Abstract
Hypertension is a major cardiovascular risk factor with a multifactorial pathogenesis, including genetic and environmental factors. In addition to hypothesis-driven strategies, unbiased approaches such as genomics, proteomics, and metabolomics are useful tools to help unravel the pathophysiology of hypertension and associated organ damage. During development of cardiovascular disease the key organs and tissues undergo extensive functional and structural changes that are characterized by alterations in the amount and type of proteins that are expressed. Proteomic approaches study the expression of large numbers of proteins in organs, tissues, cells, and body fluids. A number of different proteomic platforms are available, many of which combine two methods to separate proteins and peptides after an initial digestion step. Identification of these peptides and changes in their expression in parallel with disease processes or medical treatment will help to identify as yet unknown pathophysiological pathways. There is also potential to use proteomic signatures as biomarkers of cardiovascular disease that will contribute to population screening, diagnosis of diseases and their severity, and monitoring of therapeutic interventions.
Key messages
Proteomic strategies study the expression of proteins at a given time point and characterize the status of cells, tissues, and organs.
In cardiovascular diseases, proteomics therefore has the potential to unravel novel disease pathways, aiding in diagnosis and monitoring of therapeutic measures.
Introduction
Hypertension is one of the most important cardiovascular risk factors worldwide. The incidence and prevalence of hypertension are increasing in all societies and contribute significantly to the global burden of cardiovascular disease (Citation1). The exact pathophysiology of some rare forms of hypertension has been elucidated in detail; underlying mechanisms include alterations in renal salt and water handling, hormonal changes, and over-activity of the sympathetic nervous system. In the majority of patients with hypertension, however, a single cause cannot be found, and it appears that ‘essential hypertension’ is due to interactions between multiple pathophysiological mechanisms and genetic and environmental factors.
Traditional approaches to improve understanding of the pathophysiology of hypertension and associated cardiovascular diseases are hypothesis-driven, focusing on single mechanistic pathways such as the renin-angiotensin-aldosterone system or the sympathetic nervous system. From these mechanisms further pathways are identified and lead to understanding of their complex interplay. In contrast, the more recent ‘systems’-based methods start from a global study of a large number of molecules and then dissect individual pathways and pathogenetic principles. These ‘systems biology’ or ‘systems medicine’ strategies are based on the principle of molecular biology that DNA is transcribed to RNA and translated to proteins that control an organism's metabolism. Accordingly, the areas that study these steps are genomics, transcriptomics, proteomics, and metabolomics (Citation2). The goal of proteomics is a ‘comprehensive, quantitative description of protein expression and its changes under the influence of biological perturbations such as disease or drug treatment’ (Citation3).
Proteomics has been successfully applied to a multitude of clinical conditions, in particular cancer and chronic kidney disease, where changes in protein expression are paralleled by development of disease. These conditions are not subject to the current review and have been described previously (Citation4–9).
Proteomic strategies complement genomic strategies
There is a clear genetic component to blood pressure, with estimated heritabilities of systolic and diastolic blood pressure ranging from 15%–40% and 15%–30%, respectively (Citation10–12). These figures are influenced by non-genetic factors, including shared environment and measurement errors that help to explain the wide range across the literature. The continuous distribution of blood pressure and the associated cardiovascular risk in the general population suggests a complex polygenic inheritance of essential hypertension (Citation13).
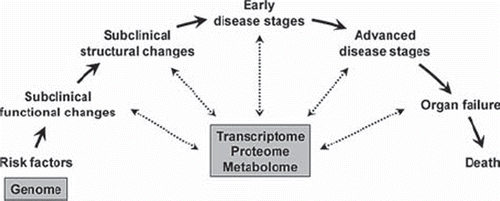
In the development of cardiovascular diseases, hypertension is a risk factor together with other risk factors such as high salt intake, hypercholesterolaemia, smoking, obesity, and sedentary life-style. The concept of the cardiovascular continuum as originally proposed by Dzau and Braunwald (Citation14) proposes that these risk factors lead to early functional and structural changes of the vasculature (e.g. endothelial dysfunction and increased vascular stiffness) that then gradually develop into early and advanced organ damage (e.g. coronary artery disease (CAD) and stroke) and failure (e.g. heart failure). During this development the genome remains largely unchanged, whereas it is clear that the protein structure of tissues and organs change substantially. It is therefore evident that while genetic factors define an organism's potential or risk early in life, the transcriptome, proteome, and metabolome are better suited to define an organism's actual ‘position’ on the cardiovascular continuum (). This description of current status will still be driven by genetic factors but takes into account the interaction with environmental factors and their modifying role over a longer period of time.
Figure 1. Systems biology in the cardiovascular continuum. Cardiovascular disease progresses from risk factors to early and advanced stages of disease (Citation14). Genetic factors do not change during this development and are therefore suited to predict an organism's potential or risk already at early stages of life. The transcriptome, proteome, and metabolome change in parallel to the disease processes and are suited to characterize an organism's current state of disease.
A principle that studies a wide range of expressed proteins is ideally suited to unravel as yet unknown pathophysiological pathways, but can also be used to diagnose disease and predict an individual's risk of developing overt organ damage. Due to its dynamic nature, the proteome will not only change due to disease processes but also in response to therapeutic measures and can therefore be used to monitor treatment effects. The potential clinical applications of proteomics are of particular interest for this review.
The complexity of the proteome poses challenges for its analysis
The results of proteomic studies are often difficult to interpret and appear on first inspection to be inconsistent between studies. In the first instance this is due to the large number of available proteomic platforms, the subject of a recent review by Tuñòn et al. (Citation15). The common principle of proteomic platforms is that proteins in a sample are digested into smaller peptides that are then separated and identified with a combination of two methods, e.g. liquid chromatography and mass spectrometry, so that conclusions about the original protein can be drawn (Citation16). The proteomic platforms are characterized by different sensitivities, allowing identification of peptides and proteins in certain ranges of molecular weight, so that even if the same samples are processed the results can be different depending on the method used. Knowledge about the performance of each platform is therefore important for the interpretation of results.
Further complexity is added by the fact that proteomic studies can be performed on a wide range of biological specimens, including whole-tissue samples but also cells and biofluids (Citation15). For clinical purposes samples that can be obtained non-invasively (urine) or minimally invasively (blood) appear ideal, although they are unlikely fully to represent the proteome of certain organs such as the heart or the vasculature. In contrast to genetic studies where DNA differs very little between cell types, allowing genomic studies on DNA extracted from a wide range of specimens with similar results, the results of proteomic studies depend heavily on the specimen used.
Finally, proteins undergo complex post-translational modifications, with phosphorylation, acetylation, and glycosylation being the most common (Citation16). These modifications can change protein function dramatically but are not easy to detect comprehensively with existing proteomic platforms. A combination of methods can be employed to detect a larger range of post-translational modifications, but interpretation of the results remains challenging. Combinations of different post-translational modifications and rarer modifications such as prenylation or S-nitrosylation add further complexity.
The dynamic nature of the proteome has direct implications for sample handling and processing. Post-translational modifications can occur ex vivo, for example due to hypoxia if a sample is not immediately snap-frozen or otherwise processed. The abundance of proteases in tissue and biofluids such as serum will also change the proteomic make-up of specimens starting immediately from the time of sampling. For clinical purposes in particular, very strict criteria for sample processing must be in place so that proteomic studies can be interpreted with confidence (Citation17).
There is concern that some of this complexity and thereby important information is removed from the proteome by depletion of albumin, immunoglobulins, and other larger proteins during sample preparation. Albumin binds lower-molecular-weight proteins and thereby protects them from clearance by the kidneys. It has been shown that albumin-associated peptides contain disease-relevant information for example in the serum proteome of women with ovarian cancer (Citation18). In particular cytokines including macrophage inflammatory protein 1α, monocyte chemotactic protein, and interleukins 6 and 8 will be recovered at significantly lower levels from albumin-depleted plasma and thereby change the information content considerably (Citation19). A new technique has recently been introduced by Tanaka et al. (Citation20), to facilitate analysis of the plasma proteome without initial depletion of major blood proteins. This one-step direct transfer technique uses a novel target plate to transfer analytes from 1D-gel electrophoresis for matrix-assisted laser desorption/ionization-mass spectrometry. The 1-DE/MS platform not only covers a wider range of peptides, including those bound to major blood proteins, but also speeds up the analysis process considerably and brings proteomics closer to clinical practice.
Clinical proteomics is a multimarker strategy
It is unlikely that the pathophysiology of a complex disorder such as hypertension can be represented by a single biomarker, irrespective of whether it is a biochemical, functional, or imaging biomarker. A more integrative approach that interrogates multiple pathways simultaneously, without being restricted to preconceived ideas of the pathophysiology, appears most promising to achieve this task. Combining multiple serological markers has been shown to improve risk stratification in some (Citation21,Citation22) but not all studies (Citation23). By assessing a range of peptides and peptide fragments, proteomic studies have the potential to detect changes in many pathways involved in the pathogenesis of cardiovascular diseases.
There are, however, important limitations of multimarker strategies that also apply to proteomics. There is a risk of over-fitting models with too many biomarkers relative to the sample size so that diagnostic models will not be transferrable to other cohorts or to the general population. Strict criteria for the use of proteomics as a clinical diagnostic tool must therefore be observed (Citation2,Citation17). Of particular importance is replication of findings in independent cohorts with the investigator being blinded to the diagnosis of study participants.
Urinary proteomics in clinical studies of coronary artery disease: general thoughts
For clinical studies in humans there are advantages of urine over blood for the purpose of proteomics. Urine contains polypeptides originating from a large number of biochemical pathways within the body. In contrast to blood, protease activity in urine is low so that urine is more stable. Furthermore, the urinary proteome or peptidome is less complex, its analysis technically less demanding, and thereby more representative when compared to the plasma proteome (Citation24). Urinary proteomics has been successfully piloted in the diagnosis of renal disease (Citation25), transplant rejection (Citation26), and cancer (Citation27,Citation28).
A number of analytical platforms are available. Work in our group focuses on capillary electrophoresis online coupled with tandem mass spectrometry. This is a relatively fast, reliable, reproducible, and cost-effective proteomic platform that is better suited for high-throughput analysis of clinical samples than other methods (Citation29). The method is characterized by its high resolution and is therefore an ideal platform for analysis of complex biofluids that contain several thousands of different peptides and proteins. There is a degree of intra-individual variability of the urinary proteome, depending for example on state of hydration and time of collection, so that the complex polypeptide patterns can only be interpreted in comparison with data on the normal urinary proteome in healthy subjects. We normalize data to levels of polypeptide markers that are consistently found in urine, based upon experience from analysis of thousands of samples in previous and on-going studies. During model development it is important to correct for multiple testing when statistical analysis is performed in order to avoid false positive findings (Citation30). We will summarize some of our own work into CAD using urinary proteomics before we review other work in this area.
Urinary proteomic markers of CAD
CAD is an ideal model for proteomic studies of cardiovascular disease. CAD is an end-point of a number of pathogenetic principles that lead to atherosclerosis; irrespective of the exact contribution of risk factors the clinical picture is very similar across patients. A gold standard diagnostic test (coronary angiography) is available and routinely performed to secure the diagnosis, so that proteomic studies can be calibrated against this standard. It is further possible to define contrasting phenotypes, such as patients with normal coronary arteries versus patients with severe CAD, which is an important first step in biomarker discovery (Citation31). The principles of proteomics of CAD, however, can also apply to other cardiovascular diseases.
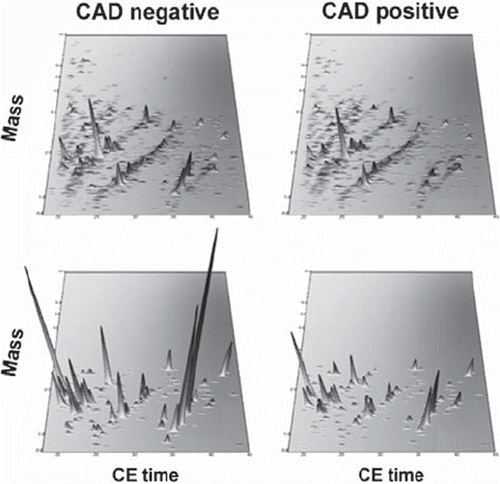
We developed proteomic signatures that are specific for CAD. Our initial small study defined a 15-biomarker panel with sensitivity and specificity to predict presence of CAD of 98% and 83%, respectively (Citation32). Subsequent work has further refined this panel (Citation33). In our most recent study we have extensively validated a 238-biomarker panel in patients and controls from different centres and filtered the panel against concomitant disease and treatment-specific effects. Thereby we were able to demonstrate robust sensitivity and specificity which now reach 79% and 88% in this large study involving samples from 623 subjects (Citation34). The most recent diagnostic panel is shown in .
Figure 2. CAD-specific urinary polypeptide panel. Capillary electrophoresis coupled to mass spectrometry profiling of urine (upper panels) resulted in the definition of 238 polypeptides constituting a CAD-specific polypeptide panel (bottom panels). Normalized molecular weight (800–20,000 Da) in logarithmic scale is plotted against normalized migration time (18–45 min). The mean signal intensity of the polypeptide peak is given in 3D-depiction. Compiled data sets of patients suffering from CAD and controls are shown. The CAD-specific polypeptide pattern is magnified with a zoom factor of 4.5. Modified from (Citation34) with permission.
Sequencing studies have identified the original proteins from which the urinary polypeptides derive. These proteins include fragments of alpha-1-antitrypsin, collagen types 1 and 3, granin-like neuroendocrine peptide precursor, membrane-associated progesterone receptor component 1, sodium/ potassium-transporting ATPase gamma chain, and fibrinogen-alpha chain (Citation34). The majority of diagnostic polypeptides, however, were fragments of collagen which would be very much in line with the substantial changes in vascular structure that define the disease process (Citation34,Citation35).
One of the most exciting features of the urinary proteome is that, unlike the genome, it can mirror progression of disease and effects of therapeutic interventions. In our initial study into CAD (Citation32) we demonstrated that, after surgical coronary revascularization, patients who are able to mount high levels of physical exercise as part of their structured rehabilitation programme show significant changes in their urinary proteome compared to patients who are less physically active. This study showed for the first time that physical exercise, which is known to improve vascular health, is associated with changes in urinary polypeptides that characterize the atherogenic disease process.
In our studies we adjusted the diagnostic proteome signatures for short-term effects that cardiovascular medication may have on urinary polypeptides (Citation32). It is therefore not surprising that in our recent study into CAD (Citation34) short-term (10 weeks) treatment with the angiotensin receptor blocker irbesartan did not change the urinary proteomic panel. In contrast, long-term treatment (2 years) with irbesartan improved the proteomic signature compared to patients treated with placebo, mirroring the antifibrotic effects of angiotensin receptor blockers. We have also demonstrated that the urinary expression of biomarkers of CAD can be modified by other interventions such as polyphenol-rich food supplements (Citation36).
Other proteomic studies into CAD
Other studies into CAD have been performed on plasma samples. Donahue et al. (Citation37) studied the plasma proteome of patients with angiographically confirmed CAD and control subjects without CAD using liquid chromatography–electrospray ionization tandem mass spectrometry. In this study the authors pooled samples to obtain large volumes of 6 litres per group for analysis. Pooling is another approach to identify signals that are present in sufficiently many samples to be characteristic of disease. Not only because of practical challenges, we prefer analysis of individual samples with subsequent rigorous statistical evaluation for one important reason: only in individual analysis can the quality of each sample be checked against established standards and prevent problems related to pre-analytical issues. Especially when stored samples are analysed, bacterial overgrowth is not a rare phenomenon; other possible pre-analytical handling errors include, amongst others, incomplete centrifugation, haemolysis, and incomplete coagulation in the case of serum proteomics. Nevertheless, Donahue et al. (Citation37) identified markers of CAD, some of which are remarkably similar to our own and other groups’ markers, including fragments of collagens, fibrinogen, α-2-antiplasmin, apolipoproteins, and complement factors.
Another approach was chosen by Dardé et al. (Citation38) who studied the plasma proteome of patients with acute coronary syndrome (ACS) using two-dimensional differential gel electrophoresis. This study highlights the dynamic nature of the proteome, as samples were analysed at days 0 (admission), 4, 60, and 180. The authors describe proteomic markers that were present at all time points and others whose expression changed during follow-up. Comparison of these markers with samples from healthy controls and patients with stable CAD illustrates disease processes that are paralleled by changes in the plasma proteome. The markers in the study by Dardé et al. (Citation38) were classified into coagulation proteins, proteins involved in metabolism and/or lipid transport, inflammation and immune response, metabolite transport, cytoskeleton, and other proteins; many of the markers, including fragments of fibrinogen and apolipoproteins, have also been described by others (Citation37). The work is further supported by a study in APOE knock-out mice by Jing et al. (Citation39) who looked at two time points in the development of CAD in this model (6 and 12 weeks). A longitudinal or prospective design helps to reduce noise in proteomic studies and highlights the changes in biomarkers in parallel to development of disease. This is not unique to proteomic studies but is also our own preferred design, for example in transcriptomics studies where paired analysis of samples can also be performed (Citation40).
Samples from patients with CAD and other heart diseases are also ideally suited to interrogate specific pathophysiological pathways by studying the proteome of tissues and cells. Cardiac tissue obtained during surgery can be subject to proteomic studies. In an elegant study, Salgado-Somoza et al. (Citation41) compared the proteome of epicardial adipose tissue with that of subcutaneous tissue. Among the differentially expressed proteins were antioxidant enzymes including catalase whose expression was reduced in epicardial adipose tissue. Reduced ability to counteract reactive oxygen species in the epicardium increases oxidative stress close to coronary arteries and could thereby contribute to the development of CAD. Protein expression can also be studied directly in coronary arteries. You et al. (Citation42) used two-dimensional gel electrophoresis and subsequent mass spectrometry to identify increased ferritin light chain protein expression in coronary arteries from patients with CAD despite reduced ferritin light chain mRNA expression in these samples, indicating up-regulation of ferritin light chain protein expression at post-transcriptional level. Finally, studies that go beyond cardiac and coronary tissue and focus on circulating mononuclear cells have shown increased superoxide production by these cells in patients with cardiovascular diseases (Citation43,Citation44). It is interesting to note that in a study by Barderas et al. (Citation45) that used the same design as the previously mentioned work by Dardé et al. (Citation38) differential expression of some proteins that are involved in oxidative stress, such as paraoxonase and glutathione transferase (Citation46,Citation47), has been found between mononuclear cells from patients with ACS along the different time points from 0 to 180 days.
The currently available proteomic platforms are often purpose-built or specifically modified by their users. Larger-scale multicentre studies could be performed on a limited number of proteomic biomarkers using appropriate multiplexing technologies. Such technologies are extremely useful for clinical studies, and marker protein microarrays have been developed and successfully applied to CAD (Citation48). It is, however, important to consider that these arrays are based on candidate proteins and are not open to new, unexpected discoveries.
Proteomics of hypertension
As discussed previously, hypertension is not a disease per se but a risk factor for cardiovascular diseases. It is therefore generally more suitable for genetic studies than for proteomic studies. Changes that go in parallel with the development of hypertension, either causally or as a consequence, however, can be studied with proteomics. Vascular remodelling in particular should be detectable by studying structural proteins of vessel walls. With recent evidence from genome-wide association studies that large numbers of genes and their variants contribute to the pathogenesis of hypertension, one can expect even larger numbers of proteins to play a causal role in this process. It has not yet been determined whether a combination of genomic information (i.e. the potential to express certain disease-related proteins) and proteomic information (i.e. the actually expressed proteins as a result of the genetic background and its interaction with environmental factors) can further refine an individual's cardiovascular risk profile. Clearly, the diagnosis of hypertension will always be made from blood pressure measurement, but proteomic strategies could help to elucidate factors that contribute to hypertension in individual patients and to discover early evidence of hypertension-associated organ damage.
Proteomic studies into hypertension
The number of proteomic studies into hypertension is currently very limited. The multifactorial pathogenesis of the condition has already been a challenge for genomic studies, and one would expect similar complexities in proteomic studies. Valuable insights do, however, derive from proteomic studies into specific forms of hypertension and particularly into complications of hypertension (Citation49).
Thongboonkerd et al. (Citation50) studied the renal proteome of Sprague-Dawley rats that had been exposed to either episodic or sustained hypoxia for 14 or 30 days. This study identified alterations in the renal kallikrein pathway where episodic hypoxia (a model of obstructive sleep apnoea) was associated with impaired kallikrein-kallistatin-mediated vasodilatation, whereas sustained hypoxia appeared to induce compensatory changes in this pathway that prevented the development of hypertension. This work highlights the potential of proteomics to identify not only single markers but disease pathways. It should also be mentioned that in certain forms of hypertension proteomic platforms can be used outwith their original context of marker discovery specifically to detect single peptides. Reid et al. (Citation51) used immuno-matrix-assisted laser desorption/ionization to detect angiotensin I that can be developed into a method for high-throughput and accurate assessment of plasma renin activity—superior to conventional analysis by conventional radioimmunoassay.
A number of studies have been conducted into hypertension-related organ damage. It is useful in this context to establish strain-specific proteomic profiles of experimental models before disease-specific changes will be studied. Grussenmeyer et al. (Citation52) therefore studied the differences in the left ventricular proteome between Dahl salt-sensitive and salt-resistant strains. Generally the authors found a good correlation in the proteome between the two strains, but a number of differentially expressed markers between the strains could be interesting candidates for future studies into left ventricular hypertrophy and heart failure in high-salt experiments. In earlier work by Jin et al. (Citation53) spontaneously hypertensive rats (SHR) and Wistar-Kyoto rats were used to study cardiac protein expression at early (4 weeks) and advanced stages (20 weeks) of hypertension. They found 13 proteins at the early time point and 7 proteins modulated at the later time point; about the latter the authors conclude that their expression is secondary to already established hypertension at this point, whereas the earlier time point indicates proteins that affect left ventricular remodelling in SHR. Interestingly a number of oxidative stress-related proteins were found in this model, including glutathione transferase omega 1. Both left ventricular hypertrophy and expression of hypertrophy-associated proteins regressed with antihypertensive treatment. Other authors have also analysed the cardiac proteome in studies into the prevention (Citation54) and regression (Citation55) of left ventricular hypertrophy by antihypertensive treatment in models of hypertension.
Another phenotype associated with hypertension is arterial wall remodelling. Delbosc et al. (Citation56) studied aortic protein patterns in two rat strains, Fischer and Brown-Norway, in N(Ω)-nitro-L-arginine methyl ester-induced hypertension. The authors identified strain differences with much more prominent arterial wall remodelling in Fischer rats that was paralleled by changes in ubiquitin, smooth muscle 22α, thymosin β4, and C-terminal fragment of filamin A expression in the secretome of aortic conditioned media.
Preeclampsia is a model disease for proteomic studies into human essential hypertension
We and others have performed studies into preeclampsia as a model disease for human essential hypertension. Preeclampsia complicates 3%–5% of pregnancies in the Western world and is associated with considerable maternal and foetal morbidity and mortality worldwide. The clinical features of preeclampsia include hypertension and proteinuria occurring after 20 weeks’ gestation in women who were not previously known to be hypertensive (Citation57). Preeclampsia shares a number of features with essential hypertension. Both conditions are thought to be of multifactorial origin with contribution of a complex genetic background and its modification by environmental factors; both conditions are associated with organ damage, including renal damage and endothelial dysfunction, and both conditions share risk factors and are associated with future cardiovascular disease (Citation58). In contrast to essential hypertension the development of preeclampsia is within a defined period of time, starting in early stages of pregnancy. Development of hypertension is accelerated in preeclampsia, as opposed to the small annual increases in blood pressure that lead to essential hypertension. The relatively low incidence of preeclampsia is one of the drawbacks of investigating the condition. We conducted a study that was specifically designed to define a preeclampsia-specific urinary proteomic signature that could be used for predictive and diagnostic purposes. We had to study more than 2,400 women in order to obtain a sufficient number of samples from women who subsequently developed preeclampsia during their pregnancy (Citation59).
Proteomic studies of preeclampsia
In our study (Citation59) we were interested in proteomic biomarkers that could predict the development of preeclampsia at an early, asymptomatic stage of pregnancy. Whilst we were able to define such a proteomic signature for samples obtained at gestational week 28, we were not able to find predictive markers in the first trimester of pregnancy, a time where preventative measures and more intensive monitoring could be initiated. We were able to show that the diagnostic proteomic signature changed significantly from gestational week 16 to 28 towards the disease-specific constellation in women who subsequently develop preeclampsia, whereas the panel in women with uncomplicated pregnancy remained unchanged or even ‘improved’ slightly. Again, sequencing studies identified the original proteins from which the urinary polypeptides derive. These proteins include fibrinogen alpha, collagen alpha-1 (I), collagen alpha-1 (III), collagen alpha-2 (I), collagen alpha-3 (IX) chain, and uromodulin fragments (Citation59).
Other investigators have also studied the proteome of preeclampsia. Blumenstein et al. (Citation60) examined the plasma proteome of women at 20 weeks’ gestation who subsequently developed preeclampsia. They reported overexpression of fibrinogen gamma chain and alpha-1-antichymotrypsin in women who went on to develop preeclampsia. It is interesting to note that we also detected fibrinogen alpha in our study into proteomics of preeclampsia (Citation59) and that these proteins were also highlighted in our recent study into proteomics of CAD (Citation34), indicating the potential overlap in pathogenesis between the two conditions. Alpha-1-antichymotrypsin (SERPINA1) also featured prominently in a urinary proteomic study by Buhimschi et al. (Citation61). Although the prospective part of this study was small, the authors point out that the SERPINA1 fragments could have the potential to improve accurate diagnosis of preeclampsia. In fact, differentiation of preeclampsia from other, less dangerous hypertensive conditions in pregnancy could be an invaluable first step to establish a clinical use of proteomics in preeclampsia. We interpreted our own study in a similar way (Citation59). Very recent work into the urinary proteome of women with preeclampsia, gestational hypertension, and normotensive pregnancy by Chen et al. (Citation62) identified 113 peptides that were differentially expressed between women with preeclampsia and healthy controls, but also 31 peptides that were differentially expressed across all three groups. Low urinary angiotensinogen levels were one of the most striking findings in preeclamptic women in this study. A further prospective study by Rasanen et al. (Citation63) using serum proteomics again showed, among others, that fragments of fibrinogen alpha chain are part of a proteomic signature that is characteristic for subsequent development of preeclampsia. Interestingly, the authors demonstrated distinct differences between the serum proteome of women with established preeclampsia compared to the proteome at an earlier, still clinically asymptomatic state. This again illustrates the dynamic changes of the proteome in parallel with the development of disease.
As discussed above, the plasma and serum proteome changes if high-abundance proteins, namely large molecules such as albumin, are removed in pre-analytical steps. Novel methods to study the serum proteome without depletion of major blood proteins (Citation20) have recently been employed for proteomic studies into pregnancy-induced hypertension (PIH). Araki et al. (Citation64) studied the serum proteome of 11 women with PIH and 13 controls and described 23 characteristic proteins for the condition. It is reassuring that some of these markers, including fibrinogen alpha chain, have been described previously (Citation59,Citation63), but indeed the authors found lower levels of the characteristic peptides after an albumin and IgG depletion step. Larger studies with this technique especially at earlier stages of the condition are required to develop its potential fully. It would further be interesting to study plasma instead of serum as significant changes to the protein content of blood occur during the coagulation process.
Future directions
In our view, the future of systems biology will be driven by three major developments. First, proteomic studies will be used to understand comprehensively the effect of specific pathophysiological pathways on blood pressure and blood pressure-related organ damage. Proteomic studies of knock-out models are one way to achieve this goal. For example, Zlatkovic et al. (Citation65) studied the effect of Kir6.2 K+ (KATP) channel knock-out on the cardiac proteome and thereby identified the cardiac KATP subproteome. In their study proteomic analysis predicted functional and structural changes of the heart due to KATP knock-out including hypertrophy and fibrosis, which were then validated echocardiographically and histologically. Secondly, and this is also exemplified by the study by Zlatkovic et al. (Citation65), methods to analyse and understand the full information of proteomic data will have to be developed. The ‘interactome’ that describes the interaction of differentially expressed proteins (and other molecules including genes and metabolites) and clusters them into functional groups and pathways is a particularly attractive concept. Network analysis to study protein–protein interactions will help to predict the effects of changes in the expression of large number of proteins. And thirdly, proteomic data should not be interpreted in isolation from other systems biology approaches including genomics, transcriptomics, and metabolomics. Bioinformatic tools to analyse such complex data are still in their infancy, but promising data are already available from experimental models where effects of well-defined perturbations, namely knock-out of key genes, can be studied. A recent review by Arrell et al. (Citation66) summarizes these concepts in detail and focuses on network analysis, its use for diagnosis of heart failure, and how such complex information could guide treatment.
Summary and perspectives
Proteomics remains a novel research area, but with the technical developments over the last decade the number of publications is steadily increasing (Citation67). For clinical purposes the advantage of proteomics is that it is closer to the actual disease process than for example genomic data. The potential of clinical proteomics therefore lies in its ability to screen for the presence of disease, to diagnose disease accurately, and to monitor the effect of therapeutic approaches. There are promising examples of studies that have defined biomarkers of cardiovascular disease. Studies into the proteome of preeclampsia are the closest to proteomics of hypertension that is currently available in the literature, but we expect to see more proteomic studies of hypertension and its complications in the near future.
A number of challenges, however, remain when it comes to the clinical application of proteomics. There is no consensus about the most suitable sample for routine tests, and studies have been performed using serum, plasma, und urine. The high costs of proteomics as compared with other biomarkers may be seen as another limitation, although in keeping with other novel developments the costs can be expected to come down to more reasonable levels. Currently, however, all clinical proteomic work is performed in research laboratories that are not certified for clinical diagnostic procedures. The equipment is costly and not produced in large quantities, and often individual laboratories have made their own modifications to proteomic platforms. Clinical proteomics will only be widely applicable if the technology can be developed to comply with good laboratory practice and be available at least to core laboratories in larger referral centres. Despite these limitations, a technique that comprehensively studies thousands of biomarkers for clinical diagnosis and risk prediction has enormous potential in hypertension and other cardiovascular diseases.
Declaration of interest: Work in our laboratory is supported by a Strategic Research Development grant from the Scottish Funding Council, a BHF Programme grant RG/07/005/23633, and the European Union's Seventh Framework programmes EU-MASCARA and PRIORITY.
References
- Lawes CM, Vander HS, Rodgers A. Global burden of blood-pressure-related disease, 2001. Lancet. 2008;371:1513–8.
- Dominiczak AF, Herget-Rosenthal S, Delles C, Fliser D, Fournier I, Graber A, . Systems biology to battle vascular disease. Nephrol Dial Transplant. 2010;25:1019–22.
- Anderson NL, Anderson NG. Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis. 1998;19:1853–61.
- Schiffer E, Mischak H, Theodorescu D, Vlahou A. Challenges of using mass spectrometry as a bladder cancer biomarker discovery platform. World J Urol. 2008;26: 67–74.
- Schiffer E, Mischak H, Novak J. High resolution proteome/peptidome analysis of body fluids by capillary electrophoresis coupled with MS. Proteomics. 2006;6:5615–27.
- Kolch W, Mischak H, Pitt AR. The molecular make-up of a tumour: proteomics in cancer research. Clin Sci (Lond). 2005;108:369–83.
- Mullen W, Delles C, Mischak H. Urinary proteomics in the assessment of chronic kidney disease. Curr Opin Nephrol Hypertens. 2011;20:654–61.
- Mischak H, Delles C, Klein J, Schanstra JP. Urinary proteomics based on capillary electrophoresis-coupled mass spectrometry in kidney disease: discovery and validation of biomarkers, and clinical application. Adv Chronic Kidney Dis. 2010;17:493–506.
- Mischak H, Massy ZA, Jankowski J. Proteomics in uremia and renal disease. Semin Dial. 2009;22:409–16.
- Feinleib M, Garrison RJ, Fabsitz R, Christian JC, Hrubec Z, Borhani NO, . The NHLBI twin study of cardiovascular disease risk factors: methodology and summary of results. Am J Epidemiol. 1977;106:284–5.
- Mongeau JG, Biron P, Sing CF. The influence of genetics and household environment upon the variability of normal blood pressure: the Montreal Adoption Survey. Clin Exp Hypertens A. 1986;8:653–60.
- Staessen JA, Wang J, Bianchi G, Birkenhager WH. Essential hypertension. Lancet. 2003;361:1629–41.
- Padmanabhan S, Delles C, Dominiczak AF. Genetic factors in hypertension. Arch Med Sci. 2009;5(2A):S212–9.
- Dzau V, Braunwald E. Resolved and unresolved issues in the prevention and treatment of coronary artery disease: a workshop consensus statement. Am Heart J. 1991;121:1244–63.
- Tunon J, Martin-Ventura JL, Blanco-Colio LM, Lorenzo O, Lopez JA, Egido J. Proteomic strategies in the search of new biomarkers in atherothrombosis. J Am Coll Cardiol. 2010;55:2009–16.
- Gerszten RE, Asnani A, Carr SA. Status and prospects for discovery and verification of new biomarkers of cardiovascular disease by proteomics. Circ Res. 2011;109:463–74.
- Mischak H, Allmaier G, Apweiler R, Attwood T, Baumann M, Benigni A, . Recommendations for biomarker identification and qualification in clinical proteomics. Sci Transl Med. 2010;2:46ps42.
- Lowenthal MS, Mehta AI, Frogale K, Bandle RW, Araujo RP, Hood BL, . Analysis of albumin-associated peptides and proteins from ovarian cancer patients. Clin Chem. 2005;51:1933–45.
- Granger J, Siddiqui J, Copeland S, Remick D. Albumin depletion of human plasma also removes low abundance proteins including the cytokines. Proteomics. 2005;5:4713–8.
- Tanaka K, Tsugawa N, Kim YO, Sanuki N, Takeda U, Lee LJ. A new rapid and comprehensive peptidome analysis by one-step direct transfer technology for 1-D electrophoresis/MALDI mass spectrometry. Biochem Biophys Res Commun. 2009;379:110–4.
- Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Cheh C, . Multiple biomarkers for the prediction of first major cardiovascular events and death. N Engl J Med. 2006;355:2631–9.
- Zethelius B, Berglund L, Sundstrom J, Ingelsson E, Basu S, Larsson A, . Use of multiple biomarkers to improve the prediction of death from cardiovascular causes. N Engl J Med. 2008;358:2107–16.
- Blankenberg S, McQueen MJ, Smieja M, Pogue J, Balion C, Lonn E, . Comparative impact of multiple biomarkers and N-terminal pro-brain natriuretic peptide in the context of conventional risk factors for the prediction of recurrent cardiovascular events in the Heart Outcomes Prevention Evaluation (HOPE) Study. Circulation. 2006;114:201–8.
- Good DM, Thongboonkerd V, Novak J, Bascands JL, Schanstra JP, Coon JJ, . Body fluid proteomics for biomarker discovery: lessons from the past hold the key to success in the future. J Proteome Res. 2007;6:4549–55.
- Haubitz M, Wittke S, Weissinger EM, Walden M, Rupprecht HD, Floege J, . Urine protein patterns can serve as diagnostic tools in patients with IgA nephropathy. Kidney Int. 2005;67:2313–20.
- Wittke S, Haubitz M, Walden M, Rohde F, Schwarz A, Mengel M, . Detection of acute tubulointerstitial rejection by proteomic analysis of urinary samples in renal transplant recipients. Am J Transplant. 2005;5:2479–88.
- Theodorescu D, Wittke S, Ross MM, Walden M, Conaway M, Just I, . Discovery and validation of new protein biomarkers for urothelial cancer: a prospective analysis. Lancet Oncol. 2006;7:230–40.
- Theodorescu D, Schiffer E, Bauer HW, Douwes F, Eichhorn F, Polley R, . Discovery and validation of urinary biomarkers for prostate cancer. Proteomics Clin Appl. 2008;2: 556–70.
- Mischak H, Coon JJ, Novak J, Weissinger EM, Schanstra JP, Dominiczak AF. Capillary electrophoresis-mass spectrometry as a powerful tool in biomarker discovery and clinical diagnosis: an update of recent developments. Mass Spectrom Rev. 2009;28:703–24.
- Dakna M, He Z, Yu WC, Mischak H, Kolch W. Technical, bioinformatical and statistical aspects of liquid chromatography-mass spectrometry (LC-MS) and capillary electrophoresis-mass spectrometry (CE-MS) based clinical proteomics: a critical assessment. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1250–8.
- Hlatky MA, Greenland P, Arnett DK, Ballantyne CM, Criqui MH, Elkind MS, . Criteria for evaluation of novel markers of cardiovascular risk: a scientific statement from the American Heart Association. Circulation. 2009;119: 2408–16.
- Zimmerli LU, Schiffer E, Zurbig P, Good DM, Kellmann M, Mouls L, . Urinary proteomic biomarkers in coronary artery disease. Mol Cell Proteomics. 2008;7:290–8.
- von zur Muhlen C, Schiffer E, Zuerbig P, Kellmann M, Brasse M, Meert N, . Evaluation of urine proteome pattern analysis for its potential to reflect coronary artery atherosclerosis in symptomatic patients. J Proteome Res. 2009;8:335–45.
- Delles C, Schiffer E, von Zur Muhlen C, Peter K, Rossing P, Parving HH, . Urinary proteomic diagnosis of coronary artery disease: identification and clinical validation in 623 individuals. J Hypertens. 2010;28:2316–22.
- Delles C, Diez J, Dominiczak AF. Urinary proteomics in cardiovascular disease: Achievements, limits and hopes. Proteomics Clin Appl. 2011;5:222–32.
- Mullen W, Gonzalez J, Siwy J, Franke J, Sattar N, Mullan A, . A pilot study on the effect of short-term consumption of a polyphenol rich drink on biomarkers of coronary artery disease defined by urinary proteomics. J Agric Food Chem. 2011;59:12850–7.
- Donahue MP, Rose K, Hochstrasser D, Vonderscher J, Grass P, Chibout SD, . Discovery of proteins related to coronary artery disease using industrial-scale proteomics analysis of pooled plasma. Am Heart J. 2006;152:478–85.
- Darde VM, de la Cuesta F, Dones FG, Alvarez-Llamas G, Barderas MG, Vivanco F. Analysis of the plasma proteome associated with acute coronary syndrome: does a permanent protein signature exist in the plasma of ACS patients? J Proteome Res. 2010;9:4420–32.
- Jing L, Parker CE, Seo D, Hines MW, Dicheva N, Yu Y, . Discovery of biomarker candidates for coronary artery disease from an APOE-knock out mouse model using iTRAQ-based multiplex quantitative proteomics. Proteomics. 2011;11:2763–76.
- Taurino C, Miller WH, McBride MW, McClure JD, Khanin R, Moreno MU, . Gene expression profiling in whole blood of patients with coronary artery disease. Clin Sci (Lond). 2010;119:335–43.
- Salgado-Somoza A, Teijeira-Fernandez E, Fernandez AL, Gonzalez-Juanatey JR, Eiras S. Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am J Physiol Heart Circ Physiol. 2010;299:H202–9.
- You SA, Archacki SR, Angheloiu G, Moravec CS, Rao S, Kinter M, . Proteomic approach to coronary atherosclerosis shows ferritin light chain as a significant marker: evidence consistent with iron hypothesis in atherosclerosis. Physiol Genomics. 2003;13:25–30.
- Fortuno A, Olivan S, Beloqui O, San Jose G, Moreno MU, Diez J, . Association of increased phagocytic NADPH oxidase-dependent superoxide production with diminished nitric oxide generation in essential hypertension. J Hypertens. 2004;22:2169–75.
- Zalba G, Beloqui O, San Jose G, Moreno MU, Fortuno A, Diez J. NADPH oxidase-dependent superoxide production is associated with carotid intima-media thickness in subjects free of clinical atherosclerotic disease. Arterioscler Thromb Vasc Biol. 2005;25:1452–7.
- Barderas MG, Tunon J, Darde VM, de la Cuesta F, Duran MC, Jimenez-Nacher JJ, . Circulating human monocytes in the acute coronary syndrome express a characteristic proteomic profile. J Proteome Res. 2007;6:876–86.
- Delles C, Padmanabhan S, Lee WK, Miller WH, McBride MW, McClure JD, . Glutathione S-transferase variants and hypertension. J Hypertens. 2008;26:1343–52.
- McBride MW, Brosnan MJ, Mathers J, McLellan LI, Miller WH, Graham D, . Reduction of Gstm1 expression in the stroke-prone spontaneously hypertension rat contributes to increased oxidative stress. Hypertension. 2005;45:786–92.
- Wykrzykowska JJ, Garcia-Garcia HM, Goedhart D, Zalewski A, Serruys PW. Differential protein biomarker expression and their time-course in patients with a spectrum of stable and unstable coronary syndromes in the Integrated Biomarker and Imaging Study-1 (IBIS-1). Int J Cardiol. 2011;149:10–16.
- Thongboonkerd V. Genomics, proteomics and integrative “omics” in hypertension research. Curr Opin Nephrol Hypertens. 2005;14:133–9.
- Thongboonkerd V, Gozal E, Sachleben LR Jr, Arthur JM, Pierce WM, Cai J, . Proteomic analysis reveals alterations in the renal kallikrein pathway during hypoxia-induced hypertension. J Biol Chem. 2002;277:34708–16.
- Reid JD, Holmes DT, Mason DR, Shah B, Borchers CH. Towards the development of an immuno MALDI (iMALDI) mass spectrometry assay for the diagnosis of hypertension. J Am Soc Mass Spectrom. 2010;21:1680–6.
- Grussenmeyer T, Meili-Butz S, Roth V, Dieterle T, Brink M, Winkler B, . Proteome analysis in cardiovascular pathophysiology using Dahl rat model. J Proteomics. 2011;74: 672–82.
- Jin X, Xia L, Wang LS, Shi JZ, Zheng Y, Chen WL, . Differential protein expression in hypertrophic heart with and without hypertension in spontaneously hypertensive rats. Proteomics. 2006;6:1948–56.
- Liu L, Wang W, Meng X, Gao J, Wu H, Wang P, . Left ventricular hypertrophy induced by abdominal aortic banding and its prevention by angiotensin receptor blocker telmisartan—a proteomic analysis. J Physiol Biochem. 2010;66:329–38.
- Gallego-Delgado J, Lazaro A, Osende JI, Esteban V, Barderas MG, Gomez-Guerrero C, . Proteomic analysis of early left ventricular hypertrophy secondary to hypertension: modulation by antihypertensive therapies. J Am Soc Nephrol. 2006;17:S159–64.
- Delbosc S, Haloui M, Louedec L, Dupuis M, Cubizolles M, Podust VN, . Proteomic analysis permits the identification of new biomarkers of arterial wall remodeling in hypertension. Mol Med. 2008;14:383–94.
- Carty DM, Delles C, Dominiczak AF. Novel biomarkers for predicting preeclampsia. Trends Cardiovasc Med. 2008;18:186–94.
- Carty DM, Delles C, Dominiczak AF. Preeclampsia and future maternal health. J Hypertens. 2010;28:1349–55.
- Carty DM, Siwy J, Brennand JE, Zurbig P, Mullen W, Franke J, . Urinary proteomics for prediction of preeclampsia. Hypertension. 2011;57:561–9.
- Blumenstein M, McMaster MT, Black MA, Wu S, Prakash R, Cooney J, . A proteomic approach identifies early pregnancy biomarkers for preeclampsia: novel linkages between a predisposition to preeclampsia and cardiovascular disease. Proteomics. 2009;9:2929–45.
- Buhimschi IA, Zhao G, Funai EF, Harris N, Sasson IE, Bernstein IM, . Proteomic profiling of urine identifies specific fragments of SERPINA1 and albumin as biomarkers of preeclampsia. Am J Obstet Gynecol. 2008;199:551–16.
- Chen G, Zhang Y, Jin X, Zhang L, Zhou Y, Niu J, . Urinary proteomics analysis for renal injury in hypertensive disorders of pregnancy with iTRAQ labeling and LC-MS/MS. Proteomics Clin Appl. 2011;5:300–10.
- Rasanen J, Girsen A, Lu X, Lapidus JA, Standley M, Reddy A, . Comprehensive maternal serum proteomic profiles of preclinical and clinical preeclampsia. J Proteome Res. 2010;9:4274–81.
- Araki Y, Nonaka D, Tajima A, Maruyama M, Nitto T, Ishikawa H, . Quantitative peptidomic analysis by a newly developed one-step direct transfer technology without depletion of major blood proteins: its potential utility for monitoring of pathophysiological status in pregnancy-induced hypertension. Proteomics. 2011;11:2727–37.
- Zlatkovic J, Arrell DK, Kane GC, Miki T, Seino S, Terzic A. Proteomic profiling of KATP channel-deficient hypertensive heart maps risk for maladaptive cardiomyopathic outcome. Proteomics. 2009;9:1314–25.
- Arrell DK, Zlatkovic LJ, Yamada S, Terzic A. K(ATP) channel-dependent metaboproteome decoded: systems approaches to heart failure prediction, diagnosis, and therapy. Cardiovasc Res. 2011;90:258–66.
- Van Eyk JE. Overview: the maturing of proteomics in cardiovascular research. Circ Res. 2011;108:490–8.