Abstract
The main approach to obesity and type-II diabetes is to unravel the mechanisms involved in nutrient absorption and fuel allocation. In conditions of over-nutrition, cells must cope with a multitude of extracellular signals generated by changes in nutrient load, hormonal milieu, adverse cytokine/adipokine profile, and apoptosis/anti-apoptosis processes. To date studies have demonstrate that among all nutrients, lipids and carbohydrates play a major regulatory role in the gene transcription of glycolytic and lipogenic enzymes, insulin, and adipokines. These nutrients mainly exert their effects through the gene expression of sterol responsive binding protein 1 and 2 (SREBP) and the mammalian target of rapamycin (mTOR). Excess of adipose tissue is known to confer a significantly higher risk of coronary artery disease. Administration of rapamycin effectively attenuated inflammation, inhibited progression, and enhanced stability of atherosclerotic plaques in animal models. Herein we discuss the mTOR pathway and the molecular mechanisms of mTOR inhibitors, hypothesizing a possible protective role in atherosclerosis, taking into account also previous clinical studies emphasizing their opposite role.
Key messages
A surprising therapeutic approach of CAD is proposed on the basis of recently discovered mechanisms.
Metabolic syndrome and cardiovascular disease risk
Cardiovascular disease (CVD) continues to exert a high burden in terms of morbidity and mortality all over the world. The metabolic syndrome (MS) is a constellation of common metabolic disorders strictly linked to coronary heart disease (CAD). Unclassified non-alcoholic fatty liver disease (NAFLD), or hepatic steatosis (HS), is a further expression of MS (Citation1). It is now commonly accepted that low-grade chronic inflammation associated with obesity induces insulin resistance (IR). The liver–the organ that is central to regulating glucose, lipids, and protein metabolism–consequently plays a pivotal role. Low-grade chronic inflammation is characterized by the production of abnormal cytokines and adipokines such as interleukin-6 (IL-6) (Citation2), tumour necrosis factor-alpha (TNF-α), leptin, and resistin. These factors inhibit insulin signalling in hepatocytes by activating SOCS proteins, several kinases (such as IKK-beta and PKC), but mainly JNKs and protein tyrosine phosphatases (such as PTP1B and PTEN); the last-mentioned in turn impair insulin signalling at insulin receptor and substrate levels. Hepatic IR hence causes impaired suppression of glucose production by insulin in hepatocytes, leading to hyperglycaemia. An important and early complication of hepatic IR is the induction of hepatic VLDL production, via changes in the rate of apoB synthesis and degradation, and de novo lipogenesis, or increased free fatty acid (FFA) flux from adipose tissue to the liver. IR also stimulates the production of C-reactive protein (CRP) and plasminogen activator inhibitor-1 (PAI-1), both markers of an inflammatory state, the circulating levels of which are found elevated in HS (Citation3). All of the following metabolic abnormalities related to hepatic IR have been shown directly or indirectly to promote atherosclerosis. Hyperglycaemia induces a series of alterations including endothelial dysfunction, cellular proliferation, changes in extracellular matrix conformation, and impairment of low-density lipoprotein (LDL) receptor-mediated uptake, decreasing the in vivo clearance of LDL. Small dense LDLs associated with high circulating levels of VLDL have higher affinity to intimal proteoglycans, leading to the penetration of more LDL particles into the arterial wall. CRP can also accelerate atherosclerosis by increasing the expression of PAI-1 and adhesion molecules in endothelial cells, inhibiting nitric oxide formation, and increasing LDL uptake into macrophages. Tissue inflammation is a key factor underlying IR in established obesity. Several models of immunocompromised mice are protected from obesity-induced IR; however, whether inflammation triggers systemic IR or vice versa in obesity remains unanswered. The purpose of a recent study was to address these questions. The authors fed a high-fat diet (HFD) to wild-type mice and three different immunocompromised mouse models (lymphocyte-deficient Rag1 knock-out, macrophage-depleted, and hematopoietic cell-specific Jun NH(Citation2)-terminal kinase-deficient mice) and measured the time course of changes in macrophage content, inflammatory markers, and lipid accumulation in adipose tissue, liver, and skeletal muscle along with systemic insulin sensitivity. In wild-type mice, body weight and adipose tissue mass, as well as IR, were clearly increased after 3 days of HFD. Concurrently, during the short-term HFD, inflammation was selectively elevated in adipose tissue. Interestingly, however, all three immunocompromised mouse models were not protected from IR induced by the short-term HFD. On the other hand, the lipid content was markedly increased in liver and skeletal muscle on day 3 of the diet. These data suggest that the initial stage of HFD-induced IR is independent of inflammation, whereas the more chronic state of IR in established obesity is largely mediated by macrophage-induced pro-inflammatory actions. The early onset IR during HFD feeding is more likely related to acute tissue lipid overload (Citation4).
To begin with, Jun N-terminal kinases (JNKs), also known as stress-activated MAP kinases (SAPK), belong to the family of MAP kinases, which regulate basic processes in adipose tissue, liver, muscle, beta cells, and endothelium. Although several reports have unravelled the critical role of JNK in inducing apoptosis (), a wealth of evidence has also demonstrated a role for JNK in enhancing cell proliferation and survival (Citation5). Recently it has been shown that a small-molecule pan-JNK inhibitor, administered orally and compared to rimonabant and rosiglitazone, significantly impacted parameters such as adiposity, glucose levels, and insulin sensitization without any effect on liver enzymes, thus establishing the role for JNK as a useful target for MS linked to a prediabetic state (Citation6). A JNK1-specific antisense oligonucleotide was studied in ob/ob and diet-induced obese mouse models. A remarkable improvement in insulin sensitivity, adiposity, glucose, and plasma cholesterol levels was observed without any negative impact on liver laboratory parameters. Decreased body weight and adiposity were attributed to increased fuel combustion/metabolic rate and decreased lipogenesis (Citation7).
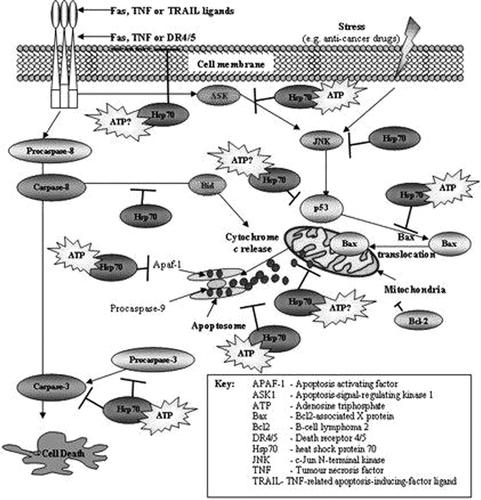
Figure 1. JNKs, apoptotic factors, chaperonins (HSP-70), and cytokines (TNF-α) involved in cell death.
What is the common mechanism by which lipid derivatives, through modulation of macrophage function, promote plaque instability in the arterial wall, impair insulin responsiveness, and contribute to inflammatory liver? In discussing the key molecular mechanism of lipid activation of pro-inflammatory pathways (JNK, nuclear factor-kappaB (NF-kappaB)), the roles played by the proliferator-activated receptor and liver X receptor α, or by the nuclear receptors–lipid sensors linking lipid metabolism and inflammation, should be taken into account (Citation8).
Atherosclerosis begins as local inflammation of artery walls at sites of disturbed flow. JNK is thought to be among the major regulators of flow-dependent inflammatory gene expression in endothelial cells in atherosclerosis. Some authors have shown that JNK activation by both the onset of laminar flow and long-term oscillatory flow is matrix-specific, with enhanced activation on fibronectin compared to basement membrane protein or collagen. Flow-induced JNK activation on fibronectin requires new integrin ligation as well as both the mitogen-activated protein kinase kinase MKK4 and p21-activated kinase. In vivo, JNK activation at sites of early atherogenesis correlates with the deposition of fibronectin. In vivo inhibition of p21-activated kinase reduces JNK activation in vascular atheroprone regions of the vasculature. These results identify JNK as a matrix-specific, flow-activated inflammatory event. Furthermore, their previous data described a network of matrix-specific pathways that determined inflammatory events in response to fluid shear (Citation9).
Visceral adipose tissue (AT) is known to confer a significantly higher risk of type 2 diabetes (T2D) and CAD. Epicardial AT expresses an inflammatory profile of proteins. Some authors have studied key mediators of the NF-kappaB and JNK pathways in paired epicardial and thigh AT from patients with CAD. Western blotting showed epicardial AT with significantly higher NF-kappaB, inhibitory-kappaB kinase (IKK)-gamma, IKK beta, and JNK-1 and -2 compared with thigh AT. Epicardial mRNA data showed strong correlations between CD-68 (another signal of impaired immunity function) and toll-like receptor-2, toll-like receptor-4, and TNF-α. Circulating endotoxin was elevated in patients with CAD compared with matched controls. These data suggest a depot-specific as well as a disease-linked response to inflammation (Citation10).
The concept of ectopic fat storage should be seen under a different light. In fact, when hepatocyte triglyceride synthesis is inhibited, FFA accumulated in the liver stimulate the fatty acid oxidizing systems that increase hepatic oxidative stress and liver damage. These findings suggest that the ability to synthesize triglycerides may, in fact, be protective in obesity (Citation11,Citation12).
This is a central point. MEK1/2 inhibition significantly increased both cellular and microsomal triglyceride mass, and mRNA levels for DGAT-1 and DGAT-2. In contrast to ERK, modulation of the phosphatidylinositol 3-kinases pathway or inhibition of the p38 MAP kinase had no effect on lipoprotein density profile (Citation13). Intracellular lipid droplets (LD) are cytosolic inclusions present in most eukaryotic cells; they contain a core of triacylglycerol and cholesteryl esters, surrounded by a phospholipid monolayer and by specific proteins, the best characterized among them belonging to the perilipin family. In the last few years, the biology of LD has received increasing interest, due to the close relationship between an excess of lipid storage in certain tissues and pathologies such as obesity, T2D, and atherosclerosis. Cellular stress has been related to the generation of LD, which might play a cytoprotective role. The biogenesis of LD induced by serum depends on group IVA phospholipase A (Citation2) (cPLA(Citation2)alpha), a regulatory enzyme that releases arachidonic acid for the production of prostaglandins and leukotrienes. Recent data suggest that cPLA(Citation2)alpha regulates the transport of tight junction and adherence junction proteins through the Golgi to cell–cell contacts in confluent endothelial cells. The expression of specific activators of different MAP kinases shows that phosphorylation of cPLA(Citation2)alpha at Ser-505 is due to JNK. This was confirmed by pharmacological inhibition and expression of a dominant-negative form of the upstream activator MEKK1. LD biogenesis was accompanied by increased synthesis of ceramide 1-phosphate. Over-expression of its synthesizing enzyme ceramide kinase increased phosphorylation of cPLA(Citation2)alpha at Ser-505 and formation of LD, and its down-regulation blocked the phosphorylation of cPLA(Citation2)alpha and LD biogenesis (Citation14).
On the other hand, the effect of glucose and palmitate on the phosphorylation of proteins is associated with cell growth and survival. Novel evidence suggests that short-term changes of MAPK and AKT signalling pathways and c-fos and JNK expressions caused by glucose are counterbalanced by palmitate through phosphatidylinositol 3-kinase inhibition via ceramide synthesis (Citation15).
Cell growth in response to nutrients and energy
As previously underlined, the mammalian target of rapamycin (mTOR), also known to be a mechanistic target of rapamycin or FK506 binding protein 12-rapamycin associated protein 1 (FRAP1), is a highly conserved ser/thr kinase that plays a key role in cell growth, proliferation, and survival (). The mammalian target of rapamycin regulates cell growth in response to growth factors, nutrients, and energy (Citation16) and is present in two functionally distinct complexes, mTORC1 and mTORC2: mTORC1 consists of mTOR, raptor, deptor, PRAS40, and mLST8; whereas mTORC2 is composed of mTOR, rictor, sin1, mLST8, deptor, and protor (Citation17). While acute exposure to rapamycin blocks mTORC1, it does not affect mTORC2. However, subsequent studies have shown that, at least in some cell lines, chronic exposure to rapamycin, while not affecting pre-existing mTORC2s, promotes rapamycin inhibition of free mTOR molecules, thus inhibiting the formation of new mTORC2 (Citation18). Despite the wealth of experimental evidence in different animal models showing that the inhibition of mTORC1 by rapamycin and its analogues blocks tumour progression, its benefits in clinical trials have been less successful than expected (Citation19). This outcome could be partly explained by the observation that the inhibition of mTORC1 results in the activation of the PI3K/Akt signalling pathway, which induces signals that sustain proliferation and survival (Citation20).
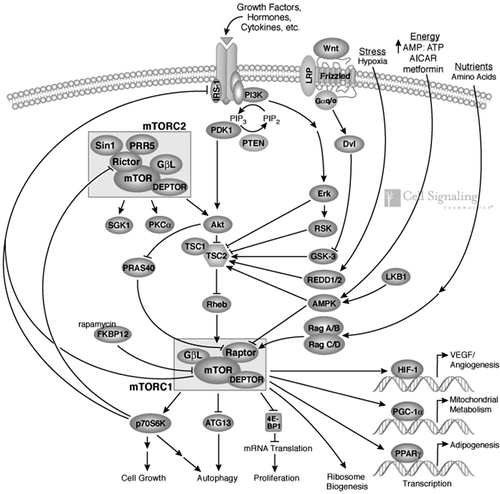
Figure 2. The mammalian target of rapamycin and its correlation to angiogenesis, mitochondrial metabolism, and adipogenesis. Pathway diagram reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
AMP-activated protein kinase (AMPK)
Mammalian target of rapamycin activity becomes repressed under conditions of energy deprivation, in which an elevated AMP/ATP intracellular ratio causes activation of AMPK. Stressful conditions, such as nutrient deprivation, hypoxia, heat shock, and ischaemia, diminish cellular energy reserves. The link between mTOR inhibition and AMPK was first demonstrated with the use of AICAR (5’-phosphoribosyl-5-aminoimidazole-4-carboxamide ribonucleoside), an AMPK activator, thus linking the amino acid- and energy-sensing functions of mTOR. Metformin, an antidiabetic drug, also activates AMPK and is hypothesized to reduce the risk of cancer in patients with T2D. Thus, targeting AMPK may be an interesting therapeutic option for cancer therapy (Citation21).
Autophagy and mTOR
Autophagy is a process by which components of the cell are degraded to maintain essential activity and viability in response to nutrient limitation. Extensive genetic studies have shown that the yeast ATG1 kinase has an essential role in autophagy induction. Furthermore, autophagy is promoted by AMPK, which is a key energy sensor and regulates cellular metabolism to maintain energy homeostasis. Conversely, autophagy is inhibited by mTOR, central cell-growth regulator that integrates growth factor and nutrient signals. Joungmok et al. demonstrated a molecular mechanism for regulation of the mammalian autophagy-initiating kinase Ulk1, a homologue of yeast ATG1. Under glucose starvation, AMPK promotes autophagy by directly activating Ulk1 through phosphorylation of Ser-317 and Ser-777. Under nutrient sufficiency, high mTOR activity prevented Ulk1 activation by phosphorylating Ulk1 Ser-757 and disrupting the interaction between Ulk1 and AMPK. This co-ordinated phosphorylation is important for Ulk1 in autophagy induction. This study has revealed a signalling mechanism for Ulk1 regulation and autophagy induction in response to nutrient signalling (Citation22).
ER stress and mTOR
Disturbance to endoplasmic reticulum (ER) homeostasis that cannot be rescued by the unfolded protein response (UPR) results in autophagy and cell death, but the precise mechanism was largely unknown. Qin et al. demonstrated that ER stress-induced cell death was mediated by autophagy, which was partly attributed to the inactivation of mTOR. Three widely used ER stress inducers, including tunicamycin, DTT, and MG132, led to the conversion of LC3-I to LC3-II, a commonly used marker of autophagy, as well as the down-regulation of mTOR concurrently. TSC-deficient cells with constitutive activation of mTOR exhibited more resistance to ER stress-induced autophagy, compared with their wild-type counterparts. Furthermore, their studies showed that ER stress-induced deactivation of mTOR was attributed to the down-regulation of AKT/TSC/mTOR pathway. Phosphatase and tensin homologue (PTEN) and AMPK as two regulators in this pathway seemed to be absent in this regulation. As a chemical chaperone helping the correct folding of proteins, 4-phenylbutyric acid (4-PBA) partly rescued the AKT/TSC/mTOR pathway in drug-induced acute ER stress. Moreover, constitutively activated mTOR-induced long-term ER stress attenuated the RTK/PI3K/AKT signalling pathway in response to the stimulation by various growth factors (), which could also be partly restored by 4-PBA (Citation23).
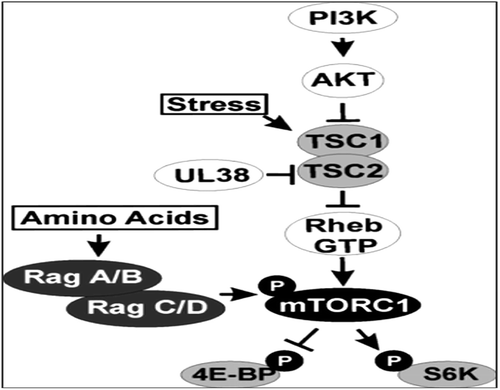
Figure 3. Likely mechanisms involving mTOR, amino acids sensing functions and ER stress.
The mammalian target of rapamycin inhibitors block cell cycle progression and inhibit lymphocyte proliferation; mTOR is a nutrient sensor and a crucial key regulator for the signal pathways of factors of cellular growth and metabolism, the p70 ribosomal S6 kinase 1 (S6K1) and the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) (Citation24). Both are implicated in protein synthesis. Furthermore, mTOR regulates transcription by either inhibiting or activating cellular processes (Citation25). Notably, rapamycin inhibition of p70S6K leads to the down-regulation of COX-2 and iNOS protein expression, but not the steady-state mRNA expression and transcription induced by catalase, suggesting that p70S6K is involved in increased COX-2 and iNOS mRNA translation by catalase. Interestingly, PI3K-dependent activation of AKT, p70S6K, JNKs, and NF-kappaB occurs in response to catalase (Citation26). From a mechanistic perspective, rapamycin showed antiangiogenic activities linked to a decreased production of vascular endothelial growth factor (VEGF) and to a markedly inhibited response of vascular endothelial cells to VEGF stimulation. Thus, the use of rapamycin, instead of cyclosporin, may reduce the chance of recurrent or de novo cancer in high-risk transplant patients (Citation27). After adjustment for age, VEGF showed a weak positive correlation with BMI, liver enzymes, CRP, and platelet count in males. In females VEGF correlated negatively with LDL-cholesterol and positively with IR and platelet count. Again, after adjustment for age, no significant correlation with carotid atherosclerosis could be detected. The plasma VEGF and soluble VEGF receptor called sFlt-1 have been correlated, albeit weakly, with cardiovascular risk factors, suggesting that circulating VEGF levels could have an impact on the development of atherosclerosis (Citation28). Noteworthily, in NAFLD patients the serum concentration of VEGF increased (Citation2). Based on this finding, some authors measured levels of VEGF, free and complexed sFlt-1 in citrated plasma from 40 patients with cardiovascular disease and 40 healthy controls. Median plasma levels of VEGF in patients were significantly higher than in controls. Free sFlt-1 was significantly lower in patients compared to controls (Citation29).
The hyperactivation of mTOR that is produced by chronic exposure to platelet-derived growth factor (PDGF), TNF-α, insulin, amino acids, or fatty acids can lead to desensitization to insulin and IR. Increased mTOR activity and subsequent over-activation of S6K1 can exert a negative feedback mechanism by the phosphorylation of the insulin receptor substrate (IRS)-1, which can negatively influence PI3K/Akt pathway signalling (Citation30). The possible IRS-1 blockade decreases the translocation of the glucose transporter 4 (GLUT-4) to the cellular membrane in adipose and muscular tissue, which itself leads to hyperglycaemia (Citation31). However, a recent publication showed that, even if rapamycin is able to block the negative feedback loop and to enhance PKB/Akt signalling, this does not influence glucose uptake (Citation32). Mammalian target of rapamycin is an essential factor for the adipocyte differentiation (Citation33) that activates PPAR-gamma (Citation34), the transcription factor with a critical role in adipogenesis and accumulation of lipids as well. The mTOR pathway is over-activated in the liver and muscles of obese rats (Citation35).
Inhibition of the mTOR pathway
Treatment with rapamycin (sirolimus), an mTOR inhibitor, produces phosphorylation in the Thr-308 and Ser-473 of Akt, a downstream target for PI3K. Insulin induces the activation of Akt through the phosphorylation of both residues in HepG2 cells; while insulin produces a temporary increase in Thr-308 phosphorylation, the effect of the hormone maintains a 30-min increase in Ser-473. Rapamycin preconditioning determines an increase in the phosphorylation of Akt in both sites and a slow decrease in the dephosphorylation of Thr-308. The inhibition of the mTOR pathway through rapamycin increases Akt signalling in HepG2 cells. However, the absence of S6K1 in a knock-out mouse model causes hypoinsulinaemia and hypersensitivity to insulin. Besides, the S6K1−/− genotype protects from obesity induced by nutrients (Citation36). As mTOR is central to the insulin signalling pathway (), mTOR inhibitors are supposedly involved in atherosclerosis, as confirmed by preliminary data. Coronary stents coated with mTOR inhibitors are already widely used in revascularization procedures following the evidence that mTOR-eluting stents reduce binary restenosis, late lumen loss, and repeat revascularization compared with standard stents (Citation37). The development of such stents was based on animal data showing that everolimus and sirolimus attenuate neointimal thickening and transplant atherosclerosis (Citation38). As a possible explanation, some authors concluded that venous endothelial cell proliferation is inhibited at concentrations needed to suppress TNF-α-stimulated IL-6 synthesis (again the main inflammatory cytokine). Furthermore, the specific suppression of basal arterial IL-6-secretion and the delayed onset of the mTOR-inhibitor effect on human coronary artery or venous endothelial cell proliferation (maximum reached after about 36 h) might be of relevance for the prevention of transplant vasculopathy in the initial stage, e.g. at the arrest of myocardial contractions (Citation39). Apart from this hypothetical mechanism, there is currently intense effort to explain the extent to which mTORs may control atherosclerosis. Animal models have indicated that both everolimus and sirolimus prevent lipid accumulation in tissues (Citation40) and help stabilize atherosclerotic plaques by selective clearance of macrophages (Citation41) and by inhibiting the local inflammatory response in arterial smooth muscle cells (Citation42). Importantly, these effects may counteract the hypercholesterolaemia and hypertriglyceridaemia associated with mTOR inhibitor therapy described by some authors. Convincing data from an independent group have been generated showing that mTOR inhibitors limit atherosclerotic plaque size and progression in animal models (Citation43). A dose-dependent reduction in atherosclerotic lesions following the administration of everolimus to mice with cholesterol-induced atherosclerosis has been observed (Citation44). Oral administration of rapamycin effectively attenuated inflammation, inhibited progression, and enhanced stability of atherosclerotic plaques in rabbits, without altering serum lipid levels. These findings suggest a novel approach to the treatment of atherosclerosis (Citation45).
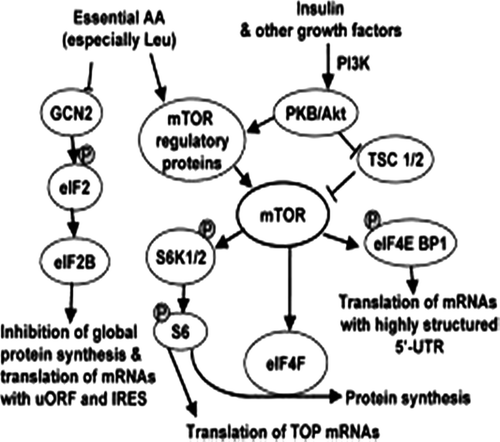
Figure 4. Main substances activating the mTOR pathway cascade.
In contrast, there is some uncertainty over the effect of immunosuppression on weight gain, which could be ultimately considered a drawback. Obesity is common in the liver transplant population, but it seems to be unrelated to any specific immunosuppressive drug. The greatest weight gain occurs after the first 6 months, and intervention with dietary advice at this point could be implemented to minimize the long-term morbidity and mortality risks associated with obesity. Several studies have observed a decreased incidence of obesity after liver transplantation (OLTx) in patients taking tacrolimus compared with those on cyclosporin (Citation46). Dietary mistakes and lack of physical activity may play a major role in the weight increase in OLTx. Despite a striking proportion of overweight and obese patients in the group studied, the number of cardiovascular episodes seems to match that in the general population (Citation47). Cyclosporin was found to cause increased weight gain compared with tacrolimus only in the first year after transplantation, but not in the long term (Citation48). A recent study demonstrates that treatment with mTOR inhibitors in otherwise healthy animals reduces body weight, adipocyte diameter, and insulin sensitivity. Furthermore, the influence of mTOR inhibition on body weight of transplant patients resembles the observations made in animals. A possible explanation may be the effects of sirolimus on metabolic regulation and cell growth (Citation49). As previously mentioned activation of mTOR is associated with increased cell survival and is a potential target for chemotherapy for some malignancies. In tumour cell lines, activation of WNT signaling proteins has been shown to activate the mTOR pathway via inhibiting glycogen synthase kinase 3 (GSK3), whereas the inhibition of mTOR by rapamycin blocks Wnt-induced cell growth (Citation50). Interestingly, in these models GSK3 inhibits the mTOR pathway by phosphorylating TSC2 in a manner dependent on AMPK-priming phosphorylation.
Mammalian target of rapamycin and cardioprotection
Conflicting results regarding the role of mTOR are also observed in cardioprotection and might depend on rapamycin concentration: some studies show a protective effect of rapamycin, while others imply a cardioprotective effect of the mTOR pathway. A recent report on an ischaemic model of injury, using low doses of rapamycin, pleads in favour of a cardioprotective effect of the mTOR pathway and suggests that mTOR integrates signals from the Wnt pathway via GSK3 to control the development of cardioprotection (Citation51). Another work recently published shows that inhibition of the canonical Wnt signalling pathway and induction of the renin-angiotensin system might be implicated in the remodelling of sc adipose tissue during the initial phase of weight gain induced by over-feeding in humans (Citation52). Wnts (network of proteins) regulate diverse processes including cell proliferation, differentiation, cell polarity, and migration and play a key role in normal (embryonic development) as well as pathological processes (carcinogenesis and inflammation disorders). Wnts have been implicated in vascular biology, specifically angiogenesis, vessel remodelling, and transendothelial migration of monocytes. As a result, the Wnt signalling pathway would be involved in the process of atherosclerosis.
Calcineurin-free immunosuppression
Another issue of great interest in relation to the kidney transplant population is the occurrence of CAD and cerebrovascular accidents, which represent the most frequent fatal cardiovascular disease events. Overall, the annual risk of a cardiovascular event is up to 50-fold higher in kidney transplant patients compared to the general population (Citation53). Kidney transplant patients face three main categories of cardiovascular risk factors. As in the non-transplant population, diabetes, hyperlipidaemia, hypertension, and smoking increase the risk of CAD. The introduction of CNIs resulted in a dramatic reduction in acute rejection and short-term graft survival rates for two decades (Citation54). Additionally, their use permitted a valuable decrease in corticosteroid doses, reducing the impact of steroid-related cardiovascular complications such as T2D, arterial hypertension, hyperlipidaemia, and obesity (Citation55). Disappointingly, however, it became apparent that maintenance CNI therapy is also associated with some important cardiovascular risk factors. The use of CNIs is associated with the onset of de novo T2D, hypertension, and hyperlipidaemia; in addition, of all the risk factors present in transplant patients, T2D confers the greatest CAD risk, with an estimated 3-fold increase in men and over 5-fold in women more than 1 year post-transplant (Citation56). CNI therapy, particularly tacrolimus, adversely affects glucose metabolism, increases the risk of new-onset T2D (Citation57), and raises cholesterol and triglyceride levels, although whether this effect is time-dependent remains unclear (Citation58).
Attempts to achieve entirely CNI-free immunosuppression have generally been associated with an unacceptable rate of acute rejection or a high rate of discontinuation because of adverse events. Current strategies attempt to minimize CNI exposure, rather than replace it completely. One of the best-addressed and most successful approaches is to employ the mTOR inhibitor agents everolimus or sirolimus with the aim of withdrawing CNI immediately after the high-risk post-transplant period or facilitating low-exposure CNI maintenance therapy (Citation59). CNI discontinuation or reduced-exposure CNI therapy achieved by the use of mTOR inhibitors could also offer an opportunity to improve cardiovascular risk following kidney transplantation, as the CNI and mTOR inhibitor classes are associated with different safety profiles. The potential advantage of mTOR inhibitor-based regimens in terms of improving CVD risk following kidney transplantation falls into two broad categories: first, the reduction of CNI-related complications and, second, possible cardioprotective effects of the mTOR inhibitor class of drugs to be used as broadly as possible.
Endoplasmic reticulum stress and obesity
Addressing the topic from a new perspective, obesity leads to chronic ER stress in adipose tissue. ATF6-regulated chaperones are increased in subcutaneous fat from obese individuals. Increased eIF2a phosphorylation in enlarged fat depots possibly reflects PERK activation. The correlation of ER stress with insulin sensitivity is lost after adjusting for body mass index (Citation60), raising doubts as to whether ER stress is indeed a direct cause of IR. IRE1 activation was found in the adipose tissue of obese, compared with non-obese, volunteers, with JNK1 activation and up-regulation of XBP1s mRNA, calreticulin, calnexin, and protein disulfide isomerase (Citation61). In a study comparing various fat depots, BiP and XBP1 expression was higher in visceral compared with subcutaneous fat, and more so in severe obesity (Citation62). One year after gastric bypass surgery, adipocyte XBP1s and BiP mRNA levels as well as JNK and eIF2a phosphorylation were significantly decreased, as compared to pre-surgery levels (Citation63). The increased expression of IRE1- and ATF6-dependent chaperones in adipocytes from obese individuals is mimicked in vitro by adipocyte exposure to Lipopolysaccharides (LPS), saturated FFAs, or glucose (Citation64). FFA-induced ER stress in adipocytes is pro-inflammatory via PERK-dependent IKK activation (Citation65). In addition to contributing to inflammation, ER stress can modify FFA and adipokine secretion. Adipocyte ER stress induces basal lipolysis through down-regulation of perilipin (Citation66). Decreased ER disulfide-bond A oxidore-ductase-like (DsbA-L) protein expression in obesity impairs adiponectin folding and multimerization, and causes ER stress (Citation67). Insulin receptor expression and leptin secretion are also decreased during ER stress, whereas IL-6 is strongly induced (Citation68). It appears that ER stress in adipocytes might initially lead to changes resembling early prediabetic stages, at least partly supporting the regulation of systemic energy homeostasis. ER stress also inhibits resistin transcription in murine adipocytes through the up-regulation of transcriptional repressor CAAT/enhancer binding protein homologous protein-10 (CHOP). ER stress is a potent regulator of resistin, suggesting that it may underlie the local down-regulation of resistin mRNA and protein in fat in murine obesity. The paradoxical increase in plasma resistin may exist because of various systemic abnormalities associated with obesity and IR (Citation69). Attempts to modulate the adipose tissue UPR have generated surprising results. BiP heterozygosity protects mice from HFD-induced IR in white fat; this has been attributed to the development of an adaptive unfolded protein response (UPR) in BiP+/– mice characterized by lesser translational attenuation and enhanced XBP1 splicing and ERAD. High-fat-diet (HFD)-induced obesity and T2D are improved in Grp78+/– mice. Adaptive UPR in white adipose tissue could contribute to this improvement, linking ER homeostasis to energy balance and glucose metabolism (Citation70). Interestingly, recent results showed that a swimming protocol reduces pro-inflammatory molecules (JNK, IκB, and NF-kappaB) in adipose and hepatic tissues. In addition, exercise decreases ER stress, by reducing PERK and eIF2α phosphorylation. Thus, exercise can reduce ER stress, improving IR in the same tissues (Citation71). Coming back to the liver, it is one of the most important secretory organs in the body. It synthesizes and secretes bile acids, lipoproteins, and all the major plasma proteins including the albumin, globulins, fibrinogen, and proteins responsible for blood clotting. In keeping with this secretory function, the liver physiologically activates the UPR. The three major arms of the mammalian UPR include 1) protein kinase RNA (PKR)-like ER kinase (PERK), 2) inositol-requiring protein-1 (IRE1α), and 3) activating transcription factor-6 (ATF6) pathways. A circadian rhythm of IRE1α signalling is closely linked to circadian regulation of mouse liver lipid metabolism. The resulting aberrant circadian lipid metabolism in mice devoid of the circadian clock could be involved in the appearance of the associated MS (Citation72). Physiological ER stress is detected in the liver of rodents that are refed after fasting; this resolves within hours (Citation73). By contrast, chronic hepatic ER stress has been described in obese animals and humans with NAFLD (Citation74). Defective autophagy in obesity may favour ER stress by insufficiently clearing dysfunctional organelles, demonstrating that autophagy is an important regulator of organelle function and insulin signalling and that the loss of autophagy is a critical component of the defective insulin action seen in obesity (Citation75). ER stress and ER stress sensors play an important role in liver lipid metabolism and in the onset of HS and IR. Recent findings demonstrate that GRP78 inhibits both insulin-dependent and ER stress-dependent SREBP-1c proteolytic cleavage, and explain the role of ER stress in HS in obese rodents (Citation76). The BiP protein, a stress response protein, plays an important role in the proper folding and assembly of nascent protein and in the scavenging of misfolded proteins in the ER lumen. Hepatic adenoviral BiP over-expression in ob/ob mice decreases SREBP-1c activation, hepatic triglyceride and cholesterol contents, and improves insulin sensitivity. Interestingly, SREBP is associated with BiP and, as is the case for ATF6, the export of SREBP towards the Golgi. The Akt kinase is a critical effector in growth factor signalling. Activation of Akt driven by the growth factor-dependent PI3K (phosphatidylinositol-3-OH kinase) is coupled to the plasma membrane translocation and phosphorylation of Akt on two sites by PDK1 (phosphoinositide-dependent protein kinase-1) on Thr-308 and by mTORC2 on Ser-473. In a recent study some authors examined the subcellular localization of mTORC2 and identified that this kinase complex predominantly resides on ER. Their immunostaining analysis did not show a substantial co-localization of the mTORC2 component rictor with Golgi, lysosome, clathrin-coated vesicles, early endosomes, or plasma membrane but indicated a strong co-localization of rictor with ribosomal protein S6 and ER marker. Their biochemical study also identified the mTORC2 components rictor, SIN1, and mTOR as the highly abundant proteins in the ER fraction, whereas only small amounts of these proteins are detected in the plasma membrane and cytosolic fractions. They found that growth factor signalling does not alter the ER localization of mTORC2, nor does it induce its translocation to the plasma membrane. Based on this study they suggest that the mTORC2-dependent phosphorylation of Akt on Ser-473 takes place on the ER surface (Citation77).
Conclusion
The aforementioned findings offer clinical investigators interesting suggestions. To date, most studies on transplanted humans have investigated cardiovascular effects, i.e. atherosclerosis, only in terms of incidence, and only rarely as a primary or main secondary end-point. Generally, the onset of T2D or lipid abnormalities is highlighted collaterally when other data are recorded during follow-up. These data have sometimes been used to argue in favour of a given immunosuppressant, but in most studies the focus is on immunological details, graft survival, and renal or hepatic function and not primarily on CAD. In addition, measurement techniques of CVD vary considerably from those used in pure cardiovascular studies. As a consequence, most of the data from observational studies are sometimes of questionable impact. Undertaking clinical trials using cardiovascular end-points in organ transplant recipients to extend them possibly to other populations is a priority. Observational studies, however, have the advantage of generating new hypotheses which ought to be proven.
Acknowledgements
G.T. conceived the research, thoroughly analysed literature data, and drafted the manuscript. D. C. critically revised the content of manuscript and helped search the literature data.
Declaration of interest: The authors state that there is no conflict of interest.
References
- Tarantino G, Saldalamacchia G, Conca P, Arena A. Non-alcoholic fatty liver disease: further expression of the metabolic syndrome. J Gastroenterol Hepatol. 2007;22:293–303.
- Tarantino G, Conca P, Pasanisi F, Ariello M, Mastrolia M, Arena A, et al. Could inflammatory markers help diagnose nonalcoholic steatohepatitis?Eur J Gastroenterol Hepatol. 2009;21:504–11.
- Barbato A, Iacone R, Tarantino G, Russo O, Sorrentino P, Avallone S, et al.; Olivetti Heart Study Research Group. Relationships of PAI-1 levels to central obesity and liver steatosis in a sample of adult male population in southern Italy. Intern Emerg Med. 2009;4:315–23.
- Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60:2474–83.
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–9.
- Cho H, Black SC, Looper D, Shi M, Kelly-Sullivan D, Timofeevski S, et al. Pharmacological characterization of a small molecule inhibitor of c-Jun kinase. Am J Physiol Endocrinol Metab. 2008;295:E1142–51.
- Yu XX, Murray SF, Watts L, Booten SL, Tokorcheck J, Monia BP, et al. Reduction of JNK1 expression with antisense oligonucleotide improves adiposity in obese mice. Am J Physiol Endocrinol Metab. 2008; 295:E436–45.
- Prieur X, Roszer T, Ricote M. Lipotoxicity in macrophages: evidence from diseases associated with the metabolic syndrome. Biochim Biophys Acta. 2010;1801:327–37.
- Hahn C, Orr AW, Sanders JM, Jhaveri KA, Schwartz MA. The subendothelial extracellular matrix modulates JNK activation by flow stress. Circ Res. 2009;104:995–1003.
- Baker AR, Harte AL, Howell N, Pritlove DC, Ranasinghe AM, da Silva NF, et al. Epicardial adipose tissue as a source of nuclear factor-kappaB and c-Jun N-terminal kinase mediated inflammation in patients with coronary artery disease. J Clin Endocrinol Metab. 2009;94:261–7.
- Choi SS, Diehl AM. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol. 2008;19:295–300.
- Tarantino G, Savastano S, Colao A, Capone D, Tarantino M, Grimaldi E, et al. Serum Bcl-2 concentrations in overweight-obese subjects with nonalcoholic fatty liver disease?World J Gastroenterol. 2011;17:5280–8.
- Tsai J, Qiu W, Kohen-Avramoglu R, Adeli K. MEK-ERK inhibition corrects the defect in VLDL assembly in HepG2 cells: potential role of ERK in VLDL-ApoB100 particle assembly. Arterioscler Thromb Vasc Biol. 2007;27:211–8.
- Gubern A, Barceló-Torns M, Barneda D, López JM, Masgrau R, Picatoste F, et al. JNK and ceramide kinase govern the biogenesis of lipid droplets through activation of group IVA phospholipase A2. J Biol Chem. 2009; 284:32359–69.
- Nogueira TC, Graciano MF, Anhê GF, Curi R, Bordin S, Carpinelli AR. Short-term modulation of extracellular signal-regulated kinase 1/2 and stress-activated protein kinase/c-Jun NH2-terminal kinase in pancreatic islets by glucose and palmitate: possible involvement of ceramide. Pancreas. 2009;38:585–92.
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84.
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22.
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68.
- Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–88.
- Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22:169–76.
- Petroulakis E, Mamane Y, Le Bacquer O, Shahbazian D, Sonenberg N. mTOR signaling: implications for cancer and anticancer therapy. Br J Cancer. 2006;94;195–99.
- Joungmok K, Mondira K, Benoit V, Kun-Liang. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk. Nature Cell Biology. 2011;13:132–41.
- Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6:239–47.
- Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–26.
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603.
- Jang BC, Paik JH, Kim SP, Shin DH, Song DK, Park JG, et al. Catalase induced expression of inflammatory mediators via activation of NF-kappaB, PI3K/AKT, p70S6K, and JNKs in BV2 microglia. Cell Signal. 2005;17:625–33.
- Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8:128–35.
- Sandhofer A, Tatarczyk T, Kirchmair R, Iglseder B, Paulweber B, Patsch JR, et al. Are plasma VEGF and its soluble receptor sFlt-1 atherogenic risk factors?Cross-sectional data from the SAPHIR study. Atherosclerosis. 2009;206:265–9.
- Belgore FM, Blann AD, Lip GY. Measurement of free and complexed soluble vascular endothelial growth factor receptor, Flt-1, in fluid samples: development and application of two new immunoassays. Clin Sci (Lond). 2001;100:567–75.
- Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, et al. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000;14:783–94.
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenes. J Cell Biol. 2004;167:399–403.
- Ginion A, Auquier J, Benton CR, Mouton C, Vanoverschelde JL, Hue L, et al. Inhibition of the mTOR/p70S6K pathway is not involved in the insulin-sensitizing effect of AMPK on cardiac glucose uptake. Am J Physiol Heart Circ Physiol. 2011;301:H469–77.
- Cho HJ, Park J, Lee HW, Lee YS, Kim JB. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem Biophys Res Commun. 2004;321:942–8.
- Kim JE, Chen J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. 2004;53:2748–56.
- Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146:1473–81.
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–5.
- Morice MC, Serrys PW, Sousa JE, Fajadet J, Ban Hayashi E, Perin M, et al.; RAVEL Study Group. Randomized Study with the Sirolimus-Coated Bx Velocity Balloon-Expandable Stent in the Treatment of Patients with de Novo Native Coronary Artery Lesions. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N Engl J Med. 2002;346:1773–80.
- Ma KL, Ruan XZ, Powis SH, Moorhead JF, Varghese Z. Anti-atherosclerotic effects of sirolimus on human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292:H2721–8.
- Schreml S, Lehle K, Birnbaum DE, Preuner JG. mTOR-inhibitors simultaneously inhibit proliferation and basal IL-6 synthesis of human coronary artery endothelial cells. Int Immunopharmacol. 2007;7: 781–90.
- Morrisett JD, Abdel-Fattah G, Hoogeveen R , Mitchell E, Ballantyne CM, Pownall HJ, et al. Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J Lipid Res. 2002;43:1170–80.
- Baetta R, Granata A, Canavesi M, Ferri N, Arnaboldi L, Bellosta S, et al. Everolimus inhibitors monocyte/macrophage migration in vitro and their accumulation in carotid lesions of cholesterol-fed rabbits. J Pharmacol Exp Ther. 2009;328:419–25.
- Gouëffic Y, Potter-Perigo S, Chan CK, Johnson PY, Braun K, Evanko SP, et al. Sirolimus blocks the accumulation of hyaluronan (HA) by arterial smooth muscle cells and reduces monocyte adhesion to the ECM. Atherosclerosis. 2007;195:23–30.
- Waksman R, Pakala R, Burnett MS, Gulick CP, Leborgne L, Fournadjiev J, et al. Oral rapamycin inhibits growth of atherosclerotic plaque in apo E knock-out mice. Cardiovasc Radiat Med. 2003;4:34–8.
- Mueller MA, Beutner F, Teupser D, Ceglarek U, Thiery J. Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR2/2 mice despite severe hypercholesterolemia. Atherosclerosis. 2008;198:39–48.
- Chen WQ, Zhong L, Zhang L, Ji XP, Zhang M, Zhao YX, et al. Oral rapamycin attenuates inflammation and enhances stability of atherosclerotic plaques in rabbits independent of serum lipid levels. Br J Pharmacol. 2009;156:941–51.
- Neal DAJ, Gimson AES, Gibbs P, Alexander AJM. Beneficial effects of converting liver transplant recipients from cyclosporin to tacrolimus on blood pressure, serum lipids and weight. Liver Transpl. 2001;7:533–9.
- Wawrzynowicz-Syczewska M, Karpińska E, Jurczyk K, Laurans L, Boroń-Kaczmarska A. Risk factors and dynamics of weight gain in patients after liver transplantation. Ann Transplant. 2009;14:45–50.
- Richards J, Gunson B, Johnson J, Neuberger J. Weight gain and obesity after liver transplantation. Transpl Int. 2005;18:461–6.
- Rovira J, Marcelo Arellano E, Burke JT, Brault Y, Moya-Rull D, Bañón-Maneus E, et al. Effect of mTOR inhibitor on body weight: from an experimental rat model to human transplant patients. Transpl Int. 2008;21:992–8.
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–68.
- Vigneron F, Dos Santos P, Lemoine S, Bonnet M, Tariosse L, Couffinhal T, et al. GSK-3β at the crossroads in the signalling of heart preconditioning: implication of mTOR and Wnt pathways. Cardiovasc Res. 2011;90:49–56.
- Alligier M, Meugnier E, Debard C, Lambert-Porcheron S, Chanseaume E, Sothier M, et al. Subcutaneous adipose tissue remodeling during the initial phase of weight gain induced by overfeeding in humans. J Clin Endocrinol Metab. 2012;97:E183–92.
- Aakhus S, Dahl K, Wideroe TE. Cardiovascular disease in stable renal transplant patients in Norway: morbidity and mortality during a 5-yr follow-up. Clin Transplant. 2004;18:596–604.
- Kramer NC, Peters TG, Rohr MS, Thacker LR, Vaughn WK. Beneficial effect of cyclosporine on renal transplantation. A multicenter long-term study. Transplantation. 1990;49:343–8.
- Jindal RM, Zawada Jr ET. Obesity and kidney transplantation. Am J Kidney Dis. 2004;43:943–52.
- Kasiske BL, Chakkera HA, Roel J. Explained and unexplained ischemic heart disease risk after renal transplantation. J Am Soc Nephrol. 2000;11:1735–43.
- Elmagd MM, Bakr MA, Metwally AH, Wahab AM. Clinicoepidemiologic study of posttransplant diabetes after living-donor renal transplant. Exp Clin Transplant. 2008;6:42–7.
- Tarantino G, Palmiero G, Polichetti G, Perfetti A, Sabbatini M, Basile V, et al. Long-term assessment of plasma lipids in transplant recipients treated with tacrolimus in relation to fatty liver. Int J Immunopathol Pharmacol. 2010;23:1303–8.
- Augustine JJ, Hricik DE. Minimization of immunosuppression in kidney transplantation. Curr Opin Nephrol Hypertens. 2007;16:535–41.
- Sharma NK, Das SK, Mondal AK, Hackney OG, Chu WS, Kern PA, et al. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab. 2008;93:4532–41.
- Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, et al. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57:2438–44.
- Vendrell J, Maymó-Masip E, Tinahones F, García-España A, Megia A, Caubet E, et al. Tumor necrosis-like weak inducer of apoptosis as a proinflammatory cytokine in human adipocyte cells: up-regulation in severe obesity is mediated by inflammation but not hypoxia. J Clin Endocrinol Metab. 2010;95:2983–92.
- Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700.
- Alhusaini S, McGee K, Schisano B, Harte A, McTernan P, Kumar S, et al. Lipopolysaccharide, high glucose and saturated fatty acids induce endoplasmic reticulum stress in cultured primary human adipocytes: salicylate alleviates this stress. Biochem Biophys Res Commun. 2010; 397:472–8.
- Jiao P, Ma J, Feng B, Zhang H, Alan Diehl J, Eugene Chin Y, et al. FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKb pathways. Obesity (Silver Spring). 2011; 19:483–91.
- Zhou QG, Zhou M, Hou FF, Peng X. Asymmetrical dimethylarginine triggers lipolysis and inflammatory response via induction of endoplasmic reticulum stress in cultured adipocytes. Am J Physiol Endocrinol Metab. 2009;296:E869–78.
- Zhou L, Liu M, Zhang J, Chen H, Dong LQ, Liu F. DsbA-L alleviates endoplasmic reticulum stress-induced adiponectin downregulation. Diabetes. 2010;59:2809–16.
- Xu L, Spinas GA, Niessen M. ER stress in adipocytes inhibits insulin signaling, represses lipolysis, and alters the secretion of adipokines without inhibiting glucose transport. Horm Metab Res. 2010;42:643–51.
- Lefterova MI, Mullican SE, Tomaru T, Qatanani M, Schupp M, Lazar MA. Endoplasmic reticulum stress regulates adipocyte resistin expression. Diabetes. 2009;58:1879–86.
- Ye R, Jung DY, Jun JY, Li J, Luo S, Ko HJ, et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes. 2010;59:6–16.
- da Luz G, Frederico MJ, da Silva S, Vitto MF, Cesconetto PA, de Pinho RA, et al. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur J Appl Physiol. 2011;111:2015–23.
- Cretenet G, Le Clech M, Gachon F. Circadian clock-coordinated 12 hr period rhythmic activation of the IRE1a pathway controls lipid metabolism in mouse liver. Cell Metab. 2010;11:47–57.
- Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2a enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7;520–32.
- Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–76.
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–78.
- Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009: 119;1201–15.
- Boulbés DR, Shaiken T, Sarbassov dos D. Endoplasmic reticulum is a main localization site of mTORC2. Biochem Biophys Res Commun. 2011;413:46–52.