Abstract
The advent of the obesity epidemic has highlighted the need to re-assess more closely the pathophysiology of obesity-related hypertension with the aim of identifying new therapies. In this article, we review the role of the renin-angiotensin-aldosterone system, sympathetic nervous system, and inflammation in relation to the pathophysiology of this condition. We also discuss the potential role of the perivascular adipose tissue in the context of obesity-related hypertension.
| Abbreviations | ||
| ACE | = | angiotensin-converting enzyme |
| ADRF | = | adipose-derived relaxing factor |
| AMPK | = | 5’ adenosine monophosphate-activated protein kinase |
| BMI | = | body mass index |
| BP | = | blood pressure |
| CNS | = | central nervous system |
| DEXA | = | dual-energy X-ray absorptiometry |
| eNOS | = | endothelial nitric oxide synthase |
| G6PD | = | glucose-6-phosphate dehydrogenase |
| IRS | = | insulin receptor substrate |
| MCP-1 | = | monocyte chemotactic protein-1 |
| MR | = | mineralocorticoid receptor |
| NADPH | = | nicotinamide adenine dinucleotide phosphate |
| NO | = | nitric oxide |
| NOS | = | nitric oxide synthase |
| OSA | = | obstructive sleep apnoea |
| PAME | = | palmitic acid methyl ester |
| PVAT | = | perivascular adipose tissue |
| RAAS | = | renin-angiotensin-aldosterone system |
| ROS | = | reactive oxygen species |
| SHR | = | spontaneously hypertensive rat |
| SNS | = | sympathetic nervous system |
| WC | = | waist circumference |
Key messages
Obesity-related hypertension is a major and growing public health concern.
The pathophysiology of obesity-related hypertension is complex and involves the renin-angiotensin-aldosterone system, sympathetic nervous system, oxidative stress, and adipokine dysregulation.
Perivascular adipose tissue damage in obesity may be a contributing factor to the development of hypertension in obesity, and rescuing its function may offer therapeutic potential.
Trends in obesity and hypertension
The obesity epidemic has emerged as a major challenge to our increasingly stressed healthcare systems. Globally, obesity has more than doubled in the past 30 years. In 2008 there were 1 billion overweight and 500 million obese adults worldwide (Citation1). In England nearly a quarter and in the US close to a third of all adults have been classified as obese (Citation2,Citation3). Public health initiatives and education have had an impact on diet in England as less saturated and trans fats were consumed in 2008/09 compared to 10 years ago, but, despite this, 38% of adults had a raised waist circumference in 2009 as compared to 23% in 1993, and those with a raised waist circumference were at least twice as likely to have high blood pressure (Citation2).
Whilst it is widely accepted that obesity and its associated disorders are major risk factors for cardiovascular events, a recent study has reported that body mass index (BMI), waist circumference, and waist-to-hip ratio do not improve cardiovascular disease risk prediction substantially when information regarding other aspects of the medical history, such as blood pressure, lipid profile, and a history of diabetes,is available (Citation4). However this does not understate the importance of obesity and metabolic syndrome as predictors of future cardiovascular events. The heterogeneity of body fat distribution has prompted the American Heart Association to stress the importance of measuring waist circumference (WC) as well as BMI. In the case of those with disproportionately high WC for a given BMI, assessment of further cardiometabolic risk factors is encouraged (Citation5). A more streamlined assessment of body composition is fraught with difficulties as there are no internationally agreed cut-offs for measurements such as waist circumference for a given BMI, or dual-energy X-ray absorptiometry (DEXA) and bioelectric impedance values (Citation5).
Hypertension affects nearly one-third of the US population (Citation6), and globally around 40% of adults had raised blood pressure(BP) in 2008 (Citation7). The co-occurrence of obesity and hypertension has focused minds on understanding the pathophysiology of obesity-related hypertension. Data from the Framingham Heart Study implicate obesity as a contributory factor in 60%–70% of essential hypertension (Citation8), and obese individuals have a 3.5-fold increase in the likelihood of suffering from hypertension (Citation9).
According to a 2011 National Health Service survey in England, high blood pressure was recorded in 48% of men and 46% of women in the obese group, compared with around 30% of those in the overweight and 15% of those in the normal weight category (Citation2).
In this review, we aim to discuss briefly the pathophysiology of obesity-related hypertension and highlight the role of perivascular adipose tissue in this context.
What causes hypertension in obesity?
There are many mechanisms via which obesity can lead to hypertension (). Gosmanov et al. have reported the acute effects of high fat ingestion by normotensive obese individuals (Citation10). Their work suggested that both bolus oral ingestion and the intravenous infusion of fat result in a significant rise in systolic blood pressure, attenuated endothelial function (assessed by flow-mediated dilatation), increased oxidative stress markers, and activation of the sympathetic nervous system as measured by heart rate variability.
Figure 1. The complex pathophysiology of obesity-related hypertension. (OSA = obstructive sleep apnoea; RAAS = renin-angiotensin-aldosterone system; SNS = sympathetic nervous system).
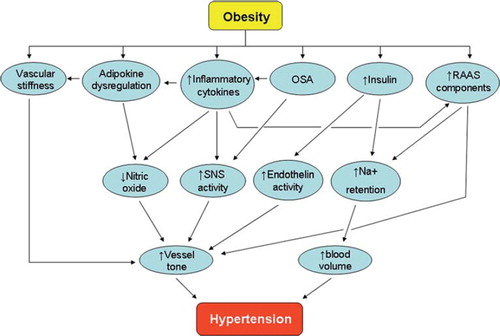
Clearly, obesity is a chronic disorder with a complex aetiology. Genetic causes of obesity are rare, and the majority of cases encountered in clinics are a consequence of indulgence in readily available and calorie-rich food which provides a large amount of fat as well as a significant proportion of the recommended daily allowance of salt. High-salt diets accelerate the development of hypertension in diet-induced obese rats without raising the ceiling of the systolic blood pressure beyond that observed in diet-induced obese rats fed a low-salt diet (Citation11). This effect may be propagated by an increase in oxidative stress levels in the vasculature as the same study also demonstrated a significant increase in superoxide levels within aortic rings of high-fat and high-salt diet fed animals.
There are numerous facilitators of obesity-related hypertension. These include the renin-angiotensin-aldosterone system (RAAS), the over-active sympathetic nervous system, metabolic dysregulation including hyperinsulinaemia, adipokine imbalance, and potentially perivascular adipose tissue (PVAT) damage, defined as a disturbance in the normal metabolic and vasoactive function of the adipocytes surrounding blood vessels (). There is currently no direct evidence to suggest that a loss of PVAT anticontractile function leads to systemic hypertension, but the theory behind such a proposition will be discussed in this review.
The contributions of the renal system are significant and manifold;they include sodium retention and impaired pressure natriuresis (Citation12). The kidneys contribute to the disease process only to fall victim to its detrimental effects as end-organ damage ensues in the form of chronic kidney disease, thus leading to a vicious cycle.
There are three major factors that highlight the involvement of the renal system in obesity-related hypertension: increased activity of the renal sympathetic nervous system (SNS), activation of the RAAS, and physical compression of the kidneys by intrarenal fat deposition and extracellular matrix modifications (Citation13).
Obesity has a differential effect on local SNS activity. A significant increase in renal sympathetic activity is observed in obese individuals, butcompared with lean normotensive subjects theobese normotensive individuals exhibit suppression of their cardiac sympathetic activity, whilst the hypertensive obese individuals show an increased sympathetic activity in both cardiac and renal nerves (Citation14). The degree of renal sympathetic stimulation in obesity is similar in both normotensive and hypertensive cohorts, thus emphasizing the significance of other contributory factors such as the suppression of cardiac sympathetic drive in the normotensive individuals (Citation15). A lower cardiac sympathetic tone in obesity can be viewed as a protective factor, and it may be over-activity that tips the balance in favour of the development of hypertension, but it is not clear which factors dictate the differential activity of local SNS. Also there is evidence of central stimulation of the SNS by reactive oxygen species in obesity. There are raised levels of oxidative stress markers such as the superoxide anion, and animal studies suggest that nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent oxidative stress in the brain may be a cause of increased sympathetic tone leading to hypertension in high-fat fed animals (Citation16).
In obesity, the full complement of the RAAS components is raised. In part this is as a consequence of the intrinsic RAAS system within the adipocyte and includes angiotensin-converting enzyme (ACE), angiotensin type 1 and type 2 receptors. In addition adipocytes secrete angiotensinogen, the levels of which are raised in obesity (Citation17), as well as renin; though the source of the adipocyterenin activity remains controversial (Citation18). Raised plasma aldosterone levels also seen in obesity correlate with the degree of visceral adiposity and waist-to-hip ratio (Citation19–21). The elevated aldosterone levels will not only contribute to increased blood volume by increasing sodium reabsorption, but also lead to the generation of reactive oxygen species (ROS) (). Aldosterone activates NADPH oxidase, thus increasing ROS levels and leading to oxidative posttranslational changes to guanylyl cyclase, rendering it NO-insensitive (Citation22). The generated ROS also react with nitric oxide to reduce its bioavailability by forming molecules such as peroxynitrite, thus contributing to endothelial dysfunction. There is also evidence of a vicious cycle with ROS stimulating the mineralocorticoid receptor (MR) (Citation23), thereby theoretically contributing to further elevations in ROS levels. Aldosterone also decreases endothelial glucose-6-phosphate dehydrogenase (G6PD) activity. G6PD is a cytosolic enzyme and the main source of intracellular NADPH which functions to limit ROS activity (Citation24). There are two aldosterone receptor antagonists in clinical use:spironolactone is a non-selective aldosterone receptor antagonist, whereas eplerenone is a selective aldosterone receptor antagonist which has a lower degree of cross-reactivity with sexsteroid hormones and a longer half-life than spironolactone (Citation25). Spironolactone leads to increased expression of G6PD and its activity, as well as elevated NADPH levels leading to diminished ROS generation in aortas of aldosterone-treated mice (Citation24). Eplerenone, a mineralocorticoid receptor antagonist, attenuates hypertension associated with diet-induced obesity in dogs, despite only modestly elevated levels of plasma aldosterone. It is not clear to what extent the blood pressure reduction is a consequence of reductions in blood volume and cardiac output secondary to reduced sodium reabsorption, or due to a reduction in sympathetic activity through the direct central nervous system (CNS) effect of aldosterone (Citation26,Citation27).
Figure 2. Perivascular adipose tissue as relevant to vessel tone and adipokines implicated in obesity-related hypertension. (eNOS = endothelial nitric oxide synthase; Kv = voltage-gated potassium channel; CNQ = a member of the Kv family; MR = mineralocorticoid receptor; NO = nitric oxide; O22 = superoxide anion; ONOO2 = peroxynitrite; PAME = palmitic acid methyl ester; VSMC = vascular smooth muscle cell).
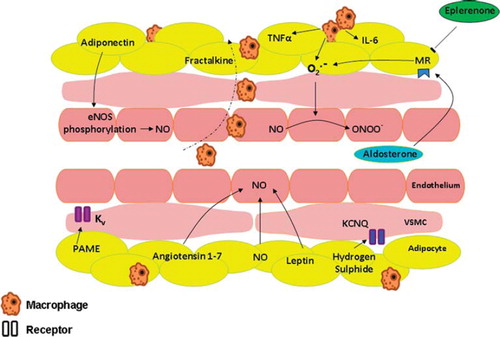
Sodium reabsorption in the kidneys is a major contributor to disease progression. The raised angiotensin II levels in obesity raise blood pressure by direct vasoconstrictor action and also by increasing sodium reabsorption either via direct action on the kidneys or by stimulation of aldosterone release. Insulin also acts on renal tubules to increase salt reabsorption, leading to retention of water and blood volume expansion (Citation28,Citation29). Insulin resistance in obesity leads to a state of hyperinsulinaemia, and there is a suggestion that renal tubular cells may possess a different insulin receptor substrate (IRS) to that of adipocytes and skeletal muscle cells (IRS1); thus renal tubular cells may well remain sensitive to the increased levels of plasma insulin in obesity via IRS1-independent stimulation (Citation30).
Another important contributing factor in the pathophysiology of obesity-related hypertension is vessel stiffness. Obesity has long been associated with arterial stiffness and increased pulse wave velocity independently of age, blood pressure, and ethnicity. However, the association is stronger for waist circumference and visceral adiposity, rather than global obesity as measured by BMI (Citation31). Obesity is a complex multifaceted disorder, and dysregulation of any number of factors can affect vascular stiffness. Leptin has been linked with impairment of arterial distensibility, and its raised levels in obesity may be a significant contributing factor in arterial stiffness (Citation32). Hyperinsulinaemia is another contributor to vascular stiffness. In lean individuals, insulin reduces central arterial stiffness before it exerts its slow vasodilatory effect on peripheral small arteries. In obese individuals, its effect on arterial stiffness is severely blunted; this attenuation correlates with the degree of obesity (Citation33).
Inflammation, oxidative stress and monocyte recruitment all play their part in initiating endothelial dysfunction in obesity. There is also disruption to the fine balance between the vasoconstrictor action of endothelin-1 and the vasodilator effect of NO in endothelial cells. In health, insulin activates phosphoinositide 3-kinase, leading to increased NO production secondary to endothelial nitric oxide synthase (eNOS) phosphorylation(Citation29). Postprandial physiological surge in insulin concentrations leads to dilatation of precapillary arterioles, thus improving blood flow and delivery of nutrients to tissues, a process known as nutritive flow (Citation34). In obesity, NO-mediated vasorelaxation is impaired, leading to vasoconstriction via unopposed endothelin-1 action (Citation29,Citation34). Reduced endothelial nitric oxide bioavailability in obesity is a significant consequence of the reactions between free radicals and the vasodilator gas. Reactive oxygen species such as the superoxide anion react with nitric oxide to produce peroxynitrite and deplete endothelial NO levels (). The role of nitric oxide in vessel tone modulation and its fate in inflammatory diseases have been extensively reviewed by Jin and Loscalzo (Citation35).
There is a close correlation between obesity, obstructive sleep apnoea (OSA), and hypertension. OSA has been identified as both a cause and a consequence of a number of the physiological and metabolic sequelae of obesity. A prospective study of 709 individuals with a follow-up period of 4 years has reported a dose-response relationship between sleep-disordered breathing and hypertension, independent of confounding factors (Citation36). There is a significant degree of OSA in about 40% of obese individuals, and nearly 70% of OSA individuals suffer from obesity (Citation37). Fat deposition around the upper airway in obesity is thought to be the most significant contributor to the development of OSA in obesity. Almost half of all hypertensive patients suffer from sleep apnoea, and half of all sleep apnoea patients are hypertensive (Citation19). There are a number of potential mechanisms linking OSA with hypertension, including endothelial dysfunction, CNS stimulation, oxidative stress, and inflammation (Citation37). It is hypothesized that the most significant causal factor is the elevated oxidative stress levels initiated by intermittent hypoxia, coupled with hyperleptinaemia (Citation38) with its direct stimulatory effects on the sympathetic nervous system. There is also the suggestion that elevated levels of aldosterone exist in OSA and that this correlates with severity of OSA. Clearly, elevated aldosterone levels would lead to blood pressure elevations, and there is a suggestion that it mayeven worsen upper airway resistance by contributing to pharyngeal oedema via fluid retention (Citation39).
PVAT function and damage in relation to obesity-related hypertension
Adipocytes surround almost every blood vessel in the body and secrete a number of adipokines with metabolic and vasoactive properties. These predominantly white adipocytes form the perivascular adipose tissue (PVAT) ().
Healthy PVAT exerts an anticontractile effect on adjacent small arteries when subject to vasoconstrictors (Citation40). The exact mechanism via which PVAT has this effect has been the subject of many recent publications and yet remains controversial. Experimental protocols have identified both endothelium-dependentand -independent (Citation41) mechanisms, and a number of molecules have been implicated which will be briefly discussed here. It is important to highlight that white and brown adipocytes have slightly different secretion profiles (Citation42), but the anticontractile property of PVAT has been documented in both tissues (Citation43,Citation44).
Adiponectin is the most abundant adipokine with a significant vasorelaxant effect on small arteries and is able to reverse endothelial dysfunction in diet-induced obese rats via the 5’ adenosine monophosphate-activated protein kinase (AMPK)-eNOS pathway (Citation45). Clinical studies have shown its levels to be low in hypertension and to improve with antihypertensive treatment (Citation46). It has been shown that adiponectin secreted from murine PVAT modulates the tone of the adjacent vessel by functioning as an adipose tissue-derived relaxant factor (Citation47). Moreover, data from our group have clearly demonstrated that adiponectin receptor type-1 blockade abolishes PVAT anticontractile effect on adjacent small arteries from healthy human tissue (Citation48), clearly demonstrating that adiponectin is also an adipose-derived relaxing factor (ADRF) in humans. Recently we have reviewed in detail the properties of this adipokine and its role as an ADRF (Citation49).
Apart from adiponectin, PVAT secretes a number of other molecules with vasorelaxant properties; these include angiotensin 1–7 (Ang1–7), nitric oxide (NO), leptin, hydrogen sulphide, and palmitic acid methyl ester (PAME).
Angiotensin 1–7 is secreted from PVAT and exerts its anticontractile effects by stimulating the release of endothelial NO, thus activating calcium-dependent potassium channels (Citation43) in arteries and voltage-dependent potassium channels in veins (Citation50). In support of this finding, Ang 1–7 receptor antagonists have been shown to attenuate PVAT anticontractile function (Citation51). Ang 1–7 is also able to function via AT2 and Mas receptors to decrease the nerve-stimulated overflow of noradrenaline(Citation52). This may be of particular therapeutic interest as sympathetic over-activity contributes to pathophysiology of obesity-related hypertension. Recently, an oral preparation of Ang 1 – 7 has been prepared to assess its cardioprotective effects in infarcted rats (Citation53); the effect of this product on small vessel tone remains to be assessed.
In health, white adipose tissue (Citation54) and PVAT itself (Citation55) are known to be sources of nitric oxide(NO). Insulin (Citation56) and leptin (Citation57) have been shown to stimulate NO production in adipocytes, and, in theory, the elevated levels of leptin and insulin in obesity should enhance NO levels in PVAT. In early diet-induced obesity, there is evidence of improved NO bioavailability in mesenteric PVAT of rats (Citation55), but factors such as elevated superoxide levels in chronic obesity would lead to a reduction in NO bioavailability via mechanisms already discussed in this review.
Leptin is secreted from white adipocytes, and its plasma levels are increased in obesity. It acts centrally on the hypothalamus to reduce appetite and also to increase SNS activity (Citation58), and locally it has a direct endothelial nitric oxide-dependent vasorelaxant effect in health. It is proposed that leptin plays a major role in pathophysiology of obesity-related hypertension. Leptin-deficient ob/ob mice develop severe obesity but remain normotensive (Citation59). In the vasculature, leptin stimulates the release of endothelial NO, so an acute rise in leptin levels does not significantly affect BP despite SNS activation. However, in obesity, its plasma levels are chronically raised, and endothelial dysfunction means a reduction in NO bioavailability (Citation60), thus the vasopressor effects of leptin become more apparent.
Hydrogen sulphide and palmitic acid methyl ester (PAME) are the most recent additions to the list of potential ADRFs.Hydrogen sulphide is thought to function via KCNQ and at least partly contribute to the PVAT anticontractile effect (Citation61); whilst PAME functions via Kv channels, independent of nitric oxide and endothelium (Citation62), and its release is calcium-dependent. PAME is implicated in the pathophysiology of hypertension. There is a reduction in its release from PVAT of 20-week-old spontaneously hypertensive rats (SHR) as compared with pre-hypertensive SHR and normotensive Wistar Kyoto Rats, and exogenously applied PAME has a reduced vasorelaxant effect on de-endothelialized aortic rings of SHR as compared with its significant vasorelaxant effect on preconstricted vessels from prehypertensive SHR and normotensive rats (Citation62).
There may be more than one substance released from PVAT that satisfies all the criteria for ADRF. We have shown that adiponectin is the ADRF from human subcutaneous PVAT (Citation48). The identity of the relaxant factor may vary according to species and site of the PVAT compartments being studied.
In obesity, the anticontractile function of PVAT is attenuated or lost completely. A number of explanations and theories exist as to the cause of this loss of function. Amongst the most likely are the effects of oxidative stress and inflammation, as well as adipokine dysregulation and increased sympathetic nervous system action.
We have shown that incubation of healthy PVAT with TNF-α and IL-6 leads to significant attenuation of PVAT anticontractile function similar to that observed in the obese phenotype (Citation48). Macrophages secrete a number of inflammatory cytokines including TNF-α, IL-6, and free radicals such as the superoxide anion. Following experimental hypoxia, which approximates obesity-induced PVAT damage, macrophage recruitment and activation in adipose tissue is an essential step leading to the loss of PVAT anticontractile function (Citation63). Interestingly, the PVAT surrounding rat thoracic aorta expresses brown adipose tissue genes and appears to resist inflammation and macrophage infiltration in diet-induced obesity (Citation64).
The contribution of aldosterone to obesity-related hypertension has been discussed previously in this article. It has been shown that aldosterone increases the expression of TNF-α from macrophages. Moreover, activation of the mineralocorticoid receptor results in generation of reactive oxygen species (ROS), and blockade of the receptor, using eplerenone, leads to a reduction of ROS and increased levels of adiponectin in obese and diabetic mice (Citation65). The superoxide anions generated by macrophages and MR stimulation also contribute to PVAT damage. Work by Gao et al. has shown that superoxide generated by NADPH oxidase in response to electric field stimulation enhances the contractile response of adjacent small arteries (Citation66). It has been shown that candesartan (angiotensin II type 1 receptor antagonist) reduces this PVAT-mediated potentiation of electric field stimulation-induced contractile response, thus providing another potential explanation for the increased vascular resistance in obesity where there is both increased sympathetic nerve activity and increased angiotensin II levels (Citation67).
In view of the significant role macrophages play in PVAT damage, their recruitment from blood to the perivascular adipose tissue is of paramount importance. Monocyte chemotactic protein-1 (MCP-1) levels are increased in adipose tissue and plasma of genetically obese and diet-induced obese mice (Citation68), as well in obese humans (Citation69). Insulin increases the secretion of MCP-1 in insulin-resistant 3T3-L1 adipocytes and in ob/ob mice (Citation70), thus the hyperinsulinaemic state in obesity leads to macrophage recruitment in PVAT and subsequent release of cytokines which attenuate its anticontractile function. Fractalkine (CX3CL1) is a recently identified chemokine secreted from adipocytes that promotes monocyte adhesion to human adipocytes (Citation71). Its levels are increased in inflammatory states such as diabetes, HIV, and rheumatoid arthritis, as well as in obesity (Citation72). There is also direct evidence for involvement of fractalkine in hypertension. The expression of CX3CL1 receptor gene in blood leukocytes from patients with arterial hypertension has been shown to be significantly increased (Citation73). The increased levels of MCP-1 and fractalkine in obesity can be considered as facilitators for the initiation of the macrophage-induced loss of PVAT anticontractile function.
Adipose tissue depots have unique inflammatory profiles. PVAT from murine aortic arch expresses lower levels of adipocyte-associated genes when compared to subcutaneous and visceral fat, and this becomes even more pronounced after 2 weeks of high-fat feeding whilst proinflammatory genes become up-regulated (Citation74). Visceral adipose tissue exhibits a more inflammatory profile with a higher macrophage content than subcutaneous fat (Citation75). This may somewhat explain the stronger link between central obesity and hypertension than between BMI and raised blood pressure (Citation76).
Treatments
There is no robust guidance with regard to the use of antihypertensive drugs in the treatment of obesity-related hypertension, and we will not cover the topic in this review. An important way to tackle the growing problem of obesity-related hypertension is to treat obesity using therapies that result in sustained weight loss. Weight reduction in obese hypertensive patients does indeed lead to an improvement in blood pressure (Citation77).
Diet and exercise are the first line in both prevention and treatment of obesity. Studies have shown that the combination of intense exercise and moderate caloric restriction can lead to major reductions in markers of inflammation such as high-sensitivity C-reactive protein as well as improving insulin sensitivity and enhancing adiponectin levels (Citation78). Exercise and weight loss can also individually help lower BP. A study of 133 overweight and sedentary individuals with either unmedicated high normal BP or stage 1 to2 hypertension has reported a reduction of 7 mmHg in systolic BP of those assigned to behavioural weight management strategies and a 4 mmHg reduction in systolic BP of those who took up aerobic exercise three to four times per week over the 6-month study period (Citation79), thus clearly demonstrating that simple life-style measures should not be ignored.
Apart from antihypertensive treatment and life-style modifications, there are a number of therapeutic interventions that have either been shown to be effective or show promise in the treatment of obesity-related hypertension.
Bariatric surgery was first performed in 1953, and with the advent of the obesity epidemic the procedure has become increasingly popular (Citation80). In the US the number of procedures has risen from 16,200 in 1992 to 220,000 in 2008 (Citation81).
Life-style changes including diet and exercise are often the first line in the treatment of obesity. However, for the morbidly obese, bariatric surgery is able to achieve a significantly greater degree of sustained weight loss. The remission rate of hypertension is also significantly higher after bariatric surgery than after life-style changes (Citation82). A 1-year follow-up of 88 hypertensive obese patients who underwent gastric banding has shown that 59% were normotensive and 33% had reduced BP with a reduction in doses of medication (Citation83). A recent systematic review of the literature has shown that, out of 16,867 patients, 49% had hypertension before the operation and 68% of these cases were either improved or resolved completely after an average follow-up period of 34 months (Citation80).
A review of 18 studies looking at bariatric surgery outcomes has shown that blood adiponectin levels increased by nearly 70% post gastric bypass and by around 36% post gastric banding procedures. The greatest increase was achieved after losing 35% of the original body weight. They also demonstrated a strong correlation between percentage increase in adiponectin levels and percentage decrease in BMI (Citation84). This is not true of weight loss by liposuction, where, despite a 10% weight reduction, no improvements in adiponectin or insulin resistance have been noted (Citation85). This is likely due to the differing qualities of adipose tissue depots, with visceral fat exhibiting a more inflammatory profile as compared with subcutaneous adipose tissue (Citation86).
Bariatric surgery has also been shown to improve the inflammatory profile of obese individuals (Citation87). In subcutaneous adipose tissue, the expression of IL-6 and TNF-α mRNA decreases significantly, and expression of adiponectin and its receptors increases after dramatic weight loss post surgery (Citation88). The significant degree of weight loss, together with improvements in adipokine and inflammatory cytokine profile, as well as resolution or improvement in diabetes status (Citation89), makes this an invaluable procedure in those suffering from morbid obesity and its sequelae.
The over-activity of the renal SNS has already been discussed in this review. In 1995, Kassab et al. had shown that renal denervation leads to a reduction in sodium retention and lower blood pressures in dogs fed a high-fat diet (Citation90). More recently,the Symplicity HTN-2 trial has shown that catheter-based renal denervation can reduce BP by 32/12 mmHg in treatment-resistant hypertension (Citation91) and has already been performed in the private sector in England in November 2010 (Citation92).
The oxidative stress-induced damage to the endothelium and PVAT in obesity would suggest that antioxidants should serve as suitable therapeutic agents to reverse this damage and possibly lower blood pressure in obesity. Indeed, administration of desmethyltirilazad (Lazaroid), a potent antioxidant, to SHR rats for 3 weeks has been shown significantly to ameliorate blood pressure in these animals whilst having no effect in control animals (Citation93).
We have already described that MR activation by aldosterone results in the generation of reactive oxygen species. Data from our group have also shown that MR blockade using eplerenone is able to reduce macrophage activation and rescue aldosterone-induced and hypoxia-induced PVAT damage (Citation63).
Well-designed and large-scale clinical trials are required to assess the potential antihypertensive actions of traditional antioxidants such as vitamin C or scavengers of ROS such as superoxide dismutase. It has been recently proposed that prevention of ROS generation using NADPH oxidase (NOX1 and NOX2) inhibitors may be a better way of tackling the oxidative stress problem rather than attempting to scavenge the free radicals once they have been generated (Citation94).
Vitamin D is known to function as a negative modulator of RAAS by suppressing renin transcription via a vitamin D receptor (VDR)-mediated mechanism. It has been shown that VDR-null mice have increased renin and angiotensin II levels and develop hypertension and cardiac hypertrophy (Citation95); thus vitamin D analogues can potentially serve as antihypertensive agents in the context of obesity. However, vitamin D is also implicated in energy expenditure and adipocyte biology. Compared with WT animals, VDR-null transgenic mice, fed a high-fat diet, remain lean and exhibit smaller adipocytes and lower leptin and triglyceride levels, as well as higher energy expenditure and oxygen consumption leading to lower fat mass (Citation96). These findings are a result of global ablation of the vitamin D receptor and do not identify the organ system or tissues responsible for the effects. A recent publication has shown that overexpression of VDR in adipocytes leads to suppression of lipolysis, reduction in fatty acid beta-oxidation and thermogenesis, and overall reduction in energy expenditure, leading to obesity (Citation97).
The current evidence suggests that use of vitamin D analogues to treat obesity-related hypertension would be fraught with difficulties. Vitamin D is a highly fat-soluble compound and is readily deposited within adipose tissue leading to lower plasma levels of the vitamin. This leads to attenuation in the negative regulation of renin expression. Higher levels of vitamin D deposited in adipocytes would also lead to reduced lipolysis and energy expenditure and contribute to further weight gain (Citation98).
Conclusion
Obesity-related hypertension has a complex aetiology and pathophysiology. Development of new therapies to treat this disorder requires a better understanding of the physiological dysfunction and the intricate interplay between obesity and hypertension. Rescuing the damaged PVAT function in obesity may be a new target for treatment of hypertension associated with obesity.
Declaration of interest: Dr Aghamohammadzadeh is a British Heart Foundation and NIHR Manchester Biomedical Research Centre clinical research fellow. Work within Professor Heagerty's group is supported by Wellcome Trust Clinical Research Facility.
References
- WHO. Obesity and Overweight: Fact Sheet number 311. 2011 [updated 2011; cited]; Available from: http://www.who.int/mediacentre/factsheets/fs311/en/index.html (accessed 8 November 2009).
- The NHS Information Centre LS. Statistics on obesity, physical activity and diet: England, 2011. Available from: http://www.ic.nhs.uk/pubs/opad11. Publisher: The health and social care information centre. (accessed 24 February 2011).
- CDC. U.S. Obesity Trends. 2011 [updated 2011; cited 18 August 2011]. Available from: http://www.cdc.gov/obesity/data/trends.html#State.
- ; Emerging Risk Factors CollaborationWormser D, Kaptoge S, Di Angelantonio E, Wood AM, Pennells L, . Separate and combined associations of body-mass index and abdominal adiposity with cardiovascular disease: collaborative analysis of 58 prospective studies. Lancet. 2011;377: 1085–95.
- Cornier MA, Despres JP, Davis N, Grossniklaus DA, Klein S, Lamarche B, . Assessing adiposity: a scientific statement from the American Heart Association. Circulation. 2011;124:1996–2019.
- CDC. Hypertension. 2011 [updated 2011; cited 18 August 2011]. Available from: http://www.cdc.gov/nchs/fastats/hyprtens.htm.
- WHO. Blood pressure. 2011 [updated 2011; cited 17 July 2011]. Available from: http://www.who.int/gho/ncd/risk_factors/blood_pressure_prevalence/en/index.html.
- Henry SL, Barzel B, Wood-Bradley RJ, Burke SL, Head GA, Armitage JA. The developmental origins of obesity-related hypertension. ClinExpPharmacol Physiol. 2011 Aug 1. [Epub ahead of print].
- Kotchen TA. Obesity-related hypertension: epidemiology, pathophysiology, and clinical management. Am J Hypertens. 2010;23:1170–8.
- Gosmanov AR, Smiley DD, Robalino G, Siquiera J, Khan B, Le NA, . Effects of oral and intravenous fat load on blood pressure, endothelial function, sympathetic activity, and oxidative stress in obese healthy subjects. Am J Physiol Endocrinol Metab. 2010;299:E953–8.
- Dobrian AD, Schriver SD, Lynch T, Prewitt RL. Effect of salt on hypertension and oxidative stress in a rat model of diet-induced obesity. Am J Physiol Renal Physiol. 2003;285: F619–28.
- Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, . Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285:17271–6.
- Hall JE, Brands MW, Henegar JR. Mechanisms of hypertension and kidney disease in obesity. Ann N Y Acad Sci. 1999;892:91–107.
- Rumantir MS, Vaz M, Jennings GL, Collier G, Kaye DM, Seals DR, . Neural mechanisms in human obesity-related hypertension. J Hypertens. 1999;17:1125–33.
- Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension. 2006;48:787–96.
- Nagae A, Fujita M, Kawarazaki H, Matsui H, Ando K, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in obesity-induced hypertension. Circulation. 2009;119:978–86.
- Van Harmelen V, Ariapart P, Hoffstedt J, Lundkvist I, Bringman S, Arner P. Increased adipose angiotensinogen gene expression in human obesity. Obesity. 2000;8:337–41.
- Engeli S, Negrel R, Sharma AM. Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension. 2000;35:1270–7.
- Goodfriend TL, Calhoun DA. Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy. Hypertension. 2004;43:518–24.
- Goodfriend TL, Egan BM, Kelley DE. Aldosterone in obesity. Endocr Res. 1998;24:789–96.
- Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res. 1999;7: 355–62.
- Maron BA, Zhang YY, Handy DE, Beuve A, Tang SS, Loscalzo J, . Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinylthiol oxidation in vascular smooth muscle cells. J Biol Chem. 2009;284: 7665–72.
- Wang H, Shimosawa T, Matsui H, Kaneko T, Ogura S, Uetake Y, . Paradoxical mineralocorticoid receptor activation and left ventricular diastolic dysfunction under high oxidative stress conditions. J Hypertens. 2008;26: 1453–62.
- Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, . Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–97.
- Maron BA, Leopold JA. Aldosterone receptor antagonists: effective but often forgotten. Circulation. 2010;121: 934–9.
- de Paula RB, da Silva AA, Hall JE. Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration. Hypertension. 2004;43:41–7.
- Rahmouni K, Barthelmebs M, Grima M, Imbs JL, De Jong W. Involvement of brain mineralocorticoid receptor in salt-enhanced hypertension in spontaneously hypertensive rats. Hypertension. 2001;38:902–6.
- DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest. 1975;55: 845–55.
- Kotsis V, Stabouli S, Papakatsika S, Rizos Z, Parati G. Mechanisms of obesity-induced hypertension. Hypertens Res. 2010;33:386–93.
- Zheng Y, Yamada H, Sakamoto K, Horita S, Kunimi M, Endo Y, . Roles of insulin receptor substrates in insulin-induced stimulation of renal proximal bicarbonate absorption. J Am SocNephrol. 2005;16:2288–95.
- Safar ME, Czernichow S, Blacher J. Obesity, arterial stiffness, and cardiovascular risk. J Am SocNephrol. 2006;17 (Suppl 2):S109–11.
- Singhal A, Farooqi IS, Cole TJ, O'Rahilly S, Fewtrell M, Kattenhorn M, . Influence of leptin on arterial distensibility: a novel link between obesity and cardiovascular disease? Circulation. 2002;106:1919–24.
- Westerbacka J, Vehkavaara S, Bergholm R, Wilkinson I, Cockcroft J, Yki-Jarvinen H. Marked resistance of the ability of insulin to decrease arterial stiffness characterizes human obesity. Diabetes. 1999;48:821–7.
- Yudkin JS, Eringa E, Stehouwer CD. “Vasocrine” signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet. 2005;365:1817–20.
- Jin RC, Loscalzo J. Vascular nitric oxide: formation and function. J Blood Med. 2010;2010:147–62.
- Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342:1378–84.
- Wolk R, Shamsuzzaman AS, Somers VK. Obesity, sleep apnea, and hypertension. Hypertension. 2003;42:1067–74.
- Yang R, Sikka G, Larson J, Watts VL, Niu X, Ellis CL, . Restoring leptin signaling reduces hyperlipidemia and improves vascular stiffness induced by chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2011;300: H1467–76.
- Dudenbostel T, Calhoun DA. Resistant hypertension, obstructive sleep apnoea and aldosterone. J Hum Hypertens. 2010 Jun 9. [Epub ahead of print].
- Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. ClinExpHypertens A. 1991;13:277–96.
- Gao YJ, Lu C, Su LY, Sharma AM, Lee RM. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007;151:323–31.
- Galvez-Prieto B, Bolbrinker J, Stucchi P, de Las Heras AI, Merino B, Arribas S, . Comparative expression analysis of the renin-angiotensin system components between white and brown perivascular adipose tissue. J Endocrinol. 2008;197:55–64.
- Lu C, Su LY, Lee RM, Gao YJ. Alterations in perivascular adipose tissue structure and function in hypertension. Eur J Pharmacol. 2011;656:68–73.
- Dubrovska G, Verlohren S, Luft FC, Gollasch M. Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am J Physiol Heart Circ Physiol. 2004;286: H1107–13.
- Deng G, Long Y, Yu YR, Li MR. Adiponectin directly improves endothelial dysfunction in obese rats through the AMPK-eNOS Pathway. Int J Obes (Lond). 2010;34:165–71.
- Yilmaz MI, Sonmez A, Caglar K, Celik T, Yenicesu M, Eyileten T, . Effect of antihypertensive agents on plasma adiponectin levels in hypertensive patients with metabolic syndrome. Nephrology (Carlton). 2007;12:147–53.
- Fesus G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC, . Adiponectin is a novel humoral vasodilator. Cardiovasc Res. 2007;75:719–27.
- Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, . Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119: 1661–70.
- Aghamohammadzadeh R, Withers SB, Lynch FM, Greenstein AS, Malik R, Heagerty AM. Perivascular adipose tissue from human systemic and coronary vessels: the emergence of a new pharmacotherapeutic target. Br J Pharmacol. 2012;165:670–82.
- Lu C, Zhao AX, GaoYJ, Lee RM. Modulation of vein function by perivascular adipose tissue. Eur J Pharmacol. 2011;657:111–6.
- Lee RM, Bader M, Alenina N, Santos RA, Gao YJ, Lu C. Mas receptors in modulating relaxation induced by perivascular adipose tissue. Life Sci. 2011;89:467–72.
- Byku M, Macarthur H, Westfall TC. Inhibitory effects of angiotensin (1 - 7) on the nerve stimulation-induced release of norepinephrine and neuropeptide Y from the mesenteric arterial bed. Am J Physiol Heart Circ Physiol. 2010;298: H457–65.
- Marques FD, Ferreira AJ, Sinisterra RD, Jacoby BA, Sousa FB, Caliari MV, . An oral formulation of angiotensin- (1 – 7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension. 2011;57:477–83.
- Ribiere C, Jaubert AM, Gaudiot N, Sabourault D, Marcus ML, Boucher JL, . White adipose tissue nitric oxide synthase: a potential source for NO production. BiochemBiophys Res Commun. 1996;222:706–12.
- Gil-Ortega M, Stucchi P, Guzman-Ruiz R, Cano V, Arribas S, Gonzalez MC, . Adaptativenitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology. 2010;151:3299–306.
- Ribiere C, Jaubert AM, Sabourault D, Lacasa D, Giudicelli Y. Insulin stimulates nitric oxide production in rat adipocytes. BiochemBiophys Res Commun. 2002;291:394–9.
- Mehebik N, Jaubert AM, Sabourault D, Giudicelli Y, Ribiere C. Leptin-induced nitric oxide production in white adipocytes is mediated through PKA and MAP kinase activation. Am J Physiol Cell Physiol. 2005;289:C379–87.
- Rahmouni K, Correia ML, Haynes WG, Mark AL. Obesity-associated hypertension: new insights into mechanisms. Hypertension. 2005;45:9–14.
- Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17(Pt 2):1949–53.
- da Silva AA, do Carmo J, Dubinion J, Hall JE. The role of the sympathetic nervous system in obesity-related hypertension. CurrHypertens Rep. 2009;11:206–11.
- Schleifenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, . Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens. 2010;28:1875–82.
- Lee YC, Chang HH, Chiang CL, Liu CH, Yeh JI, Chen MF, . Role of perivascular adipose tissue-derived methyl palmitate in vascular tone regulation and pathogenesis of hypertension. Circulation. 2011;124:1160–71.
- Withers BS, Agabiti-Rosei C, Linvingstone DM, Little MC, Aslam R, Malik RA, . Macrophage activation is responsible for loss f anticontractile function in inflamed perivascular fat. ArteriosclerThrombVasc Biol. 2011;31: 908–13.
- Fitzgibbons TP, Kogan S, Aouadi M, Hendricks GM, Straubhaar J, Czech MP. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am J Physiol Heart Circ Physiol. 2011;301: H1425–37.
- Guo C, Ricchiuti V, Lian BQ, Yao TM, Coutinho P, Romero JR, . Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-gamma, and proinflammatory adipokines. Circulation. 2008;117: 2253–61.
- Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM, . Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res. 2006;71: 363–73.
- Lu C, Su LY, Lee RM, Gao YJ. Mechanisms for perivascular adipose tissue-mediated potentiation of vascular contraction to perivascular neuronal stimulation: the role of adipocyte-derived angiotensin II. Eur J Pharmacol. 2011; 634:107–12.
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, . MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505.
- Kim CS, Park HS, Kawada T, Kim JH, Lim D, Hubbard NE, . Circulating levels of MCP-1 and IL-8 are elevated in human obese subjects and associated with obesity-related parameters. Int J Obes. 2006;30:1347–55.
- Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. ProcNatlAcadSci USA. 2003;100:7265–70.
- Shah R, Hinkle CC, Ferguson JF, Mehta NN, Li M, Qu L, . Fractalkine is a novel human adipochemokine associated with type 2 diabetes. Diabetes. 2011;60:1512–8.
- Sirois-Gagnon D, Chamberland A, Perron S, Brisson D, Gaudet D, Laprise C. Association of common polymorphisms in the fractalkine receptor (CX3CR1) with obesity. Obesity (Silver Spring). 2010;19:222–7.
- Timofeeva AV, Goryunova LE, Khaspekov GL, Kovalevskii DA, Scamrov AV, Bulkina OS, . Altered gene expression pattern in peripheral blood leukocytes from patients with arterial hypertension. Ann N Y Acad Sci. 2006;1091: 319–35.
- Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, . Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circ Res. 2009;104:541–9.
- Dorresteijn JA, Visseren FL, Spiering W. Mechanisms linking obesity to hypertension. Obes Rev. 2012;13:17–26.
- Lee CM, Huxley RR, Wildman RP, Woodward M. Indices of abdominal obesity are better discriminators of cardiovascular risk factors than BMI: a meta-analysis. J ClinEpidemiol. 2008;61:646–53.
- Reisin E, Abel R, Modan M, Silverberg DS, Eliahou HE, Modan B. Effect of weight loss without salt restriction on the reduction of blood pressure in overweight hypertensive patients. N Engl J Med. 1978;298:1–6.
- Ahmadi N, Eshaghian S, Huizenga R, Sosnin K, Ebrahimi R, Siegel R. Effects of intense exercise and moderate caloric restriction on cardiovascular risk factors and inflammation. Am J Med. 2011;124:978–82.
- Blumenthal JA, Sherwood A, Gullette ECD, Babyak M, Waugh R, Georgiades A, . Exercise and weight loss reduce blood pressure in men and women with mild hypertension: effects on cardiovascular, metabolic, and hemodynamic functioning. Arch Intern Med. 2000;160:1947–58.
- Heneghan HM, Meron-Eldar S, Brethauer SA, Schauer PR, Young JB. Effect of bariatric surgery on cardiovascular risk profile. Am J Cardiol. 2011;108:1499–507.
- Nicholas C, May R. HealthGrades Fifth Annual Bariatric Surgery Trends in American Hospitals Study; 2010 Contract No.: Document Number. Available from: http://www.healthgrades.com/media/DMS/pdf/HealthGradesBariatricSurgeryTrendsStudy2010.pdf. Publisher: HealthGrades Inc. (accessed 8 September 2010).
- Hofso D, Nordstrand N, Johnson LK, Karlsen TI, Hager H, Jenssen T, . Obesity-related cardiovascular risk factors after weight loss: a clinical trial comparing gastric bypass surgery and intensive lifestyle intervention. Eur J Endocrinol. 2010;163:735–45.
- Frezza EE, Wei C, Wachtel MS. Is surgery the next answer to treat obesity-related hypertension? J ClinHypertens (Greenwich). 2009;11:284–8.
- Butner KL, Nickols-Richardson SM, Clark SF, Ramp WK, Herbert WG. A review of weight loss following Roux-en-Y gastric bypass vs restrictive bariatric surgery: impact on adiponectin and insulin. Obes Surg.2010;20:559–68.
- Compher C, Badellino KO. Obesity and inflammation: lessons from bariatric surgery. JPEN J Parenter Enteral Nutr. 2008;32:645–7.
- Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev. 2009;11:11–18.
- Forsythe LK, Wallace JM, Livingstone MB. Obesity and inflammation: the effects of weight loss. Nutr Res Rev. 2008;21:117–33.
- Moschen AR, Molnar C, Geiger S, Graziadei I, Ebenbichler CF, Weiss H, . Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor {alpha} expression. Gut.2010;59: 1259–64.
- Blackburn GL, Wollner SB, Jones DB. Bariatric surgery as treatment for type 2 diabetes. CurrDiab Rep. 2010;10: 261–3.
- Kassab S, Kato T, Wilkins FC, Chen R, Hall JE, Granger JP. Renal denervation attenuates the sodium retention and hypertension associated with obesity. Hypertension. 1995;25(Pt 2):893–7.
- ; SymplicityHTN-2 InvestigatorsEsler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, . Renal sympathetic denervation in patients with treatment-resistant hypertension (The SymplicityHTN-2 Trial): a randomised controlled trial. Lancet. 2010;376:1903–9.
- The 1st renal denervation procedure to control hypertension conducted in the private sector. The Harley Street Clinic; 2010 [updated 2010; cited]. Available from: http://www.theharleystreetclinic.com/news/the-1st-renal-denervation-procedure-to-control-hypertension-conducted-in-the-private-sector (accessed 20 November 2011).
- Vaziri ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL. Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats. Hypertension. 2000;36: 957–64.
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–71.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110: 229–38.
- Wong KE, Szeto FL, Zhang W, Ye H, Kong J, Zhang Z, . Involvement of the vitamin D receptor in energy metabolism: regulation of uncoupling proteins. Am J PhysiolEndocrinolMetab. 2009;296:E820–8.
- Wong KE, Kong J, Zhang W, Szeto FL, Ye H, Deb DK, . Targeted expression of human vitamin D receptor in adipocytes decreases energy expenditure and induces obesity in mice. J Biol Chem. 2011;286:33804–10.
- Wortsman J, Matsuoka LY, Chen TC, Lu Z, Holick MF. Decreased bioavailability of vitamin D in obesity. Am J ClinNutr. 2000;72:690–3.