggest new treatments to avoid target-organ damage.
Introduction
Our group became interested in mechanisms by which angiotensin (Ang) II elicits target-organ damage 20 years ago. The target organs are the heart, the brain, the vasculature, and the kidneys. Our initial model was the 2-kidney, 1-clip Goldblatt hypertensive rat. Our focus was on the unprotected kidney (Citation1). We found that the hypertension resulted in an early expansion of the kidney interstitial volume by 37%, whereas hypertensive vascular changes and glomerular injury did not become evident until 3 weeks after clipping. We observed an early interstitial accumulation of collagens and fibronectin. In contrast, the glomeruli showed only mild-to-moderate changes. Staining for proliferating cell nuclear antigen as a marker of cell replication was increased in tubular epithelial but not interstitial or glomerular cells. A progressive infiltration of macrophages and T lymphocytes in the cortical interstitium had already occurred early in the course of the model. On the other hand, only macrophages increased in number within the glomeruli. We concluded that renovascular hypertension leads to an early tubular cell proliferation, mononuclear cell recruitment, and deposition of matrix proteins primarily within the interstitium. As immune cells seemed to drive the process, we directed our attention to mechanisms important to innate immunity. Since this initial study, we have conducted a series of investigations of hypertensive models and have focused on immune mechanisms. We have learned that the renin-angiotensin-aldosterone system interacts with innate immunity, acquired immunity, cell-mediated immunity, and even functional antibody production (). In the process, we have also encountered some surprising mechanisms involving immune cells. We draw attention to the fact that Rodriguez-Iturbe et al. made similar observations and have been seminally active in this field. The group has made a series of contributions that are surely equal to any contributions we may have made. Readers are referred to their papers and a recent review (Citation2–4). In this review, we necessarily focus primarily on studies from our laboratory.
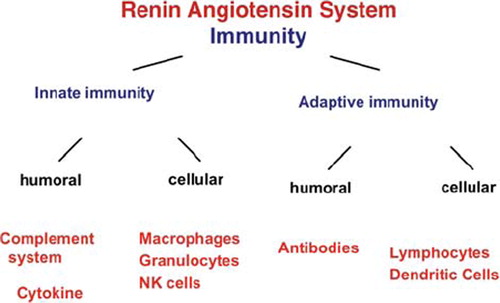
Figure 1. Surprisingly, the renin-angiotensin-aldosterone system touches on all aspects of innate and acquired immunity, including antibody formation. Complement, various immune cells, affectors, and effector mechanisms are represented.
Innate immunity and non-specific immunosuppression
To explore immune mechanisms further, we directed our attention to a unique, double-transgenic rat model (dTGR). These rats harbor the human renin and human angiotensinogen genes. Since the rat and the human renin-angiotensin systems are relatively species-specific, these rats serve as a model of high human-renin hypertension. The animals develop target-organ damage at a young age and die at age 8 weeks (Citation5). The heart shows necrosis and fibrosis, whereas the kidney pathology resembles the hemolytic-uremic syndrome vasculopathy. Surface adhesion molecules are expressed early on the endothelium, while the corresponding ligands are found on circulating leukocytes. Leukocyte infiltration in the vascular wall accompanies plasminogen activator inhibitor-1 (PAI-1), monocyte chemoattractant protein-1 (MCP-1), and vascular endothelial growth factor (VEGF) expression. The expression of transforming growth factor-beta (TGF-β) and deposition of extracellular matrix proteins follow, accompanied by fibrinoid vasculitis in small vessels of the heart and kidneys. Angiotensin-converting enzyme inhibitors and Ang II (AT1) receptor blockers each lowered blood pressure and shifted pressure natriuresis partially leftward by different mechanisms. When combined, they normalized blood pressure, pressure natriuresis, and protected from vasculopathy completely. Renin inhibition lowered blood pressure partially (remikiren), but protected from vasculopathy completely. Endothelin receptor blockade had no influence on blood pressure, but protected from vasculopathy and improved survival.
We next investigated the effects of immunosuppression in dTGR. We investigated whether or not dexamethasone (DEXA) immunosuppression ameliorates renal damage (Citation6). DEXA reduced albuminuria, renal fibrosis, vascular reactive oxygen stress, and prevented mortality, independent of blood pressure. In dTGR kidneys, p22phox immunostaining co-localized with macrophages and partially with T cells. The dTGR dendritic cells expressed major histocompatibility complex II and CD86, indicating maturation. DEXA suppressed major histocompatibility complex II +, CD86 +, dendritic, and T cell infiltration. In additional experiments, we treated dTGRs with mycophenolate mofetil to inhibit T and B cell proliferation. Reno-protective actions of mycophenolate mofetil and its effect on dendritic and T cells were similar to those obtained with DEXA. We then investigated whether or not Ang II directly promotes dendritic cell maturation in vitro. Ang II did not alter CD80, CD83, and MHC II expression, but increased CCR7 expression and cell migration. To explore the role of tumor necrosis factor-alpha (TNF-α) on dendritic cell maturation in vivo, we treated dTGRs with the soluble TNF-α receptor, etanercept. This treatment also had no effect on blood pressure, but decreased albuminuria, nuclear factor-kappaB (NF-κB) activation, and infiltration of all immunocompetent cells. These data suggested that immunosuppression prevents dendritic cell maturation and T cell infiltration in a non-immune model of Ang II-induced renal damage. Ang II induced dendritic migration directly, whereas in-vivo TNF-α is involved in dendritic cell infiltration and maturation. Thus, Ang II may initiate events leading to innate and acquired immune response.
NF-κB
We next focused our attention on the nuclear factor kappa-light-chain-enhancer of activated B cells, NF-κB, a pivotal protein complex that controls DNA transcription important to innate immunity. NF-κB is found in almost all animal cell types and is involved in cellular responses to stimuli such as stress, cytokines, free radicals, ultraviolet irradiation, oxidized LDL, and bacterial or viral antigens. NF-κB plays a key role in regulating the immune response to infection. Kappa light chains are critical components of immunoglobulins. Incorrect regulation of NF-κB has been linked to cancer, inflammatory and autoimmune diseases, septic shock, viral infection, and improper immune development. We showed evidence that Ang II stimulates oxidative stress directly or indirectly via endothelin 1 and that NF-κB is up-regulated in our dTGR model (Citation7). We speculated that the transcription factors NF-κB and activator-protein-1 (AP-1) were involved with initiating chemokine and cytokine expression, leading to the above cascade. We showed pharmacologically that NF-κB inhibition largely ameliorated target-organ damage in this model, with little or no effect on blood pressure (Citation8).
We subsequently tested whether or not endothelial cell-specific NF-κB suppression would be ameliorative (Citation9). We generated Cre/lox transgenic mice with endothelial cell-restricted NF-κB super-repressor I-κBalphaDeltaN (Tie-1-DeltaN mice) overexpression. To induce hypertension with target-organ damage in the mice, we fed a high-salt diet and N(omega)-nitro-l-arginine-methyl-ester (L-NAME) and infused Ang II. Tie-1-DeltaN mice developed a milder renal injury, reduced inflammation, and less albuminuria. RT-PCR showed significantly reduced expression of the NF-κB targets VCAM-1 and ICAM-1, compared with control mice. We concluded that in-vivo NF-κB suppression in endothelial cells stops a signaling cascade leading to reduced hypertension-induced renal damage despite high blood pressure.
Complement
We tested whether or not complement activation participates in Ang II-induced vasculopathy (Citation10). We used the dTGR model. We found that although the dTGR had no albuminuria early, C-reactive protein (CRP) was elevated. Furthermore, macrophages, T cells, TNF-α, C1q, C3, C3c, and C5b-9 expression preceded albuminuria. C1q, C3, C3c, and C5b-9 were observed in the dTGR vessel media in heart and kidney. C5b-9 co-localized with interleukin (IL)-6. Losartan and aliskiren reduced albuminuria and complement expression. We also studied vascular smooth muscle cells (VSMC) from dTGR compared to VSMC from SD. C3 and IL-6 mRNA were analyzed after Ang II, TNF-α, and CRP stimulation. VSMC from dTGR showed increased proliferation and C3 expression compared with controls. Thus, complement activation and cell infiltration occurred before the onset of albuminuria in Ang II-mediated renal damage. TNF-α and CRP played a major role in C3 activation. Our data showed that, in this Ang II-induced model, complement activation is a major participant and suggest that TNF-α and CRP may play a role in its induction.
Immune cells
The ‘David Harrison’ laboratory made a very important observation (Citation11). They demonstrated that mice lacking T and B cells (RAG-1-/-mice) are resistant to Ang II infusions in terms of increasing their blood pressures. Adoptive transfer of T, but not B, cells restored these abnormalities. Ang II is known to stimulate reactive oxygen species production via the nicotinamide adenosine dinucleotide phosphate (NADPH) oxidase in several cells, including some immune cells. Accordingly, adoptive transfer of T cells lacking the angiotensin type I receptor or a functional NADPH oxidase resulted in blunted Ang II-dependent hypertension and decreased aortic superoxide production. Ang II increased T cell markers of activation and tissue-homing in wild-type, but not NADPH oxidase-deficient, mice. Ang II markedly increased T cells in the perivascular adipose tissue (periadventitial fat) in their model. They also found that etanercept prevented the hypertension and the increase in vascular superoxide caused by Ang II. The Harrison laboratory group identified a previously undefined role for T cells in the genesis of hypertension, supporting a role of inflammation in the basis of this prevalent disease. Recently, the group expanded this observation by showing that T cell co-stimulation via B7 ligands is necessary for the development of hypertension (Citation12).
Our studies took a somewhat different course than that of the Harrison laboratory (Citation13). Mice deficient for the helix-loop-helix transcription factor inhibitor of differentiation (Id2) lack Langerhans and splenic CD8a + dendritic cells, have reduced natural killer cells, and have altered CD8 T cell memory. We tested the hypothesis that an alteration in the number and quality of circulating blood cells caused by Id2 deletion would ameliorate Ang II-induced target-organ damage. We conducted kidney and bone-marrow transplants. In contrast to Ang II-infused Id2(+ /-), Id2(-/-) mice infused with Ang II remained normotensive and failed to develop albuminuria or renal damage. Bone-marrow transplant of Id2(+ /-) bone-marrow to Id2(-/-) mice did not restore the blunted blood pressure response to Ang II. Transplantation of Id2(-/-) kidneys to Id2(+ /-) mice also could not prevent Ang II-induced hypertension and renal damage. We verified the Ang II resistance in Id2(-/-) mice in a model of local tissue Ang II production by crossing hypertensive mice transgenic for rat angiotensinogen with Id2(-/-) or Id2(+ /-) mice. Angiotensinogen-transgenic Id2(+ /-) mice developed hypertension, albuminuria, and renal injury, whereas angiotensinogen-transgenic Id2(-/-) mice did not. We also found that vascular smooth muscle cells from Id2(-/-) mice showed an antisenescence phenotype. Our bone-marrow and kidney transplant experiments supported the idea that alterations in circulating immune cells or Id2 in the kidney are not responsible for Ang II resistance. We suggested a previously undefined role for Id2 in the pathogenesis of Ang II-induced hypertension. We have not yet completed our investigations of this complicated immune model.
We next explored a possible role for regulatory T cells. We learned that, in 1986, Mosmann et al. initially proposed a model whereby CD4 + T cells are subdivided into two independent subsets with distinct effector functions (Citation14). They suggested that T helper (TH) cells can be divided into TH1 and TH2 subsets according to their cytokine production, activities, and helper cell function. Further experiments from the Mosmann laboratory and from other groups showed that interleukin (IL)-2, IL-3, TNF-α, and most notably interferon-gamma (IFN-γ) are classically secreted by TH1 cells and control cell-mediated functions such as the activation of macrophages, whereas TH2 cells secrete IL-4, IL-5, and IL-13, which leads to the stimulation of humoral immunity by aiding B cell activation and class switching (Citation13).
Our work has focused on Ang II. However, other related mechanisms deserve attention. The Harrison laboratory extended their work on inflammation to mineralocorticoid receptor-related mechanisms, as has our group. Space does not allow a comprehensive review, and we have necessarily focused on our own activities. However, we wish to draw the readership's attention to an entire showcase involving inflammation and neuroregulatory mechanisms not mentioned here. These mechanisms extend back to C. elegans and involve pathways that were recently reviewed (Citation15). Their importance must be underscored because it far extends what is presented here, to other aspects of pathophysiology (Citation16).
A few upstream master genes in ‘higher hierarchy’ control the expression of many downstream genes and integrate the signaling pathways underlying the pathogenesis of cardiovascular diseases with or without autoimmune inflammatory mechanisms. The forkhead (FOX) transcription factor family members are important in vascular pathology. The FOX transcription factors control regulatory T cells (Tregs). Immunological self tolerance is maintained at least in part by regulatory Treg cells that actively and dominantly control potentially hazardous self-reactive T cells in the periphery. Antigens that stimulate self-reactive T cells may also activate natural Treg cells, thereby maintaining dominant self tolerance. Conversely, genetic anomalies or environmental agents that specifically or predominantly affect Treg cells cause or predispose to autoimmunity (Citation17–19).
We conducted adoptive transfer of Treg cells into Ang II-infused hypertensive mice (Citation20). Treg cell recipients exhibited improved cardiac hypertrophy and less cardiac fibrosis despite sustained hypertension. Amelioration of cardiac morphology was accompanied by an improvement in arrhythmogenic electric remodeling, indicating the functional significance of the enhanced cardiac morphology. Delocalization of the connexin 43 gap junction protein is one of the major pathomechanisms in electric remodeling. Pronounced connexin 43 immunoreactivity was found at the lateral borders of cardiomyocytes in Ang II-treated mice. In contrast, connexin 43 was restricted to the intercalated disk regions in sham controls. Surprisingly, Ang II + Treg-treated mice showed normal connexin 43 gap junction protein localization. Adoptive Treg cell transfer resulted in a marked reduction in cardiac CD4 +, CD8 +, and CD69 + cell and macrophage infiltration. Our data suggested that immunosuppressive effects of transferred Treg cells ameliorated cardiac damage and accounted for the improved electric remodeling independently of blood pressure-lowering effects.
Another class of ‘immune cells’ are the ‘big eaters’, also known as macrophages. Macrophages function in innate immunity and in the specific defense mechanisms of adaptive immunity. Their role is to engulf and then digest cellular debris and pathogens, either as stationary or as mobile cells. They also stimulate lymphocytes and other immune cells to respond to pathogens. Macrophages attack foreign substances, infectious microbes, and cancer cells through destruction and ingestion. Macrophages can be identified by specific expression of a number of proteins including CD14, CD11b, F4/80 (mice)/EMR1 (human), lysozyme M, MAC-1/MAC-3, and CD68 by flow cytometry or immunohistochemical staining. The cells can move freely through the interstitium by an amoeboid movement. Our group identified hitherto unappreciated functions for macrophages relevant to hypertension. We found that macrophages harbor an osmo-sensitive transcription factor called tonicity-element binding protein (TonEBP). The cells can detect changes in osmolarity accompanying high-salt intake that features sodium binding to proteoglycans. The macrophages then express VEGF-C, which stimulates lymphatic vessel proliferation. This process apparently influences salt-storage capacity. If this macrophage function is disrupted, salt-sensitive hypertension ensues (Citation21).
Humoral autoimmunity involving Ang II
We observed and described Ang II receptor (AT1) autoantibodies (AT1-AA), directed at the AT1 receptor in the serum of pre-eclamptic patients. The autoantibodies bind to the second extracellular loop of the AT1 receptor and activate signaling. They function in Western blotting and co-immunoprecipitation experiments. Since pre-eclamptic women commonly have placentas that are infarcted, we explored possible relationships between AT1-AA and blood coagulation (Citation22). IgG from pre-eclamptic patients containing AT1-AA was purified with anti-human IgG columns. AT1-AA were separated from the IgG by ammonium sulfate precipitation. We transfected Chinese hamster ovary cells overexpressing the AT1 receptor with tissue factor (TF) promoter constructs coupled to a luciferase reporter gene. TF was measured by ELISA and detected by immunohistochemistry. Placentas from pre-eclamptic women stained strongly for TF, whereas control placentas showed far less staining. We proved AT1-AA specificity by co-immunoprecipitating the AT1 receptor with AT1-AA but not with non-specific IgG. Ang II and AT1-AA both activated extracellular signal-related kinase, the activator-protein-1 (AP-1), and the TF promoter transfected VSMC and Chinese hamster ovary cells, but only when the AP-1 binding site was present. We then demonstrated TF expression in VSMC exposed to either Ang II or AT1-AA. All these effects were blocked by losartan. Non-specific IgG or IgG from non-pre-eclamptic pregnant women had a negligible effect. We concluded that AT1-AA and Ang II both stimulate the AT1 receptor and initiate a signaling cascade resulting in TF expression. These results showed an action of AT1-AA on human cells that could contribute to the pathogenesis of pre-eclampsia.
We have performed additional studies to address mechanisms (Citation23). We reasoned that we should be able to immunize animals to produce functional AT1 receptor autoantibodies. We generated and purified activating antibodies against the AT1 receptor that we termed AT1-AB by immunizing rabbits against the AFHYESQ epitope of the second extracellular loop, which is the binding epitope of endogenous activating autoantibodies against AT1 from patients with pre-eclampsia. We then purified AT1-AB using affinity chromatography with the amino acid sequence, AFHYESQ, peptide. We were able to detect AT1-AB both by ELISA and a functional bioassay. We then passively transferred AT1-AB into pregnant rats, alone or combined with Ang II. AT1-AB activated protein kinase C-α and extracellular-related kinase 1/2. Passive transfer of AT1-AB alone or Ang II (435 ng/kg per minute) infused alone did not induce a pre-eclampsia-like syndrome in pregnant rats. However, the combination AT1-AB plus Ang II induced hypertension, proteinuria, intrauterine growth retardation, and arteriolosclerosis in the uteroplacental unit. We next performed gene-array profiling of the uteroplacental unit and found that hypoxia-inducible factor 1α was up-regulated by Ang II plus AT1-AB, which we then confirmed by Western blotting in villous explants. Furthermore, endothelin 1 was up-regulated in endothelial cells by Ang II plus AT1-AB and has been implicated in systemic sclerosis (Citation24). Our studies showed that AT1-AB induces Ang II sensitivity. The data are not definitive. However, they do support the existence of an ‘autoimmune-activating receptor’ that could contribute to Ang II sensitivity and possibly to pre-eclampsia.
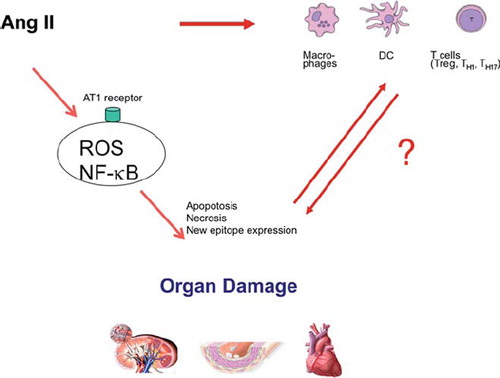
The ‘Gretchen’ questions are of course: ‘What is or are the epitopes, and how do they come to be?’ We cannot answer that question. However, we speculate that cell injury, apoptosis, and necrosis bring to bear new epitopes that are ‘prepared food’ for the immune system (). Hopefully, more complete explanations will be forthcoming in the future. In this brief review, we have focused almost solely on our own work. We are well aware that many other investigators have performed superb studies germane to this topic. Space and temporal constraints do not permit an exhaustive review here, so that not all are duly cited. However, we hope that we convey the excitement present in this area of research. Multidisciplinary expertise will be necessary to move the field forward.
Figure 2. Ang II operates through receptors. Reactive oxygen species (ROS) are generated, but other NF-κB-activating mechanisms are activated. Injured cells may survive or they undergo apoptosis, or necrosis. All these injury processes lead to expression of new epitopes that can lead to further injury. In addition, Ang II influences macrophages and dendritic cells (direct and indirect mechanisms). These activities have a substantial influence on T cell ‘fate’.
Declaration of interest: The Deutsche Forschungsgemeinschaft supported the bulk of this work. The authors are not aware of any conflicts of interest.
References
- Mai M, Geiger H, Hilgers KF, Veelken R, Mann JF, Dämmrich J, . Early interstitial changes in hypertension-induced renal injury. Hypertension. 1993;22:754–65.
- Rodríguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chávez M, Herrera-Acosta J, . Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol. 2002;282: F191–201.
- Rodriguez-Iturbe B, Johnson RJ. The role of renal microvascular disease and interstitial inflammation in salt-sensitive hypertension. Hypertens Res. 2010;33:975–80.
- Rodríguez-Iturbe B, Franco M, Tapia E, Quiroz Y, Johnson RJ. Renal inflammation, autoimmunity and salt-sensitive hypertension. Clin Exp Pharmacol Physiol. 2012;39: 96–103.
- Luft FC, Mervaala E, Müller DN, Gross V, Schmidt F, Park JK, . Hypertension-induced end-organ damage: A new transgenic approach to an old problem. Hypertension. 1999;33:212–8.
- Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, . Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–93.
- Muller DN, Mullally A, Dechend R, Park JK, Fiebeler A, Pilz B, . Endothelin-converting enzyme inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2002;40:840–6.
- Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A, . NF-kappaB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension. 2000;35:193–201.
- Henke N, Schmidt-Ullrich R, Dechend R, Park JK, Qadri F, Wellner M, . Vascular endothelial cell-specific NF-kappaB suppression attenuates hypertension-induced renal damage. Circ Res. 2007;101:268–76.
- Shagdarsuren E, Wellner M, Braesen JH, Park JK, Fiebeler A, Henke N, . Complement activation in angiotensin II-induced organ damage. Circ Res. 2005;97:716–24.
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, . Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–60.
- Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, . Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–37.
- Gratze P, Dechend R, Stocker C, Park JK, Feldt S, Shagdarsuren E, . Novel role for inhibitor of differentiation 2 in the genesis of angiotensin II-induced hypertension. Circulation. 2008;117:2645–56.
- Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57.
- Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009:418–28.
- Luft FC. Neural regulation of the immune system modulates hypertension-induced target-organ damage. J Am Soc Hypertens. 2012;6:23–6.
- Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest. 2007;117:1119–27.
- Yang XF, Fang P, Meng S, Jan M, Xiong X, Yin Y, . The FOX transcription factors regulate vascular pathology, diabetes and Tregs. Front Biosci. 2009;1:420–36.
- Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13.
- Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, . Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904–12.
- Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, . Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–52.
- Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, . AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101:2382–7.
- Wenzel K, Rajakumar A, Haase H, Geusens N, Hubner N, Schulz H, . Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension. 2011;58:77–84.
- Riemekasten G, Philippe A, Näther M, Slowinski T, Müller DN, Heidecke H, . Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis. 2011;70:530–6.