Abstract
Multiple factors including unhealthy living habits influence the life-maintaining functions of the endoplasmic reticulum (ER) and induce ER stress and metabolic abnormalities. The ER responds to the disturbances by activating mechanisms that increase the capacity to eliminate ER stress. This article elucidates the effects of ER activation that eliminates both ER stress and associated cardiovascular, type 2 diabetic (DM2), and other metabolic diseases. ER-activating compounds eliminate ER stress by lowering elevated cholesterol, regress atherosclerosis, decrease cardiovascular mortality, reduce blood glucose and insulin, and, together with the normalization of glucose–insulin homeostasis, remove insulin resistance, pancreatic β-cell failure, and DM2. A deficient cytochrome P450 activity in hepatic ER leads to cholesterol accumulation that induces stress and xanthoma formation, whereas P450-activating therapy up-regulates apolipoprotein AI and LDLR genes, down-regulates apolipoprotein B gene, and produces an antiatherogenic plasma lipoprotein profile. The ER activation reduces the stress also by eliminating hepatic fat and converting saturated fatty acids (FAs) to unsaturated FAs. Cognitive processes require gene expression modification, and preclinical studies indicate that ER-activating therapy improves cognition. Promotion of healthy lifestyle choices and indicated therapies are key factors in the prevention and elimination of ER stress and associated global health problems.
Key messages
The endoplasmic reticulum (ER) maintains vital cellular functions and responds to prolonged or excessive ER stress disturbing cellular homeostasis by activating mechanisms that eliminate both ER stress and associated cardiovascular, diabetic, and other metabolic disorders and diseases.
Positive ER stress-eliminating effects of the ER-activating therapies include lowering of elevated cholesterol, prevention and regression of atherosclerosis, normalization of glucose–insulin balance, removing of pancreatic β-cell failure, insulin resistance, and DM2, elimination of liver fat, and conversion of saturated fatty acids (FAs) to unsaturated FAs.
The studies emphasize the importance of activities that promote healthy living habits, and therapies known to alleviate and eliminate ER stress.
Introduction
The endoplasmic reticulum (ER) is a membranous synthesis, metabolism, and transport organelle that extends throughout the cytoplasm and encompasses about half of the total membrane area in a typical eukaryotic cell (Citation1,Citation2). It is continuous with the nuclear envelope and encloses a single internal space called the ER lumen. The ER has vital roles in multiple processes that are required for normal cellular function, including the synthesis and processing of proteins and lipids, regulation of glucose and calcium homeostasis, and metabolism of xenobiotic compounds (Citation1,Citation3). It is the site for the production of proteins destined for secretion and cell membranes and membrane lipid constituents (Citation4–7). P450 mono-oxygenases catalyze the biotransformation of both foreign and physiologically important substances and influence metabolic processes (Citation3,Citation8). The quality control mechanisms of the cell ensure that the newly made proteins are folded into their correct configuration according to the function and destination in the cell. A failure in the control leads to accumulation of unfolded proteins that are toxic to cells and, by disturbing cellular function, results in a state known as ER stress (Citation4,Citation5). The stress can affect the fate of proteins, lipids, and carbohydrates, cause cellular apoptosis, and promote disease processes.
The ER has capacity to adjust its functions and dimensions according to changing demands by triggering a signaling cascade called unfolded protein response (UPR) for providing new ER-folding machinery and by increasing ER proteins and membranes for restoring the metabolic balance. Clinical studies in the 1970s revealed that ER-activating compounds inducing membrane protein synthesis and proliferation, in proportion to this effect, produce a plasma lipoprotein profile indicating a low probability of atherosclerotic vascular disease (Citation8,Citation9). Consequent investigations in the 1980s revealed that the compounds inducing ER function can normalize glucose–insulin homeostasis and eliminate insulin resistance and type 2 diabetes (DM2) (Citation10,Citation11). Recent studies have emphasized the significance of disturbed ER functions in disease processes. This review article focuses on the activation of ER functions increasing ER membrane proteins and size and eliminating ER stress and major health problems including common cardiovascular, DM2, and other metabolic diseases.
UPR and elimination of ER stress
Physiological fluctuations of unfolded protein levels may cause temporary up-regulation of protein-folding machinery, while a prolonged or excessive incorrectly folded protein load can trigger full mobilization of mechanisms for the normalization of cell function and prevention of cell death. This section is a short summary of UPR mechanisms in the elimination of ER stress; for more detailed information, see recent reviews (Citation3–5,Citation12,Citation13).
ER folding mechanisms
Several ER response mechanisms can be employed to reduce unfolded protein load and alleviate ER stress, including (Citation14): 1) induction of ER chaperones to enhance folding activity in the luminal ER; 2) up-regulation of protein kinase-like ER kinase or PERK to inhibit translation and reduce the load of newly synthesized proteins; 3) activation of ER-associated degradation of unfolded proteins (ERAD); 4) activation of the apoptosis, a terminal death program to remove damaged cells in case the stress is too severe or prolonged for the recovery of cell function; and 5) up-regulation of phospholipid, protein, and membrane synthesis resulting in ER proliferation with increase of P450 (Citation15–18) and size (Citation19,Citation20). The enlarged ER alleviates ER stress and does so also independently of an increase of ER chaperone levels (Citation21). Transcriptional activation of the pro-apoptopic gene for CHOP (C/EBP homologous protein) and the IRE1-linked kinase pathway has been implicated in the ER stress-mediated apoptosis (Citation14). Autophagy is a degradation pathway under ER stress conditions that is responsible for the turnover of intracellular cargo via lysosomal degradation (Citation3).
Stress sensors and the mechanisms of UPR response
Unfolded proteins accumulated in the lumen of the ER activate three distinct ER membrane-associated stress sensors that up-regulate transcriptional and translational mechanisms to alleviate ER stress. They include inositol-requiring enzyme 1 (IRE1α), activating transcription factor (ATF6α), and PERK. Dissociation of BiP (binding protein) chaperone from the luminal domain of these sensors enables their activation.
Activated IRE1 alters the splicing of X box-binding protein 1 (XBP1), which then acts as a potent transcription factor. It up-regulates chaperones for ER protein folding and quality control, proteins involved in ER-associated protein degradation (ERAD) and in autophagy, and directs cells to construct more ER membranes for maintaining ER function (Citation19). Activated ATF6α moves to the nucleus and via transcriptional induction of ER chaperones and ERAD genes facilitates degradation of unfolded proteins (Citation22).The active form of ATF6α also induces membrane synthesis with resulting expansion of ER surface area and volume (Citation6,Citation23). Activated PERK phosphorylates translation elongation initiation factor 2α (elF2α), which results in down-regulation of general protein synthesis. It serves as a critical effector of UPR-induced growth arrest, linking stress in the ER to control of cell cycle progression (Citation24).
ER activators increase P450 and ER and liver size
ER-activating compounds up-regulate hepatic ER functions and increase phospholipids and proteins including P450 (Citation15–18) and ER size (Citation19,Citation21), and also liver size in man (Citation25,Citation26). Increased production of ER-resident P450 in hepatocytes has been shown to activate a number of cellular pathways, including those acting in membrane biogenesis, affecting the fate of cholesterol, and fatty acid (FA) metabolism () (Citation27).
Table I. Up-regulation of factors in membrane biogenesis and changes in the expression of cholesterol, chaperone, and FA desaturase genes induced by microsomal P450 overproduction in hepatocytes (Citation27).
Activated transcription factors influence ER structure and function. The active form of XBP1 induces CTP: phosphocholine cytidylyltransferase-α (CCTα) and the synthesis of phosphatidylcholine (PC), the primary phospholipid of the ER membranes (Citation6,Citation7), and co-ordinately increases membrane phospholipids and proteins, the key ingredients of ER biogenesis (Citation19). Overexpression of active ATF6α similarly induces PC synthesis and drives ER expansion and can do so in the absence of XBP1 (Citation23). ATF6α links membrane protein expression to ER expansion under conditions in which there is no indication of stress (Citation20). Activated nuclear factor kB (NF-kB) up-regulates antiapoptotic genes, ensuring survival of the hepatic cells with an expanding ER compartment (Citation27).
Activation of ER functions—effects in healthy subjects
Both endogenous and exogenous factors activate ER functions that beneficially influence lipid, protein, and carbohydrate metabolism in healthy subjects. Healthy young non-obese subjects show high plasma HDL-C, HDL2-C, and HDL-C/total cholesterol ratio together with high P450 activity in hepatic ER in vivo (Citation28) as indicated by enhanced antipyrine (AP) kinetics (Citation29). Functional cross-talk of regulatory factors links lipid and xenobiotic metabolism and P450 activity (Citation29–31). An endogenous up-regulation of apo AI synthesis raises plasma apo AI and HDL-C in familial hyper-HDL-emia which is characterized by a low death rate from coronary heart disease (CHD) and prolonged life expectancy (Citation32), while a low apo AI synthesis rate leads to hypo-HDL-emia (Citation33) that promotes atherogenesis. ER-activating drug therapy similarly increases HDL-C and HDL2-C in healthy individuals (Citation34). The positive changes in risk factor levels could result from induction of hepatic apo AI (Citation35), the key target for elevating HDL, and LDLR gene expression that associate with the increased production of P450 (Citation27). Young healthy non-obese subjects show high HDL2-C together with low plasma lipid peroxides that reflect a low level of cellular stress, and high serum glutathione peroxidase activities associate with low LDL-Cs (Citation36). Corresponding to these associations, HDLs have been found to protect cells from ER stress and autophagic response induced by oxidized LDLs (Citation37).
ER-activating therapy increasing ER P450 also influences glucose–insulin homeostasis in healthy subjects. It enhances plasma glucose disposal rate and metabolic clearance of glucose and reduces fasting immunoreactive insulin (IRI), whereas fasting blood glucose in healthy subjects remains unaltered (Citation38). These positive effects associate with enhanced P450 activity in hepatic ER in vivo, whereas P450-inhibiting therapy reduces glucose disposal rate, glucose metabolic clearance rate, and P450 activity (Citation38).
Healthy living habits including prudent diet and regular physical exercise influence ER functions and have positive metabolic effects. Diet constituents such as soy proteins and an optimal alcohol consumption induce apo AI expression and raise HDL-C, and vitamins, e.g. C and E, may have similar effects (Citation39). A healthy diet also reduces insulin secretion, increases insulin sensitivity, and prevents the occurrence of metabolic syndrome (Citation40) and also DM2 (Citation41). The factors activating ER functions and stimulating cholesterol effluxing mechanisms also prevent and regress atherosclerosis (Citation31). Moderate alcohol consumption reduces the risk of atherosclerotic disease, dementia (Citation42), CHD, and all-cause mortality (Citation43).
Regular aerobic physical exercise of endurance type up- regulates factors in lipid and apolipoprotein metabolism, including receptors such as liver X receptor α (LXRα), peroxisome proliferator-activated receptor γ (PPARγ), and LDLR, enzymes such as P450 and lecithin: cholesterol acyl transferase (LCAT), and ATP-binding cassette (ABC) transporters and apo AI (reviewed in (Citation44)). It also raises plasma apo AI, HDL-C, and HDL2-C, and reduces LDL-C, cholesterol, and triglycerides, and promotes cellular cholesterol efflux (Citation44). In addition, endurance exercise has been shown to ameliorate insulin resistance and ER stress in adipose and hepatic tissue in obese rats (Citation45) and to reduce postprandial hyperglycemia, improve glucose tolerance, and prevent the development of DM2 in man (Citation41). An anaerobic exercise, e.g. weight-lifting and sprinting, lasts a short period and increases the ability to perform tasks requiring strength and speed. It does not produce positive effect on lipid, protein, and glucose metabolism comparable to that of aerobic exercise.
ER stress in clinical disorders and diseases
Modern lifestyle, with an excess of energy-rich foods and physical inactivity, has led to a dramatic increase of clinical disorders and diseases such as obesity, atherogenic dyslipidemia, insulin resistance, metabolic syndrome, DM2, and hypertension, all of which are independent risk factors for atherosclerotic vascular disease, including CHD, myocardial infarction, and stroke (Citation46,Citation47). Atherosclerotic vascular disease has been identified as the leading cause of death in the world (Citation47,Citation48). One-third of US citizens are obese (BMI ≥ 30 kg/m2), two-thirds overweight (BMI 25 kg/m2) (Citation49), and nine out of ten diabetics in the US have DM2, the prevalence of which has tripled in the last 30 years (Citation50). Studies clarifying disease mechanisms have associated ER stress with the pathogenesis of these and many other ailments including neurodegenerative diseases and cancer.
ER stress in the liver
The liver ER has essential functions in the biosynthesis and catabolism of lipids, proteins, and carbohydrates and metabolism of xenobiotic compounds (Citation3,Citation31). P450 metabolizes in addition to xenobiotics a large number of physiologically important substances including cholesterol, FAs, steroids, prostaglandins, and bile acids. The multiple functions and effects of the liver influence metabolic processes also in other tissues.
Factors affecting hepatic function such as excessive nutrient intake and alcohol abuse can induce ER stress that activates UPR mechanisms for restoring hepatocellular functions (Citation3,Citation22,Citation51). If the cellular response to stress is insufficient, pathological consequences can ensue. A dysregulation of ER function can affect lipoprotein secretion and glucose–insulin homeostasis and promote lipid accumulation (Citation52) and hyperglycemia (Citation11), respectively. Metabolic perturbations can lead to spill-over of extra free FAs from adipose tissue and their entry to liver and other organs via the activated adipose triglyceride lipase and hormone-sensitive lipase (Citation53), induce hepatic fatty regeneration and cholestasis, and worsen the underlying causes of liver injury, such as diabetes–obesity-related liver disease (Citation22,Citation51). Both plasma lipoprotein risk factor levels (Citation18,Citation54) and glucose disposal rate (Citation55) vary in proportion to the amount of unaltered parenchyma in diseased liver as evaluated by morphometry.
ER stress and atherosclerosis
Atherosclerosis is a multifactorial disease which is characterized by the accumulation of lipids and inflammatory factors in arterial blood vessels (Citation56–59). Accumulation of apo B-containing lipoproteins in arterial intima triggers a series of maladaptive responses which are key factors in the initiation and progression of atherosclerotic lesions. Recent studies emphasize the significance of ER stress in the pathogenesis of atherosclerotic disease.
Multiple factors can induce ER stress and promote the atherosclerotic vascular process (Citation59). ER stress promotes hepatic lipogenesis, lipid accumulation, and dyslipidemia, and alleviation of ER stress can facilitate lipoprotein secretion (Citation59). Disturbed ER function also affects the fate of glucose and promotes hyperglycemia through insulin resistance, stimulation of hepatic glucose production, and suppression of glucose disposal (Citation11). The combination of stress-inducing stimuli, dyslipidemia, insulin resistance, and hyperglycemia promotes lipid accumulation, inflammation, and apoptosis and can accelerate the atherosclerotic process (Citation60). Cholesterol accumulation in ER membranes causes ER stress (Citation4) and can promote the progression of atherosclerosis (Citation61) through the activation of ER stress pathways that have been detected in atherosclerotic lesions. The ER stress–CHOP pathway is activated in macrophage-derived cells at all stages of the atherosclerotic process, and apoptosis becomes prominent at the late stage of atherosclerosis (Citation14). These processes lead to plaque formation and plaque instability and consequently to progression of atherosclerosis and possibly to infarction.
ER stress, obesity, inflammation, insulin resistance, pancreatic β-cell failure, and DM2
An imbalance in energy intake and expenditure leads to increase in body weight and obesity, a major health threat that increases the risk of DM2 and cardiovascular disease (CVD) (Citation47,Citation62,Citation63). Obese subjects show activation of ER stress pathways in metabolic tissues including adipose tissue, liver, and pancreas (Citation64,Citation65). Obesity also associates with both hepatic and peripheral insulin resistance together with elevated levels of proinflammatory cytokines in blood and tissues (Citation46,Citation66).
Obesity-induced inflammation can be described as a chronic low-grade inflammatory response initiated by excess nutrients in metabolic cells (Citation65). The three branches of the ER's unfolded protein response, IRE1, PERK, and ATF6, have been implicated in the cellular inflammatory process (Citation65). The inflammation has inhibitory effects on insulin action through the inflammatory kinases c-jun N-terminal kinase (JNK), inhibitor of k kinase (IKK), and protein kinase R (PKR) in metabolic tissues and disrupts nutrient and energy metabolism through these and other mechanisms (Citation65). Insulin resistance, a hallmark of DM2, is linked with a metabolic and cardiovascular cluster of disorders (dyslipidemia, obesity, glucose intolerance, endothelial dysfunction, hypertension), each of which is an independent risk factor for CVD. DM2 is characterized by hyperglycemia in the context of ER stress, insulin resistance, and relative insulin deficiency by β-cell failure (Citation66,Citation67).
Pancreatic β-cells have an essential role in the synthesis of insulin. Type 1 DM is an autoimmune disease with absolute insulin deficiency as a result of β-cell death. DM2 patients have relative insulin deficiency as a result of β-cell dysfunction and death consequent to effects of stressors including increased circulating glucose and saturated FAs and the development of inflammation (Citation68). Metabolic dysregulation, such as excess nutrients and insulin resistance, has been linked with the secretory burden of β-cell, leading to ER stress, severely compromising cell function (Citation69), and contributing to β-cell apoptosis (Citation70).
ER stress in neurodegenerative diseases
A common feature of neurodegenerative diseases is the accumulation of misfolded proteins in the brain (Citation71,Citation72). The aggregation of abnormal proteins can perturb cellular structure and function, lead to neuronal loss, and promote the pathogenesis of Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and many other ailments.
AD is the most common neurodegenerative disease. It also is the most common cause of dementia, the prevalence of which doubles every 5 years between 65 and 85 years of age (Citation71,Citation73,Citation74). The accumulation of aggregated proteins, extracellular amyloid β (Aβ) in the senile plaques and intracellular Tau protein in neurofibrillary tangles, is a prominent hallmark of AD (Citation74). Aβ has been considered as the main culprit in the pathogenesis of AD, resulting in synapse disruption and neuronal destruction. Several reports have indicated that UPR is activated in the neurons of AD brain (Citation75). Increased levels of the ER chaperone BiP have been found in the temporal cortex and the hippocampus of AD cases compared to non-demented control cases (Citation74). More direct evidence of UPR activation is the detection of activated PERK and phosphorylated elF2α in AD neurons (Citation74).
PD is the second most common neurodegenerative disease which affects around 2% of individuals over 65 years of age (Citation71). PD is a progressive disease with loss of dopaminergic neurons in the substantia nigra pars compacta, and with intracellular inclusion bodies termed Lewy bodies (Citation71). A major component of Lewy bodies is α-synuclein (αSyn) which could be involved in the pathogenesis of PD. Cellular studies have shown that overexpression of αSyn triggers chronic ER stress and cell death (Citation71). A mutation in the Parkin gene that induces ER stress and cell death has been linked with the pathogenesis of a juvenile-onset autosomal form of PD (Citation74). ER stress markers have also been reported in post-mortem tissue from PD cases (Citation71).
ALS is a motoneuron disease characterized by the degeneration of motor neurons in the spinal cord, cortex, and brain stem, leading to muscle atrophy and paralysis (Citation71,Citation74). Accumulating evidence suggests that ER stress contributes to ALS pathogenesis (Citation71). Mutations in the gene coding superoxide dismutase 1 (SOD1) have been linked with the pathogenesis of the disease. A mutation in ER-resident vesicle-associated membrane protein-associated protein B (VAPB), which is involved in lipid transport, causes familial ALS through interacting and inhibiting ATF6 and XBP1 and increases ER-stress-induced motoneuron vulnerability and death (Citation71,Citation76). Modifications of protein disulfide isomerase (PDI), a key ER foldase, could also contribute to the pathogenesis of ALS.
Other neurodegenerative diseases with evidence of ER stress involvement in pathogenesis include Huntington's disease, prion-related diseases, lysosomal storage diseases, myelin-related disorders, and retinitis pigmentosa (Citation71,Citation72).
ER stress, UPR, and cancer
Recent research in the cancer field has revealed that ER stress and UPR are highly activated in various tumors and closely associated with cancer cell survival and resistance to anticancer treatments (for recent reviews, see (Citation77,Citation78)). Accumulating evidence has shown that the UPR is an important mechanism required for cancer cells to maintain malignancy and therapy resistance. The UPR pathways are activated in various human tumor cells and animal models with cancer, including breast cancer, hepatocellular cancer, gastric tumor, and esophageal carcinoma (Citation78).
There are two main therapeutic approaches targeting the UPR components in cancer: induction of accumulation of misfolded protein in ER to overload the UPR response, and inhibition of UPR adaptive and antiapoptotic pathways to prevent cells from adapting stressful conditions leading to cell death. The UPR can respond by providing either survival signals for activating adaptive and antiapoptotic pathways, or death signals for inducing cell death programs (Citation78). Pharmacological activation or repression of UPR may thus have beneficial and therapeutic effects against cancer. Numerous compounds are in development as agents for UPR-targeted cancer therapy, including inhibitors of ERAD, IRE1α, HSP90, GRP78, and proteosomes such as bortezomib (Citation77,Citation78).
ER-activating therapy (ER-AC-TH) eliminates ER stress and diseases
ER-AC-TH: effects on ER, P450, liver, and risk factors, and regression of atherosclerosis
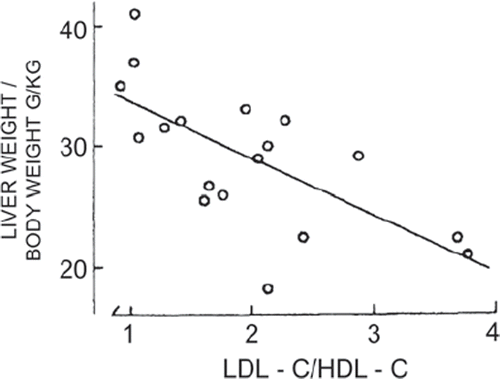
Clinical investigations performed in the 1970s evaluated the effects of drug therapies activating hepatic ER functions on key risk factors of atherosclerotic disease (Citation8,Citation9). Our original observations associated drug-caused up-regulation of ER-resident P450 with the elevation of plasma HDL-C and apo AI (Citation8,Citation9) and lowering of LDL-C (Citation29), and also with increase of hepatic protein and phospholipid concentrations (Citation17,Citation18), ER membranes (Citation8,Citation10), and of liver size in man (Citation26). Plasma apo AI and HDL-C rise in proportion to liver proteins, phospholipids, and ER P450 (Citation8,Citation18), and subjects with high P450 activity show low LDL-C and LDL-C/HDL-C ratio (Citation29). Corresponding to these changes, HDL-C rises and the LDL-C/HDL-C ratio () decreases with increasing liver size (Citation26). The favorable elevation of HDL-C and lowering of LDL-C and the LDL-C/HDL-C ratio with the increasing liver size could be linked with the induction of membrane expansion and up-regulation of apo AI (Citation35) and LDLR genes and the down-regulation of apo B gene (Citation27) expression. The beneficial effects of the therapies on risk factors prevent and regress atherosclerosis (next section).
Figure 1. Relationship between the LDL-C/HDL-C ratio and relative liver weight in patients on ER-activating drugs (r = –0.678; P 0.01). Reproduced with permission from Luoma PV, et al. Eur J Clin Pharmacol. 1985:28:615–8 (26).
The ER-activating therapy inducing hepatic ER CYP7A1 promotes cholesterol conversion to bile acids (Citation79) and can thus reduce cholesterol-induced ER stress (Citation4). Similarly, drug therapy inducing phospholipid synthesis and reducing the cholesterol/phospholipid ratio alleviates cholesterol-caused stress (Citation4).
ER-AC-TH: antiatherogenic effects of xenobiotics on lipoproteins
Numerous ER-activating compounds eliminate ER stress by lowering elevated cholesterol (Citation4) and have antiatherogenic effects. They include drugs indicated for the treatment of dyslipidemias such as statins, niacin, fibrates, cholestyramine (Citation79), and ezetimibe (Citation80). Angiotensin receptor blockers (ARBs), angiotensin-converting enzyme inhibitors (ACEIs), calcium channel blockers (CCBs), glitazones, anticonvulsants, retinoids, and alcohol also have antiatherogenic effects (Citation79).
Statins are effective antiatherogenic agents. They inhibit cholesterol synthesis, reduce hepatic cholesterol, up-regulate LDLR pathway, and reduce plasma LDL-C (Citation81). The drugs also induce apo AI synthesis in liver cells and raise plasma apo AI, HDL-C, and HDL2-C (Citation82). Statins stimulate P450 enzymes which generate PPAR-activating eicosanoids (Citation83,Citation84) and via PPAR activation up-regulate LXR, ABCA1, and ABCG1 genes in cholesterol-loaded macrophages and hepatocytes, and promote cholesterol efflux to apo AI and HDL (Citation79).
Niacin reduces cholesterol and triglycerides and raises apo AI and particularly HDL2-C (Citation85). It stimulates apo AI production, up-regulates PPARγ, LXRα, and ABCA1 genes, and promotes HDL-dependent cholesterol efflux (Citation79). Fibrates, PPARα agonists, reduce LDL-C and triglycerides and raise HDL-C and apo AI and AII (Citation86). Fibrate-caused PPARα activation up-regulates P450-FA-desaturase (Citation27,Citation87) and LXR and ABCA1 genes, and promotes cellular cholesterol efflux (Citation79). Cholestyramine binds bile acids, induces CYP7A1, and reduces cholesterol in hepatocytes, increases LDL-C elimination via the up-regulated LDLR pathway, and also induces apo AI synthesis and raises plasma apo AI and the HDL2-C subfraction (Citation88). Ezetimibe, an effective inhibitor of cholesterol absorption, reduces plasma cholesterol and apo B and raises HDL-C (Citation80). It has also been shown to decrease hepatic cholesterol, triglycerides, and steatosis, improve insulin signaling in rats, and reduce reactive oxygen species generation and ER stress in steatotic hepatocytes (Citation89).
ER-AC-TH targeting the UPR and atherosclerosis
Different kinds of molecules act on ER stress and UPR and have potential to influence the atherosclerotic vascular process. Studies on animal models of obesity, DM2, and atherosclerosis have indicated that chemical chaperones can alleviate disease symptoms by reducing ER stress (Citation90). Chemical chaperones such as 4-phenylbutyrate (PBA) and tauroursodeoxycholic acid (TUDCA) have been shown to reduce ER stress both in vitro and in vivo in rodent models (Citation91). PBA has been shown to reduce macrophage ER stress and vascular disease progression in vivo, and the reducing of the stress through macrophage lipid chaperone alleviates atherosclerosis (Citation91). Statins have also recently been shown to activate UPR and ameliorate ER stress. They up-regulate UPR target genes such as GRP78 and transcription factors XBP1 and ATF6 (Citation92). Other molecules that enhance the UPR include BiP inducer X (BIX), a small molecule inducer of endogenous GRP78 expression, and salubrinal which confers protection from the accumulation of unfolded proteins in the ER (Citation60). Valproate, an ER chaperone inducer, has been found to attenuate accelerated atherosclerosis in mice in vivo (Citation60).
ER-AC-TH eliminates pancreatic ER stress, β-cell failure, insulin resistance, and DM2
Drug therapy up-regulating ER functions in DM2 patients reduces both blood glucose and plasma IRI levels (Citation10,Citation11). Consequently, the therapy that normalizes glucose–insulin homeostasis eliminates the stress-linked glucose intolerance, insulin resistance, and DM2 (Citation10,Citation11). The normalization of glucose and IRI levels also indicates the elimination of the DM2-associated pancreatic ER stress and β-cell failure. Changes in hepatic ER function influence lipid, protein, and glucose metabolism, and, correspondingly, DM2 patients show improvement of both glucose tolerance and plasma lipoprotein profile with increasing liver phospholipid and P450 (Citation93). All three UPR transducer (IRE1α, ATF6α, PERK) responses are crucial in β-cell to alleviate ER stress and restore ER homeostasis (Citation68).
TUDCA has been shown to alleviate ER stress in obese and diabetic mouse models, normalize hyperglycemia, increase insulin sensitivity, resolve the fatty liver disease, and enhance insulin action in liver, muscle, and adipose tissues (Citation91). Studies testing TUDCA in obese subjects with insulin resistance also have produced positive results: an increase in hepatic and muscle insulin sensitivity (Citation94). Weight loss has beneficial effects. It reduces ER stress in man, together with a decrease of triglycerides in adipose tissue and liver, and blood glucose and insulin concentrations (Citation64). The activation of nuclear receptors could also modify the effect of the diet on lipids and glucose. Recent studies revealed that an agonist of the constitutive androstane receptor (CAR), which is highly expressed in the liver, reversed diet-induced obesity, reduced hepatic steatosis, improved insulin sensitivity (Citation95), and ameliorated diabetes and fatty liver disease in mice (Citation96).
ER-AC-TH eliminates hepatic steatosis and cholestasis
A dysregulation of cell function inducing ER stress and affecting lipoprotein metabolism can lead to fat accumulation in the liver, and ER-activating factors including living habits and drugs promote fat elimination. The compounds induce reparation of cell membranes and ER proliferation, reduce liver fat (Citation10) and triglycerides in man (Citation17), and can eliminate the stress-associated steatosis. The chemical chaperones, PBA and TUDCA, and molecular chaperone BiP/GRP78 have been shown to reduce hepatic steatosis in obese mice (Citation97).
The compounds inducing ER CYP7A1, the rate-limiting hepatic P450 enzyme in the synthesis of bile acids, enhance cholesterol conversion into bile acids (Citation79) and can by normalizing impaired bile acid synthesis and bile flow eliminate the ER stress-associated cholestasis.
Effects of FAs and ER-AC-TH-induced FA desaturation
Saturated and unsaturated FAs differ significantly in their effects on cellular functions. Saturated FAs including palmitate and stearate, but not their monounsaturated counterparts, are known to induce ER stress (Citation90) and contribute to the pathogenesis of obesity, the metabolic syndrome, and CVD (Citation98). FA-binding proteins (FABPs) influence the effects of FAs (Citation90). While saturated FAs are proinflammatory, unsaturated omega-3 FAs including docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) have opposite effects. A recent study identified GRP120 as an omega-3 FA receptor mediating potent anti-inflammatory, insulin- sensitizing, and antidiabetic effects of DHA and EPA (Citation99). C16:1n7 palmitoleate, a lipokine, has been identified as a potential adipose tissue-derived lipid hormone that stimulates muscle insulin action and suppresses hepatosteatosis (Citation100).
The ER-AC-TH increasing P450-desaturase activity (Citation27,Citation87) induces the conversion of saturated FAs into unsaturated FAs and thus reduces FA-induced ER stress and has beneficial eliminating effects on associated metabolic abnormalities.
ER-AC-TH in neurodegenerative diseases
Different kinds of compounds act on neuronal function and have potential as therapeutic agents in neurodegenerative diseases. Several mechanisms could be involved including activation of nuclear receptors, reducing ER stress, enhancing the UPR, and stimulation of protein degradation.
LXRs have an important function in lipid homeostasis in the brain, and the loss of the receptors leads to neurodegenerative processes (Citation101). LXR-activating compounds reduce amyloid load and improve cognitive function in mouse models of AD (Citation102). Pregnenolone-16α-carbonitrile (PCN)), a pregnane X receptor (PXR) agonist, has also been shown to reduce brain Aβ-levels (Citation103), and PBA to reduce AD-linked Tau protein and ameliorate cognitive deficit in a mouse model of AD (Citation104). PBA also restored the folding of acetylcholine receptors in a cell culture (Citation74). Arimoclomol, an inducer of molecular chaperone, has been shown to reduce neuronal loss and improve motor function in mutant SOD1 mice (Citation74). Valproate, a drug used for the treatment of epilepsy and bipolar disorder, has been found to increase BiP and other ER chaperones (Citation74) and could be effective in AD by reducing ER stress and Tau phosphorylation. An inducer of BiP, BIX, protects against ER stress and neuronal loss in an animal model, suggesting a beneficial effect in neurodegenerative disorders (Citation74). Lithium, which promotes the activation of autophagy, has been found to be effective in delaying disease progression in ALS patients, and salubrinal to act against ER stress and be protective in a PD cell model and in ischemia-induced stress in mice (Citation74).
Discussion
Advanced methods in molecular biology have identified diverse biological and clinical roles for ER in maintaining cellular homeostasis. It responds to both endogenous and exogenous stimuli and generates new signaling molecules acting in metabolic processes. ER-activating compounds induce membrane synthesis and increase ER size and also liver size in man. The liver is of critical importance in the metabolism and elimination of endogenous and exogenous compounds, and changes in hepatic structure and function influence metabolic balance also in other tissues. The ER size control has an important function in maintaining cellular homeostasis. The enlargement of ER volume and luminal space through membrane synthesis increases the ER capacity to respond to changing demands, and it is an integral yet distinct part of the cellular program to overcome ER stress (Citation21). gives a summary of the effects of ER activation presented in this article and earlier studies.
Table II. Effects of ER activation (this review, references (Citation8,Citation27,Citation44)).
The clinical studies reviewed show that the compounds activating ER functions alleviate and eliminate major global health problems. They prevent and regress atherosclerosis, reduce cardiovascular morbidity and mortality, and remove insulin resistance and DM2 together with the elimination of the DM2-linked pancreatic β-cell failure. The ER activation could also influence cognitive processes. They require gene expression modification to consolidate information, and disturbed transcriptional regulation could perturb neuronal function and cognitive performance (Citation104). Healthy living habits prevent cognitive decline and dementia in man, and preclinical studies show that ER-activating therapy improves cognition. Low HDL has been identified as a risk factor for deficit and decline in memory in midlife (Citation105), and, correspondingly, high HDL-C and apo AI concentrations associate with good cognitive performance in advanced age (Citation106). Hyperinsulinemia similarly promotes cognitive impairment and increases the risk of AD (Citation73), and removing modifiable risk factors of DM has a great reducing impact on the incidence of dementia (Citation107).
The relation of lipoprotein metabolism and glucose-insulin homeostasis to ER function has been evaluated in studies including different patient groups and also healthy individuals. The up-regulation of ER-resident P450 has been linked with the expression of genes positively affecting the fate of cholesterol, and, correspondingly, individuals with high P450-activity in hepatic ER show high HDL-C, HDL2-C, and HDL-C/LDL-C ratio, while a deficient P450 activity reduces bile acid secretion, leads to hepatic cholesterol accumulation, hypercholesterolemia (Citation108), and xanthoma formation, and promotes atherogenesis (Citation109). Studies on hepatic ER function and glucose metabolism have revealed that induction of glucokinase, a potential target for glucose-lowering therapy, reduces glucose and that restoration of low enzyme activity normalizes plasma glucose in diabetic fatty rats (Citation110,Citation111).
The recent discovery that brown adipose tissue (BAT) is present and active in adult humans has renewed interest in the study of this tissue (Citation112,Citation113). BAT is activated by exposure to cold, and the presence of active BAT is inversely associated with obesity and traits of metabolic syndrome (Citation112). The potential positive effects of BAT activation have led to increasing interest in this tissue as a target for pharmacological treatment of obesity and elimination of associated ER stress, hepatic steatosis, lipid disorders, insulin resistance, and DM2.
Dietary factors influence disease processes, and different kinds of dietary supplements have been marketed for health promotion. Lecithin or phosphatidylcholine (PC) is a commonly recommended supplement to reduce the risk of CVD. PC has important physiological functions, but no favorable effect of dietary PC on the risk of heart disease has been documented. Hence, it is of interest that a recent study reported potentially harmful, ER stress-linked effects of the metabolites of this supplement (Citation114). Gut flora converted PC to metabolites that were found to augment cholesterol accumulation in macrophages, promote the atherosclerotic process in mice, and predict the risk for CHD in a large clinical cohort. The discovery of a relationship between gut flora-dependent metabolism of dietary PC and CVD pathogenesis also implicated some therapeutic approaches to prevent and treat atherosclerosis, including the use probiotics to modulate gut flora and reduce the production of harmful metabolites.
Studies clarifying the effect of age on ER function indicate that there is impairment of the adaptive ER stress response and increase in pro-apoptopic signaling (Citation115). Many key components of the UPR could display reduced activity with aging, resulting in a dysfunctional ER (Citation115) and thus increasing the risk of cardiovascular and metabolic disease and cognitive impairment. Elderly healthy individuals have had capacity to maintain adequate ER function. Very old people possess a lipoprotein profile that is typical for a low risk of atherosclerotic disease and good cognitive health (Citation106). The subfraction HDL2 which increases with aging is the most common phenotype among people in the advanced decades of life (Citation116) and associates with low plasma insulin levels and good glucose tolerance (Citation117).
In conclusion, this article elucidates the effects of ER functions on cellular homeostasis and regulation of metabolic processes. It clarifies how ER stress leads to cellular malfunction and disease processes and how ER activation eliminates both ER stress and associated cardiovascular, diabetic, and other metabolic diseases. ER-activating compounds induce reparation of altered cellular structures consequent to ER stress and normalize and improve metabolic functions. Lifestyle factors and appropriate therapies are central in the prevention and elimination of ER stress and promotion and maintaining well-being.
Acknowledgements
The author is grateful for excellent collaboration especially with Eero Sotaniemi (†), Olavi Pelkonen, Markku Savolainen, and Vilho Myllylä from the University of Oulu, Finland; Christian Ehnholm from the National Institute of Health and Welfare, Helsinki; and Heikki Vapaatalo from the University of Helsinki. The studies have been supported by the Academy of Finland and the Paavo Nurmi Foundation, Finland.
Declaration of interest: The author reports no conflicts of interest.
References
- Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13:374–84.
- Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–73.
- Mandl J, Mèszàros T, Bànhegyi G, Hunguady L, Csala M. Endoplasmic reticulum: nutrient sensor in physiology and pathology. Trends Endocrinol Metab. 2009;20:194–201.
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29.
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30.
- Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41.
- Shaffer AL, Shapiro-Shelef M, Iwakoshi N, Lee AH, Qian SB, Zhao Hong, . XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93.
- Luoma PV. Gene activation, apolipoprotein A-I/high density lipoprotein, atherosclerosis prevention and longevity. Pharmacol Toxicol. 1997;81:57–64.
- Luoma PV, Sotaniemi EA, Pelkonen RO, Ehnholm C. Plasma high density lipoprotein and liver microsomal enzyme activity in man. In: Pilli-Sihvola AS, Laaksovirta T, editors The Medical Research Council 1977–1979. Helsinki: The Academy of Finland; 1980. p. 71.
- Sotaniemi EA, Arranto AJ, Sutinen S, Stengård J, Sutinen S. Treatment of noninsulin dependent diabetes mellitus with enzyme inducers. Clin Pharmacol Ther. 1983;33:826–35.
- Lahtela JT, Arranto AJ, Sotaniemi EA. Enzyme inducers improve insulin sensitivity in non-insulin dependent diabetic subjects. Diabetes. 1985;34:911–6.
- Hetz C, Martinon F, Rodriquez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1. Physiol Rev. 2011;91:1219–43.
- Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic disease. Diabetes Obes Metab. 2010;12(Suppl 2):108–15.
- Gotoh T, Endo M, Oike Y. Endoplasmic reticulum stress related inflammation and cardiovascular disease. Internat J Inflamm. 2011;2011: 259462.
- Orrenius S. Further studies on the induction of the drug hydroxylating enzyme system of liver microsomes. J Cell Biol. 1965;26:725–33.
- Conney AH. Pharmacological implications of microsomal enzyme induction. Pharmacol Rev. 1967;19:317–66.
- Savolainen MJ, Arranto AJ, Hassinen IE, Luoma PV, Pelkonen RO, Sotaniemi EA. Relationship between lipid composition and drug metabolizing capacity of human liver. Eur J Clin Pharmacol. 1985;27:727–32.
- Luoma PV, Savolainen MJ, Sotaniemi EA, Pelkonen RO, Arranto A, Ehnholm C. Plasma high- density lipoproteins and liver lipids and proteins in man. Acta Med Scand. 1983;214:103–9.
- Sriburi R, Bommiasamy H, Buldak GL, Robbins GR, Frank M, Jackowski S, . Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-(S)-induced endoplasmic reticulum biogenesis. J Biol Chem. 2007;282:7024–34.
- Maiuolo J, Bulotta S, Verderio C, Benfante R, Borgese N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc Natl Acad Sci U S A. 2011;108:7832–7.
- Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. 2009;187:525–36.
- Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809.
- Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, . ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci. 2009;122:1626–36.
- Brewer JB, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A. 2000; 97:12625–30.
- Pirttiaho HI, Sotaniemi EA, Pelkonen RO, Pitkänen U. Hepatic blood flow and drug metabolism in patients on enzyme-inducing anticonvulsants. Eur J Clin Pharmacol. 1982;22:441–5.
- Luoma PV, Sotaniemi EA, Pelkonen RO, Pirttiaho HI. Serum low-density lipoprotein and high-density lipoprotein cholesterol, and liver size in subjects on drugs inducing hepatic microsomal enzymes. Eur J Clin Pharmacol. 1985;28:615–8.
- Szczesna-Skorupa E, Chen CC, Liu H, Kemper B. Gene expression changes associated with the endoplasmic reticulum stress response induced by microsomal cytochrome P450 overproduction. J Biol Chem. 2004;279:13953–61.
- Luoma PV, Rautio A, Stengård J, Sotaniemi EA, Marniemi J. High-density lipoprotein subfractions, apolipoproteins and antipyrine clearance in normal subjects. Eur J Clin Pharmacol. 1990;38:625–7.
- Luoma PV, Sotaniemi EA, Arranto AJ. Serum LDL cholesterol, the LDL/ HDL cholesterol ratio and liver microsomal enzyme induction evaluated by antipyrine kinetics. Scand J Lab Invest. 1983;43:671–5.
- Xiao L, Xie X, Zhai Y. Functional crosstalk of CAR-LXR and ROR-LXR in drug and lipid metabolism. Adv Drug Deliv Rev. 2010;62:1316–21.
- Luoma PV. Cytochrome P450—physiological key factor against cholesterol accumulation and the atherosclerotic vascular process. Ann Med. 2007;39:359–70.
- Rader DJ, Lohse P, Ikewaki K, Thomas F, Harris WA, Zech LA, . Increased production of apolipoprotein A-I associated with elevated plasma levels of high-density lipoproteins, apolipoprotein A-I and lipoprotein A-I in a patient with familial hyperalphalipoproteinemia. Metabolism. 1993;42:1429–34.
- Le NA, Ginsberg HN. Heterogeneity of apolipoprotein AI turnover in subjects with reduced concentrations of plasma high density lipoprotein cholesterol. Metabolism. 1988;37:614–17.
- Miller M, Burgan RG, Osterlund L, Segrest JP, Garber DW. A prospective randomized trial of phenytoin in nonepileptic subjects with reduced HDL cholesterol. Arterioscler Thromb Vasc Biol. 1995;15:2151–6.
- Chao YU, Pickett CB, Yamin TT, Guo LS, Alberts A, Kroon PA. Phenobarbital induces rat liver apolipooprotein A-I mRNA. Mol Pharmacol. 1985;27:394–8.
- Luoma PV, Stengård J, Korpela H, Rautio A, Sotaniemi EA, Suvanto E, . Lipid peroxides, glutathione peroxidase, high density lipoprotein subfractions and apolipoproteins in young adults. J Intern Med. 1990;227:287–9.
- Muller C, Salvayre R, Nègre-Salvayre A, Vindis C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011;18:817–28.
- Lahtela JT, Gachalyi B, Eksymä S, Hämäläinen A, Sotaniemi EA. The effect of enzyme inducing and inhibiting drugs on insulin mediated glucose metabolism in man. Br J Clin Pharmacol. 1986;21:19–26.
- Dullens SPJ, Plat J, Mensink RP. Increasing apoA-I production as a target for CHD risk reduction. Nutr Metab Cardiovasc Dis. 2007;17:618–28.
- Jimènez-Gomèz J, Marin C, Pèrez-Martinèz P, Hartwich J, Malczewska-Malec M, Gobalek I, . A low-fat, high-complex carbohydrate diet supplemented with long-chain (n-3) fatty acids alters the postprandial lipoprotein profile in patients with metabolic syndrome. J Nutr. 2010;140:1595–601.
- Pan XR, Li GW, Hu YH, Wang JX, Yang WY, An ZX, . Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. Diabetes Care. 1997;20:537–44.
- Stampfer MJ. Cardiovascular disease and Alzheimer's disease: common links. J Intern Med. 2006;260:211–23.
- Poikolainen K. Alcohol and mortality: a review. J Clin Epidemiol. 1995;48:455–65.
- Luoma PV. Gene activation regresses atherosclerosis, promotes health, and enhances longevity. Lipids Health Dis. 2010;9:67.
- da Luz G, Frederico MJ, da Silva S, Vitto MF, Cesconetto PA, de Pinho RA, . Endurance exercise ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue of obese rats. Eur J Appl Physiol. 2011;111:2015–23.
- Bray GA. Medical consequences of obesity. J Clin Endocrinol Metab. 2004;89:2583–9.
- DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis. The missing links. The Claude Bernard Lecture2009. Diabetologia. 2010;53:1270–87.
- Callow AD. Cardiovascular disease 2005—the global picture. Vascul Pharmacol. 2006;45:302–7.
- Flegel KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303;235–41.
- Brown MT, LeRoth D. Overcoming challenges in type 2 diabetes management to improve patient outcomes. Expert Rev Endocrinol Metab. 2010;5741–51.
- Ji C, Kaplowitz N. ER stress: can the liver cope?J Hepatol. 2006;45: 321–33.
- Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest. 2008;118:316–32.
- Yang X, Zhang X, Heckmann BL, Lu X, Liu J. Relative contribution of adipose triglyceride lipase and hormone-sensitive lipase to tumor necrosis factor-α (TNF-α)-induced lipolysis in adipocytes. J Biol Chem. 2011;286:40477–85.
- Luoma PV, Arranto AJ, Ehnholm C, Sotaniemi EA. Liver histological changes and plasma high density lipoproteins in man. Res Commun Chem Pathol Pharmacol. 1981;33:163–73.
- Lahtela JT, Arranto AJ, Stenbäck F, Sotaniemi EA. Insulin-mediated glucose metabolism is related to liver structure and microsomal function. Scand J Gastroent. 1986;21:737–43.
- Ross R. Atherosclerosis—an inflammatory disease. New Engl J Med. 1999;340:115–26.
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–12.
- Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–50.
- Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–9.
- McAlpine CS, Bowes AJ, Werstuck GH. Diabetes, hyperglycemia and accelerated atherosclerosis: evidence supporting a role for endoplasmic reticulum (ER) stress signaling. Cardiovasc Hematol Disord Drug Targets. 2010;10:151–7.
- Thorpe E, Iwawaki T, Miura M, Tabas I. A reporter for tracking the UPR in vivo reveals patterns of temporal and cellular stress during atherosclerotic progression. J Lipid Res. 2011;52:1033–8.
- Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008;28:629–36.
- Sha H, He Y, Yang L, Qi L. Stressed about obesity: IRE1α-XBP1 in metabolic disorders. Trends Endocr Metab. 2011;22:373–81.
- Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, . Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700.
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms of obesity. Annu Rev Immunol. 2011;29:415–45.
- Özcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Özdelen E, . Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61.
- Thomas SE, Dalton LE, Daly ML, Malzer E, Marciniak SJ. Diabetes as a disease of endoplasmic reticulum. Diabetes Metab Res Rev. 2010;26:611–21.
- Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocr Metab. 2011;22:266–74.
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61.
- Laybutt RD, Preston AM, Åkerfeldt MC, Kennch JG, Busch AK, Blankin AV, . Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–63.
- Matus S, Glimcher LH, Hetz C. Protein folding stress in neurodegenerative diseases: a glimpse into ER. Curr Opin Cell Biol. 2011;23: 239–52.
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–92.
- Luchsinger JA, Gustafson DR. Adiposity, type 3 diabetes and Alzheimer's disease. J Alzheimers Dis. 2009;16:693–704.
- Scheper W, Hoozemans JJM. Endoplasmic reticulum protein quality control in neuro-degenerative disease: the good, the bad and the therapy. Curr Med Chem. 2009;16:615–26.
- Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's pathology. J Neuroinflamm. 2009;6:41.
- Suzuki H, Kanekura K, Levine TP, Kohno K, Olkkonen VM, Aiso S, . ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J Neurochem. 2009;108:973–85.
- Wang G, Yang ZQ, Zhang K. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. Am J Transl Res. 2010;2:65–74.
- Li X, Zhang K, Li Z. Unfolded protein response in cancer: the physician's perspective. J Hematol Oncol. 2011;4:8.
- Luoma PV. Gene-activators prevent and regress atherosclerosis and reduce mortality. Cardiovasc Hematol Agents Med Chem. 2009;7: 295–304.
- Knopp RH, Gitter H, Truitt T, Bays H, Manion CV, Lipka LJ, . Effects of ezetimibe, a new cholesterol absorption inhibitor, on plasma lipids in patients with primary hypercholesterolemia. Eur Heart J. 2003;24:729–41.
- Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–8.
- Asztalos BF, Le Maulf F, Dallal GE, Stein E, Jones PH, Horvath KV, . Comparison of the effects of high doses of rosuvastatin versurs atorvastatin on the subpopulations of high-density lipoproteins. Am J Cardiol. 2007;99:681–5.
- Fleming I. Vascular cytochrome P450 enzymes: physiology and pathophysiology. Trends Cardiovasc Med. 2008;18:20–4.
- Ng V, Huang Y, Reddy LM, Falck JR, Lin ET, Kroetz DL. Cytochrome 450 eicosanoids are activators of peroxisome proliferator-activated receptor. Drug Metab Dispos. 2007;35:1126–34.
- Shepherd J, Packard CJ, Patsh JR, Gotto AM Jr, Taunton OD. Effects of nicotinic acid therapy on plasma high density lipoprotein subfraction distribution and composition and on apolipoprotein A metabolism. J Clin Invest. 1979;63:858–67.
- Taskinen MR, Sullivan DR, Ehnholm C, Whiting M, Zannino D, Simes RJ, . Relationships of HDL cholesterol, apo A-I, and apo A-II with homocysteine and creatinine in patients with type 2 diabetes treated with fenofibrate. Arterioscler Thromb Vasc Biol. 2009;29:950–5.
- Guillou H, Pascal M, Jan S, D’Andrea S, Roulet A, Catheine D, . Comparative effect of fenofibrate on hepatic desaturases in wild-type and peroxisome proliferator-activatedd receptor α-deficient mice. Lipids. 2002;37:981–9.
- Shepherd J, Packard CJ.High density apolipoprotein metabolism. In: Miller NE, Miller GJ, editors. Clinical and metabolic aspects of high-density lipoproteins. Amsterdam: Elsevier; 1984. p. 247–74.
- Nomura M, Ishii H, Kawakami A, Yoshida M. Inhibition of hepatic Niemann-Pick C1-like 1 improves hepatic insulin resistance. Am J Physiol Endocrinol Metab. 2009;297:E1030–8.
- Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, . Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15:1383–91.
- Özcan U, Ylimaz E, Özcan L, Furuhashi M, Vaillancourt E, Smith RO, . Chemical chaperones reduce ER stress and restore glucose homeostais in a mouse model of type 2 diabetes. Science. 2006;313: 1137–40.
- Chen JC, Wu ML, Huang KC, Lin WW. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc Res. 2008;80:138–50.
- Luoma PV, Savolainen MJ, Sotaniemi EA, Arranto AJ, Pelkonen RO. Plasma HDL cholesterol and glucose in non-insulin-dependent diabetics related to liver lipids and microsomal enzyme activity. Acta Med Scand. 1985;217:473–9.
- Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, . Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;50:1899–905.
- Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J Biol Chem. 2009;284:25984–92.
- Dong B, Saha PK, Huang W, Chen W, Abu-Elbeiga LA, Wakil SJ, . Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc Natl Acad Sci U S A. 2009;106:18831–6.
- Flamment M, Kammoun HL, Hainault I, Ferré P, Foufelle F. Endoplasmic reticulum stress: a new actor in the development of hepatic steatosis. Curr Opin Lipidol. 2010;21:239–46.
- Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–51.
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga T, Fan W, . GRP120 is an omega-3 fatty acid receptor mediating potent anti- inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–98.
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–44.
- Wang I, Schuster GU, Hultenby K, Zhang Q, Andersson S, Gustafsson JA. Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc Natl Acad Sci U S A. 2002; 99:13878–83.
- Fan J, Donkin J, Wellington C. Greasing the wheels of Aβ clearance in Alzheimer's disease. The role of lipids and apolipoprotein E. Biofactors. 2009;35:239–48.
- Hartz AMS, Miller DS, Bauer B. Restoring blood brain barrier P-glycoprotein reduces brain amyloid-β in a mouse model of Alzheimer's disease. Mol Pharmacol. 2010;77:715–23.
- Ricobaraza A, Cuadrado-Tejedor M, Péres-Mediavila A, Frechilla D, Del Rio J, Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721–32.
- Singh-Manoux A, Gimeno D, Kivimaki M, Brunner E, Marmot MG. Low HDL cholesterol is a risk factor for deficit and decline in memory in midlife. Arterioscler Thromb Vasc Biol. 2008;28:1556–62.
- Atzmon G, Gabriely I, Greiner W, Davidson D, Schechter C, Barzilai N. Plasma HDL levels correlate highly with cognitive function in exceptional longevity. J Gerontol A Biol Med Sci. 2002;57A:M712–5.
- Ritchie K, Carriere I, Ritchie CW, Berr C, Artero S, Ancelin M-L. Designing prevention programmes to reduce of dementia: prospective cohort study of modifiable risk factors. BMJ. 2010;341:c3885.
- Pullinger CR, Eng C, Salen G, Shefer S, Barra AK, Erickson SK, . Human cholesterol 7α-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109–17.
- Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266:7779–83.
- Karvonen I, Stengård J, Saarni HU, Stenbäck F, Sotaniemi EA. Hepatic mixed function oxidase system and enzymatic glucose metabolism in rats. Diabetes Res. 1987;4:195–200.
- Torres TP, Catlin RL, Chan R, Fujimoto Y, Sasaki N, Printz RL, . Restoration of hepatic glucokinase expression corrects hepatic glucose flux and normalizes plasma glucose in Zucker diabetic rats. Diabetes. 2009;58:78–86.
- Butler PW, Mentuccia D, Celi FS. Stimulating brown fat: a potential future therapeutic approach for obesity and insulin resistance?Heart Metab. 2010;48:19–22.
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, . Functional brown adipose tissue in healthy adults. New Engl J Med. 2009;360:1518–25.
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, . Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63.
- Naidoo N. ER and ageing-protein folding and the ER stress response. Ageing Res Rev. 2009;8:150–9.
- Arai Y, Hirose N. Aging and HDL metabolism in elderly people more than 100 years old. J Atheroscler Thromb. 2004;11:246–52.
- Ostlund RE, Staten M, Kohrt WM, Schultz, J, Malley M. The waist-to-hip circumference, plasma insulin level, and glucose intolerance as independent predictors of the HDL2 cholesterol levels in older adults. N Engl J Med. 1990;322:229–34.