Abstract
Insulin resistance is a pathological condition that arises when insulin signaling is impaired, forcing β-cells to produce more insulin in order to cope with body demands and to maintain glucose homeostasis. When the pancreas is no more able to support an appropriate insulin secretion, insulin resistance becomes decompensated and hyperglycemia is detected. One of the mechanisms leading to insulin resistance is low-grade inflammation that involves a number of protagonists such as inflammatory cytokines, lipids and their metabolites, reactive oxygen species (ROS), hypoxia and endoplasmic reticulum stress, and changes in gut microbiota profiles. We review here the molecular aspects of metabolic inflammation converging to insulin resistance and secondarily to type 2 diabetes. We also discuss the place of high-sensitivity C-reactive protein (hsCRP) in the assessment of metabolic inflammation and potential therapeutic interventions aimed to impede inflammation and therefore prevent insulin resistance.
Key messages
Metabolic inflammation is a low-grade chronic inflammation that is induced during obesity.
Obesity induces a release by hypertrophied adipocytes and inflammatory cells of pro-inflammatory cytokines.
These cytokines along with increases in free fatty acids induce signaling pathways that converge to the inhibition of the insulin receptor pathway.
The signaling pathways lead to the activation of NFκB which maintains a chronic inflammatory state through induction of other inflammatory cytokines.
Gut microbiota changes play a significant role in insulin resistance through the communication with gut epithelial cells.
Endoplasmic reticulum stress contributes to increased insulin resistance.
Hydrophilic statins have beneficial effects on insulin sensitivity.
Introduction
The pathogenesis of diabetes comprises several molecular mechanisms that involve various signaling pathways leading ultimately to hyperglycemia. Increased fasting glucose levels are due either to the absence of insulin secretion by pancreatic β-cells (type 1 diabetes or T1D) that comes mainly from an autoimmune mechanism, or to decompensated insulin resistance characterized by a dysfunction in insulin action and impaired insulin secretion (type 2 diabetes or T2D). Compensated insulin resistance characterizes a pre-diabetic state where insulin action is impaired but still sufficient to maintain a euglycemic condition through an increase in circulating insulin levels. Insulin resistance is a pathological condition that is usually found in obese patients, in whom visceral white adipose tissue (VAT) secretes in abnormal proportions a number of cytokines, named adipokines. Some of these molecules have anti-inflammatory effects such as the insulin-sensitizing cytokine adiponectin which is markedly decreased during obesity, whereas others have pro-inflammatory properties such as leptin, tumor necrosis factor alpha (TNFα), and interleukin-6 (IL-6) among other molecules (Citation1). These inflammatory cytokines characterize an inflammatory state, recently named ‘metaflammation’, which is defined by a chronic low-grade inflammation initiated by metabolic and inflammatory cells in response to an excessive energetic nutrient load (Citation2).
This definition is different from classical inflammation featuring the cardinal clinical signs: redness, heat, pain, and edema. The effects of metaflammation initiated in the adipose tissue will take place in metabolic organs such as the liver, skeletal muscle, pancreas, and brain, and lead to metabolic dysfunction and insulin resistance (Citation2). Not only do the adipocytes of obese patients secrete inflammatory cytokines but they recruit in situ inflammatory cells (mainly macrophages and lymphocytes) releasing inflammatory cytokines, which contributes to enhance the tissular inflammatory burden. In this review, we will explain how this low-grade inflammation is initiated, the cellular components involved, and the molecular mechanisms that lead to insulin resistance and hyperglycemia contributing to the clinical manifestation of T2D. We will briefly discuss the clinical consequences of metaflammation and the perspectives linked to this state.
Inflammatory cells, influential protagonists in the promotion of insulin resistance
Inflammatory macrophage activation and the phenotypic transition M2 to M1
Physiologically, most metabolic tissues host resident leukocytes that participate in the response to nutritional signals. The extent of macrophage recruitment in the VAT is dynamically controlled in order to cope with the flow of lipids and can serve to inhibit lipolysis. When the latter is triggered in tandem with cytokine release, the recruited macrophages may function to favor lipid storage while inhibiting lipolysis (Citation3). One of the major consequences of abdominal obesity is the infiltration of the adipose tissue by inflammatory cells, among which macrophages play a prevailing role in the installation of chronic low-grade inflammation (Citation4,Citation5). Importantly, macrophage stimulation by type 1 helper-T lymphocyte (TH1) cytokines such as interferon gamma (IFNγ) or by bacterial products such as lipopolysaccharide (LPS) favors their maturation in the so-called ‘classical’ type M1 macrophages harboring a high pro-inflammatory profile. In contrast, macrophage stimulation with TH2 lymphocyte cytokines such as interleukins-4 and -13 (IL-4 and IL-13) induces their maturation into the ‘alternative’ type M2 phenotype with anti-parasite, anti-inflammatory, and tissue remodeling and repair functions (Citation6). During obesity, a change in infiltrating macrophage polarity occurs from M2 cells secreting the anti-inflammatory molecules IL-10 and arginase-I toward M1 macrophages that release the pro-inflammatory cytokines TNFα, IL-6, and IL-12 (Citation6,Citation7). An important study has demonstrated the crucial role of the nuclear receptor peroxisome proliferator activated-receptor gamma (PPARγ), a key factor in adipocyte differentiation, in the alternative activation of macrophages. Likewise, macrophage-specific inactivation of PPARγ leads to a defect in the activation of the protective M2 macrophages and predisposes animals to insulin resistance, glucose intolerance, and diet-induced obesity. Moreover, these animals present muscular β-oxidation abnormalities, alteration in hepatic and muscular oxidative phosphorylation (OXPHOS), and a significant decrease in the expression of metabolic regulatory genes important in mitochondrial biogenesis and function such as the PPARγ-coactivators-1α and -1β (PGC-1α and PGC-1β) (Citation8). It becomes apparent now, at least in animal models, that the beneficial actions of PPARγ agonists, thiazolidinediones (TZD), in improving systemic insulin sensitivity lie mainly in the proper expression of PPARγ in macrophages and the activation of M2 cells. Indeed, treatment of mac-PPARγ-/- mice with TZD is not efficient in restoring insulin sensitivity, already severely altered at the basal level and even more affected by a high-fat diet (Citation9). In addition, the other member of the PPAR family of nuclear receptors and metabolic regulator, PPARβ/δ, is implicated in the polarity M2 ⤑ M1 of macrophages. Hence, it was demonstrated that adipocytes were capable of secreting TH2 cytokines, mainly IL-13 and to a lesser extent IL-4, that induced the expression of PPARβ/δ via a signaling pathway involving the silencer and transducer activator of transcription-6 (STAT-6), responsible for the activation of alternative macrophages M2. Genetic ablation of PPARβ/δ impeded the transition of macrophages toward the M2 phenotype and resulted in inflammation and insulin resistance in vivo (Citation10).
Furthermore, it was shown that in response to TH2 cytokines, macrophages induced an oxidative metabolic program with enhanced expression of PGC-1β, PPARγ, and PPARβ/δ that resulted in alternative activation of M2 macrophages. Alternatively, overexpression of PGC-1β and macrophage activation via an oxidative pathway induced a stimulation of the alternative phenotype at the expense of the classical one, and a significant decrease in pro-inflammatory cytokines (Citation11). These studies demonstrated that adipose tissue inflammation and resident macrophages are key elements in the development of insulin resistance during obesity and secondarily in the pathogenesis of T2D. A combined approach aimed at preventing M2-to-M1 transition associated with an inhibition of adipose tissue macrophage infiltration would be salutary in insulin resistance prevention during obesity. For example, the NAD+ -dependent class III histone deacetylase and nutrient sensor SIRT1 (Citation12) was shown to inhibit inflammatory pathways in macrophages, prevent adipose tissue macrophage infiltration upon high-fat diet (HFD), and modulate insulin sensitivity (Citation13,Citation14). SIRT1 activation has already been shown to prevent HFD-induced obesity and insulin resistance in mice (Citation15). Recently, decreased AMP-activated protein kinase (AMPK) activity was associated with increased inflammation in VAT and with whole-body insulin resistance in morbidly obese humans (Citation16). AMPK is a well-known nutrient sensor and regulator of SIRT1 activity (Citation17). Interestingly, it has been recently shown that activation of AMPKα1 in macrophages prevented fatty acid-induced inflammation through a signaling pathway involving SIRT1, underscoring a role of the AMPK/SIRT1 axis in bridging nutrient metabolism and inflammatory pathways (Citation18).
Finally, another marker, CD11c, was shown to be implicated in the phenotypic switch of macrophages from M2 to M1. The expression of CD11c is enhanced in the adipose tissue of diet-induced obese mice and of obese patients (Citation19,Citation20). Specific genetic ablation of CD11c in macrophages induced a robust decrease in the expression of inflammatory markers and a rapid normalization of insulin sensitivity in a model of insulin-resistant obese mice (Citation21). Other studies are necessary to determine with precision the function of CD11c-expressing cells. A recent report demonstrated that HFD replacement by a normal regime induced a decrease in the expression of inflammatory markers while maintaining the same number of CD11c + cells, suggesting that CD11c + macrophages present a phenotypic plasticity allowing them to adapt their function to the nutritional environment (Citation22).
Lymphocytes and inflammation
Besides macrophages, lymphocytes (lyc) seem to be strongly implicated in inflammatory processes linked to obesity. The presence of resident lyc has been firmly established in the stomal-vascular fraction of subcutaneous adipose tissue (SCAT) and epididymal adipose tissue (EAT) of non-obese rodents. The distribution of lyc depends on the localization of adipose tissue; those of the innate immune response (natural killer (NK) cells, NKT, and Tγδ lyc) were found in the EAT, while B-lyc and Tαβ lyc of the adaptive immunity were localized in SCAT depots. These specific localizations of lyc suggest that adipose tissue uses immunological mechanisms to regulate functions that can be altered during obesity (Citation23).
It has been demonstrated that the proportion of mast cells (Citation24) and natural killer T (NKT) cells (Citation25) is augmented in obese adipose tissue as compared to lean adipose tissue and that these cells contribute to the inflammation and metabolic abnormalities observed in obesity. In addition, a positive correlation was observed between the number of lyc in human adipose tissue and the degree of adiposity, this accumulation being more pronounced in the visceral adipose tissue than the SCAT (Citation26).
As described earlier, the proportion of CD4 + helper T lyc (TH1) was shown to be increased during obesity, and these cells released pro-inflammatory cytokines that favored the phenotypic transition M2 ⤑ M1 of macrophages (Citation27). Moreover, during obesity a significant increase in the ratio of CD8 +/CD4 + was observed in the adipose tissue of animals that was proportional to their degree of adiposity, enhancing thereby the contribution of CD8 + cells in macrophage recruitment and inflammation (Citation28). In parallel to an increase in CD8 + cells, a significant decrease in immunosuppressor T cell regulators (Treg) (CD4 +/Foxp3 +/CD25 +) associated with an increased inflammation and insulin resistance was observed (Citation28). All these changes in the proportion of different T cell types participate in the activation of an immune response during obesity. These data converge to the notion that metabolic abnormalities observed during obesity stem, at least in part, from a local immunological disturbance in the adipose tissue triggered by a glut of nutrients, hence considered as an aggressive agent by the organism.
Role of inflammatory cytokines in the development of insulin resistance
Tumor necrosis factor alpha (TNFα)
The first study to report the implication of an inflammatory cytokine in human obesity demonstrated a significant increase in the expression of TNFα in the adipose tissue of 19 premenopausal obese women as compared to 18 non-obese women (Citation29). A strong correlation was shown between the expression of TNFα in the adipose tissue and the degree of hyperinsulinemia as a measure of insulin resistance (Citation29). Not only was TNFα involved in the pathogenesis of insulin resistance in obese patients, but this phenomenon could be prevented with a regime allowing weight loss (Citation29). More globally, mice lacking TNFα function (lack of TNFα and its receptors) were protected from obesity-induced insulin resistance, suggesting an important role of TNFα in mediating insulin resistance during obesity (Citation30). Alternatively, the PPARγ agonists, thiazolidinediones, which induce an increase in subcutaneous fat, were shown to block TNFα-induced inhibition of insulin signaling (Citation31). This effect was corroborated by a recent study showing that genetic lack of TNFα protected insulin sensitivity during the early phase of high-fat feeding in the absence of altered total white adipose tissue (WAT). The mechanism behind this protection may involve redirection of the fat deposition to the metabolically more inert subcutaneous depot or decreases in circulating resistin and resultant decrease in liver fat deposition (Citation32).
The mechanism of insulin resistance is complex and involves a network of phosphatases and protein kinases that act in concert to block the insulin receptor pathway and hence diminish glucose entry into cells. For instance, the tyrosine phosphatase PTP1B has been shown to tyrosine dephosphorylate and deactivate insulin receptor substrate-1 (IRS-1) (Citation33), possibly in order, physiologically, to balance the actions of kinases involved in the insulin receptor pathway. Interestingly, adipose tissue inflammation, through high-fat feeding and TNFα, induced the expression of PTP1B in adipose tissue, muscle, liver, and hypothalamic arcuate nucleus in a time- and dose-dependent manner through a mechanism involving nuclear factor kappa B (NFκB). This effect was significantly reduced by high-dose salicylate treatment (Citation34). Moreover, SIRT1 was shown to improve insulin sensitivity through PTP1B repression at the chromatin level under insulin-resistant conditions (Citation35). Recently, PTP1B inhibition was demonstrated to improve insulin resistance and obesity-related abnormalities (Citation36,Citation37). In addition, myocytes and primary brown adipocytes deficient in PTP1B were shown to be protected from TNFα-induced insulin resistance (Citation38).
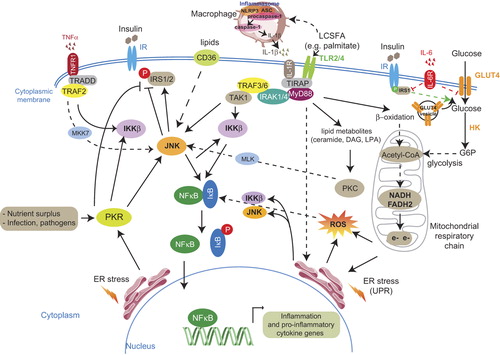
Amongst the kinases that respond to inflammatory (e.g. TNFα) and nutritional signals, Jun N-terminal kinase (JNK) and the inhibitor of nuclear factor kappa-B kinase beta (IKKβ) are activated and phosphorylate on serine residues the insulin receptor substrate-1 (IRS-1), which translates into a significant decrease in the insulin receptor signaling pathway () (Citation39,Citation40). It should be noted that several IRS-1 serine sites can be phosphorylated when stimulated by kinases that respond to cytokines or other stress stimuli. These kinases include the double-stranded RNA-dependent protein kinase (PKR) that responds to nutrient signals and endoplasmic reticulum stress (Citation41), protein kinase Cθ (PKCθ) (Citation42), mitogen-activated protein-kinase (MAPK) (Citation43), glycogen synthase kinase-3 (GSK3) (Citation44), stress-activated p38 MAPKα (Citation45), extracellular signal-regulated protein kinase (ERK), mammalian target of rapamycin (mTOR), and S6-kinase (S6K) which inactivate IRS-1 (Citation46–48). Furthermore, a seminal work by Scherer and colleagues have demonstrated that activation of the MAP kinase kinase (MEK) 1/2 p42/p44 signaling pathway is required for the TNFα-induced IRS-1 serine phoshorylation and impaired insulin signaling (Citation49). These observations reflect the complex signaling network surrounding IRS-1 serine phosphorylation and triggered by TNFα in insulin resistance installation.
Figure 1. Extracellular and intracellular signals leading to metabolic inflammation. The pro-inflammatory cytokines TNFα and IL-1β, through their respective receptors TNF1R and IL-1R, activate signaling pathways, via adaptive protein complexes, converging to stimulation of the intracellular kinases JNK and IKKβ. The latter phosphorylate the inhibitor of the pro-inflammatory transcription factor NFκB, IκB. Phosphorylated IκB releases NFκB which translocates into the nucleus where it activates the transcription of inflammation and pro-inflammatory cytokine genes. LCSFA bind to their receptors TLR2/4 and induce signaling pathways leading to excessive ROS generation, via β-oxidation and mitochondrial respiratory chain, ER stress stimulation, and lipid metabolite formation which activate JNK (lipid metabolites activate JNK via PKC stimulation). JNK is an inhibitor of the insulin receptor pathway via IRS-1/2 phosphorylation and can be activated through a lipid translocase (FAT/CD36)-mediated pathway but also through a PKR-dependent pathway. Upon HFD-induced increase in FFAs, IL-1β is cleaved and released from macrophages through the inflammasome/caspase-1 pathway. IL-6 decreases IRS-1 and GLUT4 transcription and impairs glucose transport. IRS-1 phosphorylation on serine is depicted in red and on tyrosine in green. Obesity signals, such as increased glucose entry into adipose cells, contribute to enhanced oxidative stress, through the increase in electron flux in mitochondria. All these pathways participate in the installation of low-grade inflammation.
Abbreviations: ASC = adaptor protein apoptotic speck protein containing a caspase recruitment domain; CD36/FAT = cluster of differentiation-36/ fatty acid translocase; DAG = diacyl glycerol; ER = endoplasmic reticulum; G6P = glucose-6 phosphate; GLUT = glucose transporter; HK = hexokinase; IKKβ = IκB kinase β; IL-1β = interleukin-1β; IL-6 = interleukin-6; IR = insulin receptor; IRAK = interleukin-1 receptor associated kinase; IRS = insulin receptor substrate; IκB = inhibitor of NFκB; JNK = Jun N-terminal kinase; LPA = lysophosphatidic acid; MKK7 = mitogen-activated protein kinase kinase-7; MLK = mixed-lineage kinase; MyD88 = myeloid differentiation protein-88 (a docking protein); NFκB = nuclear factor kappa B; NLRP3 = nucleotide-binding domain with leucine-rich repeats (NLR) containing pyrin domain-3; PKR = double-stranded RNA-dependent protein kinase; ROS = reactive oxygen species; TAK-1 = transforming growth factor β activated kinase-1; TLR2/4 = Toll-like receptors 2/4; TlRAP = Toll-like receptor adaptor protein; TNFα = tumor necrosis factor α; TRADD = TNF receptor associated death domain; TRAF = TNF receptor associated factor; UPR = unfolded protein response.
Following these in-vitro experiments, recent data from humans demonstrated that obese patients with features of the metabolic syndrome treated for 6 months with etanercept, a TNFα antagonist, had a significant beneficial effect on glucose homeostasis and insulin resistance as compared with obese patients given a placebo (a randomized study) (Citation50). To support further the concept of targeting low-grade inflammation in insulin resistance and diabetes prevention, it has been found that the use of a TNF inhibitor or hydroxychloroquine but not methotrexate was associated with a decreased adjusted risk of diabetes mellitus in patients with rheumatoid arthritis and psoriasis as compared to other non-biological disease-modifying drugs (Citation51). It remains to be determined in a randomized controlled trial whether these agents are able to prevent the development of diabetes among patients with systemic inflammatory diseases.
Alternatively, therapeutic venues aimed at restoring a proper IRS-1 phosphorylation status, either by inhibiting kinases which increase IRS-1 serine phosphorylation (or IRS-2 in brown adipocytes (Citation52)) or through PTP1B inhibition that will enhance IRS-1 tyrosine phosphorylation, or both, may prove beneficial in insulin resistance reversal and T2D treatment.
C-C chemokine ligand-2 (CCL-2) and its receptor CCR-2
The C-C chemokine ligand-2 (CCL-2) cytokine, also known as the macrophage chemoattractant protein-1 (MCP-1), was shown to be involved in adipose tissue accumulation of macrophages along with its receptor CCR-2. Likewise, CCL-2-deficient animals fed a HFD displayed a significant decrease in macrophage infiltration within the adipose tissue as compared to mice with intact CCL-2 under the same regime (Citation53). The expression of a dominant negative mutant for CCL-2 in genetically obese and diabetic mice (db/db) significantly decreased their insulin resistance. In contrast, CCL-2 overexpression stimulated macrophage accumulation in adipose tissue and promoted insulin resistance (Citation54). In addition, the receptor for CCL-2, CCR-2 was also shown to modulate the inflammatory and abnormal metabolic effects in diet-induced obesity (Citation55,Citation56). Importantly, acute administration of CCL-2 was shown to be sufficient for insulin resistance promotion, independently of adipose tissue infiltration by macrophages. This insulin resistance was improved by a CCR-2 antagonist, suggesting that inhibition of the CCL-2/CCR-2 axis, regardless of the CCL-2 production site, might be beneficial for the prevention of insulin resistance (Citation57). Other studies are required to identify the signaling pathway leading to macrophage-independent promotion of insulin resistance. Furthermore, CCR-2 antagonism markedly improved obesity, inflammation, insulin resistance, lipid metabolism, and kidney function in db/db mice (Citation58–60).
Interestingly, a -2518 A/G polymorphism in the MCP-1(CCL-2) gene has been associated with an increase in circulating MCP-1 levels in a Japanese population. The concentration of MCP-1 was positively correlated with insulin resistance and visceral obesity, but not with body mass index (BMI). In a subgroup analysis, T2D obese patients that carried the -2518A allele had a much higher MCP-1 concentration and a more marked insulin resistance than T2D obese patients carrying the -2518G allele (Citation61).
Of particular note, clear evidence is emerging from targeting the CCR-2 pathway in humans. Hence, a phase II clinical study has reported, in the 2011 annual meeting of the European Association for the Study of Diabetes (EASD), the safety and efficacy of the orally active CCR-2 antagonist CCX140-B on HbA1c and fasting plasma glucose lowering in type 2 diabetic patients on stable doses of metformin (Citation62).
Moreover, the other member of the family of chemokines, C-C chemokine ligand-5 (CCL-5), which is produced by the M1 pro-inflammatory macrophages, was also reported to have an important role in macrophage recruitment in the adipose tissue. However, unlike CCL-2, CCL-5 has macrophage-protective properties against apoptosis that is induced by cholesterol released from necrotic adipocytes (Citation63). It would be of importance to search for the in-vivo consequences of CCL-5 pathway blockage on insulin resistance and obesity complications.
Interleukin-1β, interleukin-6, and insulin resistance
Among the pro-inflammatory cytokines involved in insulin resistance, the obesity-induced interleukin-1β (IL-1β) seems to play an important role. Three studies illustrate this assertion: 1) The European Prospective Investigation Into Cancer and Nutrition (EPIC) study showed that obese individuals with detectable IL-1β circulating levels and elevated IL-6 concentration have an important independent risk to develop T2D (3.3-fold, 1.7–6.8) as compared to obese patients with elevated IL-6 but undetectable IL-1β levels (Citation64); 2) Treatment of T2D patients with an IL-1β receptor antagonist improved glycemia (measured by HbA1c) and β-cell function, and significantly decreased the inflammatory markers IL-6 and C-reactive protein (CRP) (Citation65); 3) Genetic variations in the IL-1 gene family (IL-1α, IL-1β, and IL-1Ra) were associated with measures of glucose homeostasis and prevalent diabetes in the Finnish population (Citation66). Most importantly, chronic (but not acute) treatment of 3T3-L1 and human adipocytes with IL-1β induced insulin resistance through IRS-1 down-regulation and subsequent decrease in IRS-1 tyrosine phosphorylation (Citation67). Interleukin-1α has also been shown to inhibit insulin signaling through a similar mechanism in 3T3-L1 adipocytes (Citation68). This effect was shown to be largely due to IL-6 production (Citation69).
As for the role of IL-6 in the pathophysiology of insulin resistance, it remains controversial, and the following conclusions can be drawn: 1) IL-6 impairs hepatic and adipocyte insulin signaling in vitro through IRS-1 inhibition (Citation70,Citation71), and IL-6 levels were found to be robustly increased in fat cells from insulin-resistant individuals (Citation71); 2) IL-6 acts both centrally and peripherally to induce energy expenditure and impair insulin signaling (Citation72,Citation73); 3) both IL-6 deficiency (Citation74) and overexpression (Citation75) result in liver inflammation and insulin resistance; 4) dietary and genetic obesity induced liver inflammation and tumorigenesis by enhancing IL-6 and TNFα expression (Citation76); 5) despite IL-6 positive effects on weight loss through increased energy expenditure (Citation72,Citation73), IL-6 should not be given chronically because of its deleterious effects on the liver and insulin signaling; 6) IL-6 levels need to be maintained to a certain threshold of production since complete IL-6 inhibition leads to liver inflammation and insulin resistance; 7) mice with metabolic tissue-specific targeting of IL-6 will probably improve our comprehension of IL-6 pathophysiological roles in whole-body animals.
Endoplasmic reticulum stress in inflammation and insulin resistance
Another mechanism that contributes to the activation of JNK and IKKβ kinases is that of the endoplasmic reticulum (ER) stress implicated in obesity and insulin resistance through JNK-mediated inhibition of the insulin receptor pathway by phosphorylating the IRS-1 (Citation77), but also via the stimulation of the inflammatory response mediators ().
ER is the main site of protein folding, and ER stress is measured by the activation of a response to protein unfolding referred to as the unfolded protein response (UPR), which is an important adaptive response of cells to an important metabolic load (Citation78). The UPR occurs through the intervention of three transmembrane molecules which transmit the ER stress to the nucleus, via a number of cytoplasmic signaling molecules and kinases, in order to activate the transcription of pro-inflammatory genes (Citation48).
The first molecule, inositol-requiring enzyme-1 (IRE-1) utilizes its kinase domain in association with the TNF receptor-associated factor-2 (TRAF-2) to activate the pro-inflammatory kinases JNK and IKKβ, stimulating thereby the transcription factors AP-1 and NFκB, respectively. The IRE-1 has also an endoribonuclease that cleaves the X-box-binding protein-1 (XBP-1) mRNA, activating the transcription of several chaperone and inflammatory genes (Citation2). The second molecule involved in the UPR is the ER membrane-localized transcription factor ATF-6 that is cleaved in response to the ER stress. The cleaved ATF-6 then enters into the nucleus to activate the transcription of chaperone genes and augment NFκB transcription (Citation2). The third ER transmembrane molecule is the PKR-like eukaryotic initiation factor 2α kinase (PERK), which inhibits protein translation by phosphorylating the transcription initiation complex eIF2α, activates the alternative translation of the ATF-4 factor which induces the production of inflammatory cytokines, and stimulates NFκB transcription, while inhibiting the translation of its inhibitor IκBα (Citation2).
The ultimate goal of UPR activation, which stimulates inflammatory pathways, is to restore ER homeostasis by favoring a correct protein folding with the assistance of chaperone proteins, inducing protein synthesis arrest, and augmenting the degradation of a number of ER proteins. In the case that ER functions were not corrected, the UPR stimulates apoptosis signaling pathways (Citation2).
From a metabolic standpoint, during obesity the ER senses the changes in the metabolic flux inside the cell and reacts accordingly. Hence, in the adipocyte, the increases in lipid stores, triglyceride-genesis, adipokines, and fatty acids act as ER stress signals. In the liver, increased protein synthesis, lipogenesis, protein transport, and gluconeogenesis have a negative influence on ER function, which in return contributes to the increase in lipogenesis (via IRE-1 and PERK signaling pathways) and gluconeogenesis (via a PERK/eIF2α/C-EBPα and -β axis) (Citation48). In addition, the increased insulin demand from β-cell islets in insulin-resistant states can contribute to the ER stress, which leads ultimately to the decrease in insulin synthesis and β-cell apoptosis through the IRE-1/JNK and PERK/eIF2α pathways (Citation48).
It should be noted that ER-induced PKR activation not only modulates JNK subsequently to activate the expression of pro- inflammatory genes but also directly induces IRS-1 phosphorylation and negatively regulates the insulin receptor pathway (Citation41). PKR assumes a role in regulation of IRS-1 phosphorylation in both a JNK1-dependent and independent manner in cultured cells. Besides ER stress, PKR is also activated in WAT and liver in conditions of nutrient surplus both in genetic obesity (ob/ob mice) and in mice fed a high-fat diet, while PKR-/- animals exhibit a marked decrease in the expression of several pro-inflammatory genes in the VAT as compared to wild-type mice (Citation41). It was suggested that PKR may represent a core component of a putative ‘metabolic inflammasome’ that consists of major elements in inflammatory signaling (e.g. JNK, IKKβ, and eIF2α) and insulin action (IRS-1), and appears to be critical for JNK activation compared to the IRE/TRAF pathway in a metabolic context. Infections are also activators of the PKR pathway (Citation79).
Altogether, these signals contribute to the installation and maintenance of an inflammatory state, favored by the bulk of circulating inflammatory cytokines and free fatty acids that activate in return ER stress in the hypothalamus and other metabolic organs, converging towards a chronic and systemic inflammation, which ultimately will lead to insulin resistance and T2D ().
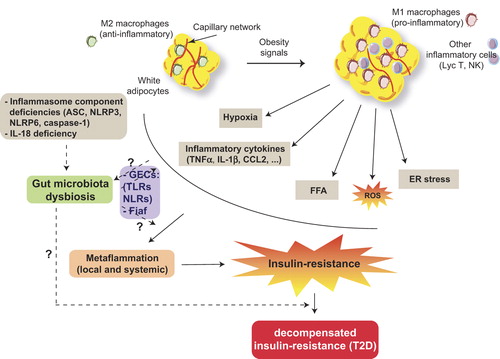
Figure 2. Obesity, a causal factor of metabolic inflammation. Overnutrition is an environmental element inducing an increase in adipocyte size and a change in resident macrophage polarity from an anti-inflammatory M2 to a pro-inflammatory M1 phenotype secreting pro-inflammatory cytokines. Autocrine and paracrine signals stimulate chemotactic factor release and other inflammatory cell recruitment, contributing to increased oxidative stress (ROS), ER stress, hypoxia, and free fatty acid release (FFA due to enhanced lipolysis). In this context, a local chronic low-grade inflammation (metaflammation) is installed followed by a systemic chronic inflammatory state inducing a systemic insulin resistance. When insulin resistance becomes decompensated, symptomatic type 2 diabetes can be diagnosed. Gut microbiota changes impact metabolic inflammation through a possible disturbance in innate immunity (e.g. TLR5 decrease) or impaired gut epithelial cell (GES) microbiota interactions through alteration in pathogen recognition receptor (PRR) TLR and/or nucleotide oligomerization domain-like receptor (NLR) signaling. Obesity has also an influence on changes in microbiota profiles. Gut microbiota inhibits the fasting-induced adipocyte factor (Fiaf), which results in energy storage in adipocytes. Inflammasome component deficiencies induce gut microbiota dysbiosis and subsequent metabolic disorders.
Abbreviations: ASC=adaptor protein apoptotic speck protein containing a caspase recruitment domain; CCL-2=C-C chemokine ligand-2; IL-1β=interleukin-1beta; NLRP3 and NLRP6=nucleotide-binding domain with leucine-rich repeats (NLR) containing pyrin domain-3 and -6, respectively.
Hypoxia and metabolic inflammation
At the onset of obesity, adipocytes, mainly those of the visceral fat, undergo a marked hypertrophy. Initially, resident M2 macrophages contribute to the tissular and vascular remodeling to help the adipose tissue adapt to the new environment of nutritional excess, in order to prevent hypoxia of the adipose tissue and the ischemia that might result. This ischemia would induce adipocyte necrosis which provokes inflammatory cell recruitment (Citation80). Obesity favors hypoxia of adipose tissue because of the decrease in the proportion of the protective M2 macrophages, while the deleterious M1 macrophages increase. Furthermore, M1 macrophages induce reactive oxygen species (ROS) and nitrogen monoxide (NO) which alter endothelial cells, compromising angiogenesis needed to counteract hypoxia (Citation80,Citation81).
The installation of a chronic low-grade inflammatory state allowed the identification, in the adipose tissue of obese individuals, of hypoxic tissular areas with enhanced expression of hypoxia genes such as the hypoxia inducible factor-1α (HIF-1α). The increased expression or overexpression of HIF-1α will stimulate the expression of inflammatory genes, which will amplify the ‘metaflammatory’ reaction, initiate adipose tissue fibrosis, and block the pro-angiogenic program which will lead to insulin resistance (Citation82,Citation83).
Role of glucose and intracellular lipid excess in metabolic inflammation
Increased glucose supply will provide enhanced NADH and FADH2 metabolites which will actively contribute to ROS formation through their electron transfer to the mitochondrial respiratory chain, contributing thereby to the activation of the pro-inflammatory NFκB transcription factor, via the activation of JNK () (Citation40). Moreover, a diet enriched with glucose provides metabolites, such as acetyl-CoA, necessary to de-novo synthesis of fatty acids such as palmitate, whose metabolites will participate in the ectopic tissular lipotoxicity by mechanisms described hereunder.
In the context of obesity, excess lipid content will be stored in adipocytes. When the adipocyte-storing capacity is reached, lipid surplus will accumulate ectopically in tissues which are not lipid-storing organs. Hence, intracellular lipids have been suggested to play an active part in the pathogenesis of insulin resistance and pancreatic β-cell dysfunction, but also in the lipotoxicity of lean metabolic organs such as the liver, skeletal muscle, and heart (Citation84). Several mechanisms involved in this lipotoxicity have been advanced:
Increased fatty liver β-oxidation (the saturated C16:0 fatty acid palmitate but not the monounsaturated C18:1 fatty acid oleate) induces an enhanced electron transfer flow through the mitochondrial respiratory chain, via increased NADH and FADH2 production, and contributes to the overproduction of ROS, which will augment insulin resistance through a prolonged JNK activation (ROS can induce inflammation through NFκB nuclear translocation). Interestingly, anti-oxidants reversed palmitate-induced ROS production, JNK activation, and insulin resistance (Citation85). In contrast, acute oxidative stress (e.g. during exercise-mediated muscle contraction) induced a decrease in cytoplasmic JNK phosphorylation due to a redistribution of JNK-specific phosphatase MKP7 from the nucleus into the cytoplasm, which resulted in restored normal insulin sensitivity and glucose uptake in insulin-resistant muscle cells, an effect that was MKP7-dependent (Citation86). Hence, it was suggested that the opposing effects of acute versus chronic oxidative stress on insulin sensitivity depend on MKP7 subcellular localization and JNK activation.
Intracellular lipids, mainly long-chain saturated fatty acids (LCSFA) which induce ER stress, provoke a prolonged JNK activation and subsequent insulin resistance, as well as β-cell alteration. LCSFA-mediated lipotoxicity is initiated after their binding to Toll-like receptors (TLR2 and TLR4) which recruit the adapting protein MyD88 and the formation of a multiprotein complex converging to the activation of JNK and IKKβ (), amplifying further their inflammatory and insulin resistance-promoting effects. Indeed, disruption of pathways involving TLR4/MyD88, IKKβ, and ER stress in neurons was shown to prevent diet-induced obesity, leptin resistance, insulin resistance, and their deleterious effects on metabolism (Citation87–89). Interestingly, genetic variants in the TLR4 have been associated with factors of the metabolic syndrome and modified the association between dietary saturated fatty acids (SFA) and fasting HDL levels (Citation90).
LCSFA active metabolites such as ceramide, diacylglycerol (DAG), and lysophosphatidic acid (LPA) contribute to JNK activation through a protein kinase C (PKC)-dependent signaling pathway (Citation40,Citation91). Besides, recently it has been demonstrated that TLR4-mediated lipid-induced insulin resistance requires de-novo ceramide synthesis through an IKKβ-dependent mechanism in mice. Of particular note, increased ceramide production is not required for TLR4-mediated inflammatory cytokine production but is mandatory for TLR4-dependent insulin resistance expression (Citation92). This discovery supports an in-vivo study showing that ceramide accumulation was strongly implicated in insulin resistance promotion (Citation93). A recent report from Scherer and colleagues have demonstrated that the insulin-sensitizing cytokine adiponectin reduced hepatic ceramide levels via activation of ceramidase, which converts ceramide into sphingosine, while adiponectin deficiency increased ceramide levels and exacerbated insulin resistance (Citation94). A pharmacologic inhibition of ceramide synthesis in obese patients would be an interesting approach to test for insulin resistance prevention.
Along the same lines but involving a different mechanism, Ting and colleagues have elegantly shown that free fatty acids (FFAs) (the saturated FA palmitate but not the unsaturated oleate) can interfere with insulin signaling through an IL-1β response via the activation of a molecular platform called the inflammasome (Citation95), consisting of the nucleotide-binding domain with leucine-rich repeats (NLR) containing pyrin domain-3 (NLRP3) associated with the adaptor protein apoptotic speck protein containing a caspase recruitment domain (ASC, a.k.a PYCARD) and procaspase-1 (Citation96). It was demonstrated that HFD-induced FFA increase can activate the NLRP3 inflammasome in macrophages by an AMPK-autophagy-ROS signaling pathway which in turn catalyzes the caspase-1-mediated cleavage and release of IL-1β (Citation96). Paradoxically, deficiencies in inflammasome components (ASC, NLRP3, NLRP6, and caspase-1) and in the pro-inflammatory cytokine IL-18 (but not IL-1) yielded gut microbiota dysbiosis and subsequent obesity, and exacerbated hepatic steatosis and inflammation through increased influx of TLR4 and TLR9 agonists in portal circulation (Citation97). This study demonstrated that altered host–gut microbiota interactions induced by defective inflammasome sensing produced metabolic abnormalities such as obesity and progression of hepatic steatosis (Citation97). Altogether, although inflammasome components can mediate lipid-induced insulin resistance in adipose tissue through the IL-1β pathway, they act as steady-state sensors and regulators of intestinal microbiota preventing its dysbiosis and subsequent metabolic abnormalities.
Lysophosphatidic acid (LPA) is considered as an inflammatory mediator inducing, via NFκB activation, the expression of the pro-inflammatory cytokine MCP-1 (CCL-2) and of cellular adhesion molecules in human endothelial cells (Citation98,Citation99). Hence, LPA contributes to the installation of low-grade inflammation through inflammatory cell recruitment and JNK activation. Antagonism of the LPA signaling pathway might reduce insulin resistance in obese individuals.
Under HFD, mice deficient in the lipid transporter fatty acid translocase (FAT/CD36) exhibit a significant reduction in adipose tissue inflammation and a marked improvement in their insulin sensitivity as compared to wild-type animals. CD36 is thought to contribute to the cross-talk between adipocytes and macrophages leading to the development of insulin resistance via a JNK-dependent pathway (Citation100,Citation101).
Role of the gut microbiota in metabolic inflammation and obesity-related disorders
Diverse microbes are known to colonize the intestine, with several health benefits to the host, a process referred to as the ‘prebiotic effect’ (Citation102). Early on after birth, this huge community of intestinal resident microbes stimulate the immune system that will later on protect our body against pathological micro-organisms (Citation103–105). In the last few years, robust evidence demonstrated that changes in gut microbiota appear to have a strong impact on metabolic inflammation that accompanies obesity, insulin resistance, and T2D (Citation106,Citation107). For instance, the proportion of two beneficial intestinal bacterial residents, namely Bacteroides and Firmicutes, has been shown to be significantly modified in mice and in obese patients as compared to lean individuals, with a relative decrease in the Bacteroides species as compared to Firmicutes that was reversed upon weight loss (Citation106,Citation108). Also, gut microbiota profiled from fecal samples of T2D patients revealed a significantly reduced proportion of Faecalibacterium prausnitzii, a member of the Firmicutes, that correlated with increased levels of circulating inflammatory markers as compared to obese non-diabetic and control patients (Citation109). Furthermore, it was demonstrated that gut microbiota profiles were different and associated with metabolic phenotypes (e.g. diabetic versus non-diabetic), regardless of the genetic background or the diet, and can change rapidly after metabolic adaptations such as a regime (HFD supplemented with dietary fiber regime containing gluco-oligosaccharide (DIO-GOS)) (Citation110) or in a starvation-like situation induced by a surgical procedure (Roux-en-Y gastric bypass (RYGB)) (Citation109), underscoring a gut microbial signature associated with the metabolic phenotype and adaptation. Restoring the proportion of bacterial species composing gut microbiota in obese and diabetic patients to levels similar to those found in lean individuals, by means of probiotics, might be beneficial in the treatment of metabolic diseases.
The impact of gut microbiota on the generation of metabolic diseases can be elucidated from the role played by microbial products in mediating the cross-talk between cells from metabolic tissues and the host intestinal epithelium represented by gut epithelial cells (GECs) (Citation111). GECs, which are known to regulate dendritic cells and macrophage and lymphocyte functions by GEC-secreted cytokines, express at their surface pathogen recognition receptors (PRRs) that can sense the presence of microbes. In this context, PRRs signal to maintain an appropriate immunosuppressive tone preventing thereby the gut mucosa from an impetuous reaction against healthy microbiota (Citation111). PRRs are represented by the well-characterized TLRs and nucleotide oligomerization domain-like receptors (NLRs) with established effects in pathogen recognition and stimulation of innate immune effectors and inflammation. Disruption in the GEC–gut microbiota relationship is thought to influence negatively the metabolic status of the body and potentially lead to metabolic diseases. For instance TLR5 deficiency induced features of the metabolic syndrome through altered gut microbiota (Citation112), whereas TLR2 and TLR4 deficiencies did not affect intestinal microbiota composition in mice (Citation113).
Interestingly, gut microbiota has been identified as an important environmental factor that affects energy harvest from the diet (e.g. digesting complex polysaccharides) and energy storage in the host through suppression of intestinal fasting-induced adipocyte factor (Fiaf), a circulating lipoprotein lipase inhibitor. This suppression has been shown to be crucial for microbiota-induced deposition of triglycerides in adipocytes (Citation114). While this property of microbiota to promote lipid storage has been used by ancient humans to get access to energy in periods of scarcity, this positive effect became detrimental and caused obesity and metabolic diseases when large portions of high-calorie diets are accessible.
All these data should invite us not to neglect gut microbiota among the multiple therapeutic strategies aimed to combat obesity and its related disorders.
Consequences of metabolic inflammation and clinical perspectives
Role of high-sensitivity C-reactive protein (hsCRP) as a marker of cardiovascular risk
In routine clinical practice, the existence of a low-grade chronic inflammation has led to the identification of an increased risk to develop a peripheric vascular disease through the quantification of high-sensitivity C-reactive protein (hsCRP) in apparently healthy individuals (Citation115). Later, it has been demonstrated that hsCRP measurement as a marker of inflammation is associated with the highest risk (4.4-fold) of cardiovascular events as compared to other markers such as IL-6, homocysteine, or LDL-cholesterol in a population of 28,263 premenopausal women followed over 3 years (Citation116). Furthermore, results from the prospective study MONICA/KORA (Augsburg 1984–1998) have shown a strong association between hsCRP and death caused by a cardiovascular disease (hazard ratio (HR) 2.15) (Citation117).
Interestingly, hsCRP was also linked to increased cardiovascular risk in T2D patients. Indeed, increased hsCRP levels were significantly associated with cardiovascular risk indicators linked to T2D such as the homeostatic model assessment index (HOMA), insulin levels, body mass index (BMI), and β-cell dysfunction. However, hsCRP was not correlated with disease duration or glycemic control (Citation118). In addition, hsCRP was shown to be connected to the risk of T2D development in patients with the metabolic syndrome, and a significant positive correlation between hsCRP and HbA1c was demonstrated in T1D and T2D patients (Citation119). Nevertheless, the role of hsCRP needs to be further clarified in pre-diabetic states.
The American Heart Association (AHA) recommends the measurement of hsCRP in individuals with an intermediate to elevated risk to develop a coronary heart disease, including diabetic patients (Citation119).
Drug development and molecules targeting chronic inflammation in cardiovascular disease prevention
Before discussing molecules that target metabolic inflammation in obese patients, it is important to mention that prevention of inflammation and insulin resistance should be initiated through a change in lifestyle/dietary habits (i.e. an appropriate nutritional diet associated with a regular physical activity), to impede an increase in weight in non-obese individuals and stimulate weight loss in obese patients. Hence, it has been demonstrated that weight loss by means of a hypocaloric regime in obese patients improved significantly their inflammatory profile. This weight loss induced a significant decrease in the expression of genes encoding inflammatory factors and an increase in the expression of anti-inflammatory genes in the adipose tissue (Citation120).
Identification of the molecular mechanisms underlying metabolic inflammation should allow the development of new pharmacologic agents (or the use of available FDA-approved drugs) aiming at reducing the risk of cardiovascular diseases in patients with intermediate or elevated hsCRP values (1–3 mg/L and 3–10 mg/L, respectively). Hence, different non-diabetic molecules have proved their efficiency in reducing hsCRP levels, such as aspirin, statins, cyclo-oxygenase-2 (Cox-2) inhibitors, and fibrates (Citation119). Recently, salsalate, a non-steroid anti-inflammatory molecule, and salicylate-related agents have demonstrated beneficial effects against inflammation and insulin resistance. In this context, these molecules displayed hypoglycemic properties (Citation121,Citation122), reversed diet-induced obesity and insulin resistance, as well as IKKβ inhibition in animal models (Citation123). These studies pursued older observations that showed the hypoglycemic properties of salicylate agents (Citation124,Citation125). However, it remains necessary to perform large-scale studies to evaluate the link between salicylate-induced hsCRP diminution and prevention of insulin resistance and cardiovascular risk in obese and diabetic patients.
Furthermore, the previously employed anti-diabetic agents, PPARγ agonists, have demonstrated a significant reduction in hsCRP and other markers of cardiovascular risk, independently of glycemia improvement (Citation119). Unfortunately, these agents have been withdrawn due to an increased cardiovascular mortality rate in diabetic patients. Nevertheless, it would be interesting to test the effect of other anti-diabetic molecules in the potential reduction of hsCRP and cardiovascular risk.
Finally, the widely used hypocholesterolemic drugs, statins, have recently demonstrated their efficacy in reducing hsCRP levels and cardiovascular risks associated with high hsCRP (Citation126,Citation127). These studies have shown that the hydrophilic drug rosuvastatin reduced the risk of the first major cardiovascular event and global mortality in normocholesterolemic healthy individuals with elevated circulating hsCRP levels. Additional studies need to establish whether statin treatment reduce hsCRP and other pro-inflammatory cytokines involved in metaflammation, and whether this potential reduction is associated with an improved insulin sensitivity. Interestingly, the hydrophilic drug pravastatin improved glucose homeostasis, enhanced adiponectin production, and increased insulin secretion from isolated islets, while reducing insulin resistance in db/db mice under high-fat/glucose conditions (Citation128,Citation129). Pravastatin increased also plasma adiponectin levels in humans as compared to placebo treatment (Citation129). Conversely, the lipophilic statin, simvastatin, inhibited dose-dependently glucose-induced insulin secretion in β-cells through blockade of L-type Ca2+ channels, an effect not observed with the hydrophilic molecule pravastatin. The inhibitory potencies of statins correlated with their lipophilicities (Citation130). From these observations, we recommend using the hydrophilic statins for their beneficial effects on glucose homeostasis and insulin resistance prevention (Citation128,Citation129) instead of the more lipophilic statins, which reduce insulin secretion (Citation130). A meta-analysis study which included four trials with simvastatin or atorvastatin treatment (both lipophilic drugs) and only one trial with atorvastatin or pravastatin therapy found a slightly increased risk for diabetes development that might be explained by the use of lipophilic statins (Citation131).
Concluding remarks and perspectives
We have presented in this review the molecular mechanisms of low-grade inflammation (the so-called ‘metaflammation’) that contribute to insulin resistance. We have mentioned the role of inflammatory cells in inflammatory cytokine secretion, some of which are also released by adipocytes, lipids (mainly long-chain saturated fatty acids and their metabolites), and oxidative and ER stresses in translating signals yielding the activation of various intracellular kinases such as PKC, JNK and IKKβ. These intracellular signaling pathways ultimately result in the inactivation of the insulin receptor pathway, through IRS phosphorylation, and the release of the pro-inflammatory transcription factor NFκB from its inhibitor IκB to induce the expression of pro-inflammatory genes, supporting thereby a chronic inflammatory state. It would be interesting to test whether appropriate inactivation of these kinases is sufficient to prevent/reverse insulin resistance. Alternatively, we could act upstream of these signaling cascades and try to prevent the transition of adipose tissue-resident macrophages from the M2 to M1 phenotype () and therefore potentially impede the chronic inflammation responsible for insulin resistance in obese individuals. We also discussed the importance of changes in gut microbiota profiles in obesity and diabetic states, and this represents a serious target in the quest for therapeutic agents against these diseases.
Finally, in view of the published studies, we have discussed the possibility to employ hsCRP as a marker of metaflammation and as a predictive factor in the occurrence of cardiovascular events in diabetic patients, but also in apparently healthy individuals who present one or several cardiovascular risk factors. We then addressed the place of available drugs used to treat other diseases (e.g. salicylates and statins) that can be considered to prevent insulin resistance in high-risk patients. We are currently investigating the molecular mechanisms of metaflammation in a cohort of non-diabetic obese patients and their non-obese controls aiming at adding another brick in the wall of insulin resistance comprehension.
Declaration of interest: The authors declare no competing financial interests.
Related Research Data
References
- Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–83.
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45.
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–7.
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30.
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808.
- Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab. 2008;4:619–26.
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84.
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20.
- Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117:1658–69.
- Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7:485–95.
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24.
- Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med. 2007;39:335–45.
- Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298:E419–28.
- Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, et al. SirT1 regulates adipose tissue inflammation. Diabetes. 2011;60:3235–45.
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–22.
- Gauthier MS, O’Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, et al. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. 2011;404:382–7.
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–60.
- Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem. 2010;285:19051–9.
- Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–92.
- Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH, et al. CD11c expression in adipose tissue and blood and its role in diet- induced obesity. Arterioscler Thromb Vasc Biol. 2010;30:186–92.
- Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–9.
- Li P, Lu M, Nguyen MT, Bae EJ, Chapman J, Feng D, et al. Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J Biol Chem. 2010;285:15333–45.
- Caspar-Bauguil S, Cousin B, Galinier A, Segafredo C, Nibbelink M, Andre M, et al. Adipose tissues as an ancestral immune organ: site-specific change in obesity. FEBS Lett. 2005;579:3487–92.
- Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15:940–5.
- Ohmura K, Ishimori N, Ohmura Y, Tokuhara S, Nozawa A, Horii S, et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol. 2010;30:193–9.
- Duffaut C, Zakaroff-Girard A, Bourlier V, Decaunes P, Maumus M, Chiotasso P, et al. Interplay between human adipocytes and T lymphocytes in obesity: CCL20 as an adipochemokine and T lymphocytes as lipogenic modulators. Arterioscler Thromb Vasc Biol. 2009;29: 1608–14.
- Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–9.
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8 + effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20.
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–15.
- Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–4.
- Peraldi P, Xu M, Spiegelman BM. Thiazolidinediones block tumor necrosis factor-alpha-induced inhibition of insulin signaling. J Clin Invest. 1997;100:1863–9.
- Bouter B, Geary N, Langhans W, Asarian L. Diet-genotype interactions in the early development of obesity and insulin resistance in mice with a genetic deficiency in tumor necrosis factor-alpha. Metabolism. 2010;59:1065–73.
- Goldstein BJ, Bittner-Kowalczyk A, White MF, Harbeck M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J Biol Chem. 2000;275:4283–9.
- Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283:14230–41.
- Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, et al. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 2007;6:307–19.
- Agouni A, Mody N, Owen C, Czopek A, Zimmer D, Bentires-Alj M, et al. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem J. 2011;438:369–78.
- Ma YM, Tao RY, Liu Q, Li J, Tian JY, Zhang XL, et al. PTP1B inhibitor improves both insulin resistance and lipid abnormalities in vivo and in vitro. Mol Cell Biochem. 2011;357:65–72.
- Lorenzo M, Fernandez-Veledo S, Vila-Bedmar R, Garcia-Guerra L, De Alvaro C, Nieto-Vazquez I. Insulin resistance induced by tumor necrosis factor-alpha in myocytes and brown adipocytes. J Anim Sci. 2008;86(14 Suppl):E94–104.
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–6.
- Solinas G, Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J. 2010;24:2596–611.
- Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 2010;140: 338–48.
- Kellerer M, Mushack J, Seffer E, Mischak H, Ullrich A, Haring HU. Protein kinase C isoforms alpha, delta and theta require insulin receptor substrate-1 to inhibit the tyrosine kinase activity of the insulin receptor in human kidney embryonic cells (HEK 293 cells). Diabetologia. 1998;41:833–8.
- De Fea K, Roth RA. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J Biol Chem. 1997;272:31400–6.
- Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci U S A. 1997;94:9660–4.
- Hemi R, Yochananov Y, Barhod E, Kasher-Meron M, Karasik A, Tirosh A, et al. p38 mitogen-activated protein kinase-dependent transactivation of ErbB receptor family: a novel common mechanism for stress-induced IRS-1 serine phosphorylation and insulin resistance. Diabetes. 2011;60:1134–45.
- Ueno M, Carvalheira JB, Tambascia RC, Bezerra RM, Amaral ME, Carneiro EM, et al. Regulation of insulin signalling by hyperinsulinaemia: role of IRS-1/2 serine phosphorylation and the mTOR/p70 S6K pathway. Diabetologia. 2005;48:506–18.
- Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. 2009;296:E581–91.
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17.
- Engelman JA, Berg AH, Lewis RY, Lisanti MP, Scherer PE. Tumor necrosis factor alpha-mediated insulin resistance, but not dedifferentiation, is abrogated by MEK1/2 inhibitors in 3T3-L1 adipocytes. Mol Endocrinol. 2000;14:1557–69.
- Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, et al. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab. 2011;96:E146–50.
- Solomon DH, Massarotti E, Garg R, Liu J, Canning C, Schneeweiss S. Association between disease-modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA. 2011;305:2525–31.
- Fasshauer M, Klein J, Ueki K, Kriauciunas KM, Benito M, White MF, et al. Essential role of insulin receptor substrate-2 in insulin stimulation of Glut4 translocation and glucose uptake in brown adipocytes. J Biol Chem. 2000;275:25494–501.
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505.
- Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281:26602–14.
- Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–24.
- Tamura Y, Sugimoto M, Murayama T, Minami M, Nishikaze Y, Ariyasu H, et al. C-C chemokine receptor 2 inhibitor improves diet-induced development of insulin resistance and hepatic steatosis in mice. J Atheroscler Thromb. 2010;17:219–28.
- Tateya S, Tamori Y, Kawaguchi T, Kanda H, Kasuga M. An increase in the circulating concentration of monocyte chemoattractant protein-1 elicits systemic insulin resistance irrespective of adipose tissue inflammation in mice. Endocrinology. 2010;151:971–9.
- Tamura Y, Sugimoto M, Murayama T, Ueda Y, Kanamori H, Ono K, et al. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler Thromb Vasc Biol. 2008;28: 2195–201.
- Kang YS, Lee MH, Song HK, Ko GJ, Kwon OS, Lim TK, et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int. 2010;78:883–94.
- Sayyed SG, Ryu M, Kulkarni OP, Schmid H, Lichtnekert J, Gruner S, et al. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 2011;80:68–78.
- Kouyama K, Miyake K, Zenibayashi M, Hirota Y, Teranishi T, Tamori Y, et al. Association of serum MCP-1 concentration and MCP-1 polymorphism with insulin resistance in Japanese individuals with obese type 2 diabetes. Kobe J Med Sci. 2007;53:345–54.
- Hanefeld M, Schell E, Gouni-Berthold I, Melichar M, Vesela I, Sullivan T, et al. Safety and efficacy of oral chemokine receptor 2 antagonist CCX140-B in a phase 2 type 2 diabetes study. 47th meeting of the European Association for the Study of Diabetes, 12–16 September 2011, Lisbon, Portugal. Abs. No, 192.
- Keophiphath M, Rouault C, Divoux A, Clement K, Lacasa D. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler Thromb Vasc Biol. 2009;30:39–45.
- Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–7.
- Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26.
- Luotola K, Paakkonen R, Alanne M, Lanki T, Moilanen L, Surakka I, et al. Association of variation in the interleukin-1 gene family with diabetes and glucose homeostasis. J Clin Endocrinol Metab. 2009;94:4575–83.
- Jager J, Gremeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. 2007;148:241–51.
- He J, Usui I, Ishizuka K, Kanatani Y, Hiratani K, Iwata M, et al. Interleukin-1alpha inhibits insulin signaling with phosphorylating insulin receptor substrate-1 on serine residues in 3T3-L1 adipocytes. Mol Endocrinol. 2006;20:114–24.
- Uno T, He J, Usui I, Kanatani Y, Bukhari A, Fujisaka S, et al. Long-term interleukin-1alpha treatment inhibits insulin signaling via IL-6 production and SOCS3 expression in 3T3-L1 adipocytes. Horm Metab Res. 2008;40:8–12.
- Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. 2002;51:3391–9.
- Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003;278:45777–84.
- Wallenius V, Wallenius K, Ahren B, Rudling M, Carlsten H, Dickson SL, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8:75–9.
- Wallenius K, Wallenius V, Sunter D, Dickson SL, Jansson JO. Intracerebroventricular interleukin-6 treatment decreases body fat in rats. Biochem Biophys Res Commun. 2002;293:560–5.
- Matthews VB, Allen TL, Risis S, Chan MH, Henstridge DC, Watson N, et al. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia. 2010;53:2431–41.
- Franckhauser S, Elias I, Rotter Sopasakis V, Ferre T, Nagaev I, Andersson CX, et al. Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia. 2008;51:1306–16.
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208.
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61.
- Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66(2 Suppl 1):S102–9.
- Goh KC, deVeer MJ, Williams BR. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000;19:4292–7.
- Rutkowski JM, Davis KE, Scherer PE. Mechanisms of obesity and related pathologies: the macro- and microcirculation of adipose tissue. FEBS J. 2009;276:5738–46.
- Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275–97.
- Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002.
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009; 29:4467–83.
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–65.
- Nakamura S, Takamura T, Matsuzawa-Nagata N, Takayama H, Misu H, Noda H, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284:14809–18.
- Berdichevsky A, Guarente L, Bose A. Acute oxidative stress can reverse insulin resistance by inactivation of cytoplasmic JNK. J Biol Chem. 2010;285:21581–9.
- Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10:249–59.
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73.
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51.
- Cuda C, Badawi A, Karmali M, El-Sohemy A. Polymorphisms in Toll-like receptor 4 are associated with factors of the metabolic syndrome and modify the association between dietary saturated fat and fasting high-density lipoprotein cholesterol. Metabolism. 2011;60: 1131–5.
- Turban S, Hajduch E. Protein kinase C isoforms: mediators of reactive lipid metabolites in the development of insulin resistance. FEBS Lett. 2011;585:269–74.
- Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:1858–70.
- Ussher JR, Koves TR, Cadete VJ, Zhang L, Jaswal JS, Swyrd SJ, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes. 2010;59:2453–64.
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63.
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid- induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15.
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–85.
- Shimada H, Rajagopalan LE. Rho-kinase mediates lysophosphatidic acid-induced IL-8 and MCP-1 production via p38 and JNK pathways in human endothelial cells. FEBS Lett. 2010;584:2827–32.
- Shimada H, Rajagopalan LE. Rho kinase-2 activation in human endothelial cells drives lysophosphatidic acid-mediated expression of cell adhesion molecules via NF-kappaB p65. J Biol Chem. 2010; 285:12536–42.
- Kennedy DJ, Kuchibhotla S, Westfall KM, Silverstein RL, Morton RE, Febbraio M. A CD36-dependent pathway enhances macrophage and adipose tissue inflammation and impairs insulin signalling. Cardiovasc Res. 2011;89:604–13.
- Nicholls HT, Kowalski G, Kennedy DJ, Risis S, Zaffino LA, Watson N, et al. Hematopoietic cell-restricted deletion of CD36 reduces high-fat diet-induced macrophage infiltration and improves insulin signaling in adipose tissue. Diabetes. 2011;60:1100–10.
- Roberfroid M, Gibson GR, Hoyles L, McCartney AL, Rastall R, Rowland I, et al. Prebiotic effects: metabolic and health benefits. Br J Nutr. 2010;104 Suppl 2:S1–63.
- Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361:512–9.
- Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–89.
- Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010; 328:1705–9.
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–3.
- Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5:e9085.
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–5.
- Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. 2010;59:3049–57.
- Serino M, Luche E, Gres S, Baylac A, Berge M, Cenac C, et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut. 2012;61:543–53.
- Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci USA. 2011; 108(Suppl 1):4607–14.
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–31.
- Loh G, Brodziak F, Blaut M. The Toll-like receptors TLR2 and TLR4 do not affect the intestinal microbiota composition in mice. Environ Microbiol. 2008;10:709–15.
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–23.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation. 1998;97:2007–11.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–43.
- Koenig W, Khuseyinova N, Baumert J, Meisinger C. Prospective study of high-sensitivity C-reactive protein as a determinant of mortality: results from the MONICA/KORA Augsburg Cohort Study,1984–1998. Clin Chem.2008;54:335–42.
- Pfutzner A, Standl E, Strotmann HJ, Schulze J, Hohberg C, Lubben G, et al. Association of high-sensitive C-reactive protein with advanced stage beta-cell dysfunction and insulin resistance in patients with type 2 diabetes mellitus. Clin Chem Lab Med. 2006;44:556–60.
- Pfutzner A, Forst T. High-sensitivity C-reactive protein as cardiovascular risk marker in patients with diabetes mellitus. Diabetes Technol Ther. 2006;8:28–36.
- Clement K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004;18:1657–69.
- Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care. 2008;31:289–94.
- Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, et al. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci. 2008;1:36–43.
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–7.
- Williamson RT. On the treatment of glycosuria and diabetes mellitus with sodium salicylate. Br Med J. 1901;1:760–2.
- Baron SH. Salicylates as hypoglycemic agents. Diabetes Care. 1982; 5:64–71.
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207.
- Ridker PM, MacFadyen J, Libby P, Glynn RJ. Relation of baseline high-sensitivity C-reactive protein level to cardiovascular outcomes with rosuvastatin in the Justification for Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER). Am J Cardiol. 2010;106:204–9.
- Abe M, Toyohara T, Ishii A, Suzuki T, Noguchi N, Akiyama Y, et al. The HMG-CoA reductase inhibitor pravastatin stimulates insulin secretion through organic anion transporter polypeptides. Drug Metab Pharmacokinet. 2010;25:274–82.
- Takagi T, Matsuda M, Abe M, Kobayashi H, Fukuhara A, Komuro R, et al. Effect of pravastatin on the development of diabetes and adiponectin production. Atherosclerosis. 2008;196:114–21.
- Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2 + signalling and insulin secretion due to blockade of L-type Ca2 + channels in rat islet beta-cells. Br J Pharmacol. 1999;126:1205–13.
- Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305:2556–64.