Abstract
Background. Sudden cardiac death (SCD) remains a major cause of death in Western countries. It has a heritable component, but previous molecular studies have mainly focused on common genetic variants. We studied the prevalence, clinical phenotypes, and risk of SCD presented by ten rare mutations previously associated with arrhythmogenic right ventricular cardiomyopathy, long QT syndrome, or catecholaminergic polymorphic ventricular tachycardia.
Methods. The occurrence of ten arrhythmia-associated mutations was determined in four large prospective population cohorts (FINRISK 1992, 1997, 2002, and Health 2000, n = 28,465) and two series of forensic autopsies (The Helsinki Sudden Death Study and The Tampere Autopsy Study, n = 825). Follow-up data were collected from national registries.
Results. The ten mutations showed a combined prevalence of 79 per 10,000 individuals in Finland, and six of them showed remarkable geographic clustering. Of a total of 715 SCD cases, seven (1.0%) carried one of the ten mutations assayed: three carried KCNH2 R176W, one KCNH2 L552S, two PKP2 Q59L, and one RYR2 R3570W.
Conclusions. Arrhythmia-associated mutations are prevalent in the general Finnish population but do not seem to present a major risk factor for SCD, at least during a mean of 10-year follow-up of a random adult population sample.
Key messages
As many as 1 in 130 individuals in Finland carries a gene mutation associated with an inherited arrhythmia disorder.
The arrhythmia-associated mutations show geographic clustering within Finland due to a founder effect.
These arrhythmia-associated mutations are present in 1% of adult victims of sudden cardiac death, and the yearly incidence of probable and possible sudden cardiac death is 0.19% in the mutation carriers and 0.18% in non-carriers.
Introduction
Sudden cardiac death (SCD) accounts for an estimated 180,000–250,000 deaths every year in the United States, and approximately half of them occur as the first manifestation of a cardiac disease (Citation1,Citation2). Coronary heart disease (CHD) underlies approximately 80% and cardiomyopathy 10%–15% of SCDs, while congenital heart disease or a primary electrical disorder account for 5%–10% of cases (Citation3). Known risk factors for CHD predispose to SCD, and myocardial infarction may manifest itself as a life-threatening ventricular tachycardia (Citation1). A positive family history of SCD is associated with increased risk of SCD even after adjustment of CHD risk factors, indicating involvement of a significant genetic component (Citation4,Citation5). Inherited arrhythmia disorders can be detected in approximately half of the families with autopsy-negative SCD victims (Citation6,Citation7). Rare arrhythmia-associated gene mutations may thus provide interesting candidates for prediction of the inherited risk of SCD.
Long QT syndrome (LQTS), arrhythmogenic right ventricular cardiomyopathy (ARVC), and catecholaminergic polymorphic ventricular tachycardia (CPVT) represent inherited arrhythmia syndromes predisposing to SCD (Citation8–10). LQTS manifests with prolonged QT interval and ventricular arrhythmias in a structurally normal heart (Citation8). LQTS is a cardiac channelopathy in which delayed repolarization occurs due to mutations in cardiac ion channels or in their regulatory proteins (Citation11,Citation12). In ARVC, the ventricular myocardium is progressively replaced by adipose and fibrous tissue leading to potentially life-threatening arrhythmias and heart failure (Citation9,Citation13). Mutations in desmosomal cell adhesion proteins have been detected in approximately half of the reported cases (Citation13). CPVT patients present with stress-induced polymorphic ventricular tachycardia without structural abnormalities of the heart (Citation10,Citation14). This disorder is caused by a defect in calcium signaling due to mutations in the cardiac ryanodine receptor gene RYR2 (Citation15,Citation16) and in the calsequestrin gene CASQ2 (Citation17).
In Finland, high prevalence of specific founder mutations has been reported in several disorders, apparently due to the unique population history involving a relatively small founder population, occurrence of population bottlenecks, and genetic isolation (Citation18). We have previously reported four founder mutations, KCNQ1 G589D, KCNQ1 IVS7-2A> G, KCNH2 L552S, and KCNH2 R176W, to account for approximately 70% of molecularly verified LQTS cases in Finland (Citation19). Five desmosomal gene mutations have been reported in Finnish ARVC cases; of these, PKP2 Q59L represents a founder mutation, whereas PKP2 Q62K, PKP2 N613K, DSG2 3059_3062delAGAG, and DSP T1373A were detected in single ARVC families (Citation20,Citation21). We have also identified the RYR2 mutation R3570W showing a gain-of-function defect in two distinct SCD victims (Citation22), while the RYR2 G2145R mutation showed a milder functional defect and was present in only one SCD proband (Citation22). The four LQTS mutations and the five ARVC mutations are strikingly prevalent in the general Finnish population (Citation21,Citation23), but the geographic distribution and clinical significance of these mutations as well as of the RYR2 R3570W mutation are presently unknown.
In the general population, prolonged QT interval is associated with an increased risk of SCD (Citation24,Citation25). T wave alternans, representing the beat-to-beat alternation in the shape, amplitude, or timing of the T wave, is another marker of abnormal myocardial repolarization, which is known to predispose to SCD and cardiovascular mortality in the Finnish population (Citation26,Citation27). Common variants in ion channel genes or their regulatory genes have been associated with QT interval duration (Citation28–30) and T wave alternans (Citation31,Citation32). Therefore, these variants provide suitable candidates for studying the common genetic variation underlying risk of SCD. We have previously reported two novel common variants, one in SCN5A and another near PITX2, to be associated with risk of SCD (Citation33). However, the risk of SCD associated with rare Finnish LQTS- and ARVC-associated gene mutations has not previously been investigated in a population-based study. The present study addresses the prevalence and geographic distribution of arrhythmic disease-associated gene mutations in Finland and their role as determinants of risk of SCD, using four large prospective population cohorts (n = 28,465) and two series of forensic autopsies (n = 825).
Materials and methods
Study materials
The material consisted of FINRISK 1992 (n = 5,342), FINRISK 1997 (n = 7,673), FINRISK 2002 (n = 8,210), and Health 2000 (n = 7,240, including the Mini-Finland Health Survey) cohorts collected from the Finnish population, as well as the Helsinki Sudden Death Study (HSDS) (n = 297) and the Tampere Autopsy Study (TASTY) (n = 528) series of forensic autopsies. The National FINRISK Study sample is collected at 5-year intervals as a sex and 10-year age group stratified random sample drawn from the population aged 25–74 years separately for each survey area (Citation34). Each FINRISK cohort was drawn independently of each other from the population register for each study area. The Health 2000 Study is a two-stage stratified cluster sample collected between 2000 and 2001 and representing the adult (≥ 30 years) Finnish population (Citation35). The Mini-Finland Health Survey was conducted between 1978 and 1980 similarly to The Health 2000 Study, and DNA samples were collected in a follow-up study (n = 985) in 2001 (Citation36). HSDS is a series of consecutive forensic autopsies of men aged 35–69 years who died suddenly out of hospital in Helsinki between 1991 and 1992 (Citation37). TASTY included consecutive medico-legal autopsies of men and women ≤ 97 years of age performed in Tampere between 2002 and 2004 (Citation38). All included studies were carried out in accordance with the Declaration of Helsinki and had appropriate ethical approvals from the Ethics Committee of the National Public Health Institute, the Epidemiology Ethics Committee of the Helsinki and Uusimaa hospital region, the Ethics Committee of the Department of Forensic Medicine, University of Helsinki, the Tampere University Hospital Ethics Committee, and/or the National Supervisory Authority for Welfare and Health (Valvira).
Clinical information
All deaths were classified as probable, possible, or unlikely SCDs, or deaths of unknown cause as described previously (Citation25,Citation33). Briefly, data from the FINRISK and Health 2000 baseline investigations and prospective clinical information from the four national health care registries (the Causes of Death Registry, the Hospital Discharge Registry, the Drug Reimbursement Registry, and the Pharmacy Database) were reviewed by two independent physicians. In cases of disagreement on the cause of death, two additional physicians reviewed the data independently, and final adjudication was achieved by consensus of all four physicians. The classification of causes of death was performed by the same physicians using the same classification scheme both in the prospective population cohorts and in the series of forensic autopsies. In the HSDS and TASTY autopsy series, the classification was based on autopsy findings and information obtained from the death certificates as no baseline or register-based information was available for these individuals. All adjudications were carried out blinded in regard to the molecular genetic data. Probable and possible SCDs were pooled for the analyses.
The diagnosis of arrhythmia was based on the Hospital Discharge Registry or the Causes of Death Registry (ICD-10 I47–I49, ICD-9 427, ICD-8 427.4–427.9) or based on special reimbursement right for arrhythmia medication (ICD-10 I47–I49) in the Drug Reimbursement Registry. The diagnosis of heart failure was based on the Hospital Discharge Registry or the Causes of Death Registry (ICD-10 I50, ICD-9 428, ICD-8 427.0–427.1) or based on special reimbursement right for heart failure medication (ICD-10 I11.0, I13, I50, I97.1, P29.0) in the Drug Reimbursement Registry. The follow-up data until the end of 2008 were based on personal ID codes and covered 100% of all study subjects living in Finland.
Genotyping
KCNQ1 G589D, KCNQ1 IVS7-2A> G (c.1129-2A> G), KCNH2 L552S, PKP2 Q59L, PKP2 Q62K, PKP2 N613K, DSG2 3059_3062delAGAG, DSP T1373A, and RYR2 R3570W were genotyped in genomic DNA of all study subjects using the Sequenom iPLEX Gold assay (MALDI-TOF mass spectrometry, MassARRAY Analyzer Compact, Sequenom Inc., San Diego, CA, USA). KCNH2 R176W could not be genotyped with this approach, and therefore three haplotype-tagging SNPs (rs4725970, rs1036145, rs1808593, D’ = 1) were selected from Illumina 610-Quad genome-wide SNP data of six KCNH2 R176W carriers (Illumina Inc., San Diego, CA, USA) and genotyped using Sequenom iPLEX Gold. These tag SNPs captured 91% of an independent test sample of 32 unrelated KCNH2 R176W carriers. Samples with the tagging haplotype (n = 854) were genotyped for KCNH2 R176W (rs36210422) using Custom TaqMan SNP Genotyping Assay (Applied Biosystems, Foster City, CA, USA) and 7900HT Real Time PCR System (Applied Biosystems). For each mutation, ≥ 90% genotyping success and Hardy–Weinberg equilibrium P > 0.05 was required. For each study subject, ≥ 80% genotyping success was required.
Statistical analyses
Mutation prevalences in the Finnish population aged ≥ 25 years were estimated in the pooled FINRISK and Health 2000 population cohorts excluding the Mini-Finland Health Survey (total n = 27,670) using the function ‘svyratio’ from R package ‘survey’ with survey-specific sampling weights. The estimation was stratified by study, sex, study region, and 10-year age group. The prevalences were also calculated separately in the population samples representing Southern (n = 6,376), Western (n = 6,259), Eastern (n = 9,927), and Northern (n = 5,108) Finland. Association with SCD (probable/possible SCD versus unlikely SCD/unknown cause of death/alive) was analyzed using Fisher's exact test and age at death using Mann–Whitney test. Subjects aged > 80 or < 25 years at baseline were excluded from the analyses of SCD risk. The data are presented as mean ± SD. The statistical analyses were performed with SPSS 17.0 (SPSS Inc., Chicago, IL, USA) and R 2.14 (R Foundation for Statistical Computing, Vienna, Austria). Two-tailed P value < 0.05 was considered statistically significant.
Results
Prevalence and regional clustering
Demographic characteristics of the study samples as well as the numbers of probable, possible, and unlikely SCDs and deaths of unknown cause are presented in . Upon screening for the ten arrhythmia-associated mutations (KCNQ1 G589D, KCNQ1 IVS7-2A> G, KCNH2 L552S, KCNH2 R176W, PKP2 Q59L, PKP2 Q62K, PKP2 N613K, DSG2 3059_3062delAGAG, DSP T1373A, and RYR2 R3570W) in the population samples (n = 28,465), a total of 215 mutation carriers were identified. Accordingly, the combined prevalence of these mutations was 79 per 10,000 individuals (). The four Finnish LQTS founder mutations KCNQ1 G589D, KCNQ1 IVS7-2A> G, KCNH2 L552S, and KCNH2 R176W had a combined prevalence of 36 per 10,000, and the four Finnish ARVC mutations PKP2 Q59L, PKP2 Q62K, DSG2 3059_3062delAGAG, and DSP T1373A a combined prevalence of 39 per 10,000. The most prevalent of these mutations, PKP2 Q59L, was detected in 29 per 10,000 individuals. The rare ARVC mutation PKP2 N613K was not detected in the population samples. RYR2 R3570W, identified previously in two unrelated SCD cases (Citation22), had a prevalence of 2.6 per 10,000 individuals. The estimated mutation prevalences and their 95% confidence intervals (CI) in the Finnish population aged ≥ 25 years are shown in .
Figure 1. Prevalence and geographic clustering of the ten arrhythmia-associated mutations in Finland. A: Prevalence per 10,000 individuals and 95% confidence interval for each mutation in Finland. B: Division of Finland into four geographic regions. C: Prevalence and 95% confidence interval for each mutation in the different geographic regions shown in B.
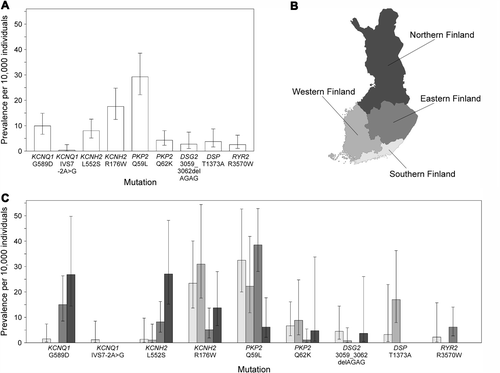
Table I. Demographic characteristics of the study samples.
Table II. Prevalence and clinical information of mutation carriers.
To study the geographic clustering of the ten arrhythmia-associated mutations, Finland was divided into four geographic regions: Southern Finland consisting of the Helsinki University Central Hospital region, Western Finland consisting of the Turku University Hospital and the Tampere University Hospital regions, Eastern Finland consisting of the Kuopio University Hospital region, and Northern Finland consisting of the Oulu University Hospital region (). Substantial regional differences occurred in the prevalences of the three LQTS mutations KCNQ1 G589D, KCNH2 L552S, and KCNH2 R176W, as well as of the ARVC mutation PKP2 Q59L (). KCNQ1 G589D had the highest prevalence in Northern and Eastern Finland and KCNH2 L552S in Northern Finland. KCNH2 R176W occurred most frequently in Western and Southern Finland and PKP2 Q59L in Eastern and Southern Finland.
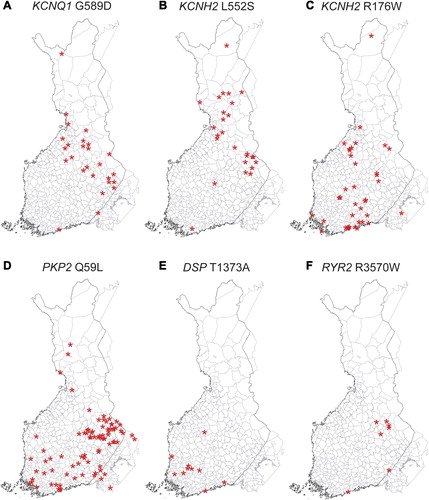
Because regional differences were detected in the prevalences of the mutations based on the places of residence during study sampling (), we also studied the geographic clustering based on municipality of birth in order to discover a possible founder effect. shows the municipality of birth for all individuals included in the population cohorts and for the carriers of the six selected mutations. Similar to the places of residence, the birthplaces of KCNQ1 G589D and KCNH2 L552S carriers clustered in Northern and Eastern Finland. The birthplaces of the KCNH2 R176W carriers did not show clear clustering, but most of the PKP2 Q59L carriers were born in Eastern Finland. In addition, DSP T1373A showed clustering in South-western Finland and RYR2 R3570W in Eastern Finland.
Figure 2. Birthplaces of all FINRISK 1992, 1997, 2002, and Health 2000 participants. The dots are randomly located within the municipality of birth of the participants.
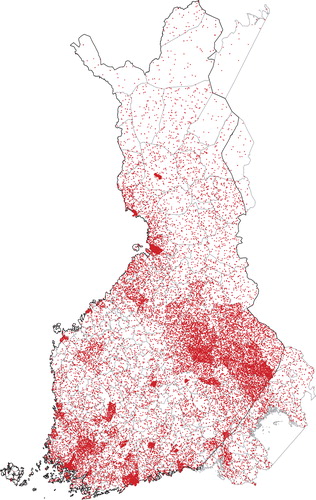
Figure 3. Birthplaces of the carriers of the six selected mutations. A: KCNQ1 G589D; B: KCNH2 L552S; C: KCNH2 R176W; D: PKP2 Q59L; E: DSP T1373A; F: RYR2 R3570W. The asterisks are randomly located within the municipality of birth of each mutation carrier. The corresponding distribution of birthplaces of all participants is shown in .
Clinical findings and SCDs
In the FINRISK and Health 2000 population samples, the mean age of mutation carriers was 49 ± 14 years at the moment of recruitment to the study. Of the mutation carriers, 48% were females. The carriers were followed for a mean of 9.8 ± 3.7 years through national health care registries, and the clinical follow-up data are summarized in . Fourteen (6.5%) mutation carriers had a diagnosis of arrhythmia. Eight of them (57%) had received this diagnosis before recruitment to the study, and six developed arrhythmia during follow-up. Two carriers, both with PKP2 Q59L, had a diagnosis of ventricular tachycardia. Seven carriers (3.3%) had a diagnosis of heart failure, and three of them (43%) had received this diagnosis before recruitment to the study.
A total of 493 SCDs occurred in the population samples during a mean follow-up time of 10.0 ± 3.9 years. Sixteen mutation carriers in the population samples died during follow-up, with four of them encountering a probable or possible SCD (incidence 0.19% per year). The corresponding incidence of probable or possible SCD in non-carriers was 0.18%. Among the mutation carriers, three probable SCD cases were identified: a 46-year-old male with PKP2 Q59L described in our previous report (Citation21), a 69-year-old female with KCNH2 R176W and suffering from cardiomegaly and Parkinson's disease, and a 57-year-old male with RYR2 R3570W and with diagnoses of paroxysmal supraventricular tachycardia, chronic ischemic heart disease, old myocardial infarction, heart failure, and diabetes. A 79-year-old female with PKP2 Q59L had diagnoses of atrial arrhythmia, sick sinus syndrome, and heart failure, and encountered a possible SCD. The HSDS and TASTY autopsy series (n = 825) included a total of 222 probable or possible SCD cases. Three probable SCD victims (1.4%) carried one of the ten arrhythmia-associated mutations screened for: a 44-year-old male with KCNH2 R176W and alcoholic cardiomyopathy, a 58-year-old male with KCNH2 R176W, acute transmural myocardial infarction, and atherosclerotic heart disease, and a 63-year-old male with KCNH2 L552S, old myocardial infarction, and atherosclerotic heart disease. Combining the population studies and the autopsy series, the study materials included a total of 715 SCD cases. Seven (1.0%) of them carried one of the ten mutations assayed, but none of the individual mutations was associated with a significantly increased risk of SCD (P > 0.05). The mean age at SCD was 60 ± 13 for the mutation carriers and 65 ± 10 for the non-carriers (P = 0.25).
Discussion
We detected a high combined prevalence of the ten arrhythmia-associated mutations KCNQ1 G589D, KCNQ1 IVS7-2A> G, KCNH2 L552S, KCNH2 R176W, PKP2 Q59L, PKP2 Q62K, PKP2 N613K, DSG2 3059_3062delAGAG, DSP T1373A, and RYR2 R3570W in four Finnish population samples comprising a total of 28,465 individuals. One in 130 Finns was found to carry one of these mutations predisposing to LQTS, ARVC, or CPVT. KCNQ1 G589D, KCNH2 L552S, KCNH2 R176W, and PKP2 Q59L showed substantial prevalence differences between different geographic regions of Finland. In addition, the birthplaces of DSP T1373A and RYR2 R3570W carriers showed marked geographic clustering as an indication of a founder effect. However, the penetrance of cardiac symptoms, at least as assessed by data collected in the national health registers, was relatively low, and none of the mutations was associated with a significantly increased risk of SCD during a mean of 10-year follow-up.
The prevalence of the four Finnish LQTS founder mutations was found to be 36 per 10,000, which is considerably higher than the previously published estimates for LQTS prevalence ranging approximately from 1 to 5 per 10,000 (Citation39,Citation40). Using a significantly larger population sample than previously, this study confirms the reported prevalence of 40 per 10,000 for the four LQTS founder mutations (Citation23). The prevalence of ARVC is estimated to range from 2 to 10 per 10,000 individuals (Citation41,Citation42). We report here a prevalence of 39 per 10,000 individuals for the four ARVC mutations PKP2 Q59L, PKP2 Q62K, DSG2 3059_3062delAGAG, and DSP T1373A in the general Finnish population. This is similar to the prevalence estimate of 48 per 10,000 that we previously reported in a smaller Finnish population sample (Citation21), but higher than the estimated ARVC prevalence in other populations. Considering that the prevalence estimate in the present study was calculated in the adult (> 25 years) Finnish population and involves only four known ARVC mutations, the total prevalence of all desmosomal mutations in the general population is expected to be even higher. The prevalence of the Finnish ARVC founder mutation PKP2 Q59L is especially high, amounting to 29 per 10,000. Studies in affected families suggested a penetrance of about 20% for this mutation (Citation20), which is in accordance with the register-based clinical findings reported in the present study (). Recurrent PKP2 founder mutations have also been reported in other populations (Citation43–45), but their prevalence in the general population remains to be studied.
Due to its unique population history featuring geographic and cultural isolation, Finland represents a genetic isolate ideal for genetic studies (Citation18). In addition, the small size of the initial founder population and genetic drift has decreased the genetic diversity of the Finnish population, which is reflected by the high prevalence of the LQTS and ARVC founder mutations. Northern, Eastern, and Central Finland represent the late settlement region inhabited in the sixteenth century by a small founder population originating from South-eastern Finland (Citation18). This internal migration has given rise to regional subisolates within Finland. The present study shows that KCNQ1 G589D and KCNH2 L552S are especially prevalent in Northern and Eastern Finland, probably due to a founder effect. Similarly, KCNH2 R176W and PKP2 Q59L showed the highest prevalence in South-western and South- eastern Finland, respectively. Due to increasing internal migration in the twentieth century, the birthplaces of the mutation carriers are expected to reflect the founder effect more clearly than the places of residence during the study sampling. Indeed, the birthplaces of the DSP T1373A and RYR2 R3570W carriers clustered in South-western and Eastern Finland, respectively, indicating that the mutation carriers may be descendants of a founder individual who resided in the corresponding area hundreds of years ago.
Functional and clinical data show a pathogenetic role for the LQTS (Citation46–48) and ARVC (Citation20,Citation21,Citation49) mutations investigated in the present study, as well as for the RYR2 R3570W mutation (Citation22). However, a significantly reduced disease penetrance has been associated with these mutations in Finnish families (Citation19–22,Citation46,Citation47). This less high pathogenicity might partly explain their high prevalence in the Finnish population. In the present study, only 6.5% of the mutation carriers had a diagnosis of cardiac arrhythmia, and 3.3% had a diagnosis of heart failure according to the national health care registries. Because these diagnoses were only based on hospitalization, medication with special reimbursement, or reported cause of death, and because extensive cardiologic examinations were probably not performed for all mutation carriers, the true proportion of symptomatic mutation carriers is expected to be higher. During the mean follow-up of ten years, four mutation carriers encountered a probable or possible SCD: two with PKP2 Q59L, one with KCNH2 R176W, and one with RYR2 R3570W. In addition, among the 222 SCD cases in the series of forensic autopsies, three (1.4%) carried one of the ten arrhythmia-associated mutations: two with KCNH2 R176W and one with KCNH2 L552S. None of the individual mutations was significantly associated with increased risk of SCD. However, as the population samples were collected from the adult Finnish population (≥ 25 years), we cannot estimate the potential risk of SCD presented by these mutations to children and young adults.
The limitations of the present study include heterogeneity of the study populations consisting of four prospective population cohorts and two series of consecutive forensic autopsies. Future studies with a longer follow-up of a more homogeneous study population are needed to evaluate further the risk of SCD presented by these arrhythmia-associated mutations. As the time between the onset of symptoms and death cannot reliably be determined in a population-based study due to lack of witness reports, the classification of SCDs could not be performed according to the definition developed by the National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop (Citation50).
In previous studies, sequencing of LQTS genes in victims of unexplained SCD has revealed mutations in up to 20% of cases (Citation51–54), and mutations in the RYR2 gene associated with CPVT have been detected in occasional victims of SCD (Citation22,Citation55). Identification of arrhythmia-associated mutations in SCD cases may assist in determination of arrhythmia risk in surviving relatives. Since mutations in the LQTS genes occur also in healthy individuals (Citation39,Citation56), it is important to study the phenotypic effects of these variants in epidemiological and functional studies. To our knowledge, this is thus far the largest study investigating the prevalence and clinical significance of rare arrhythmia-associated gene mutations at the population level.
Conclusions
As many as 1 in 130 individuals in Finland carries a gene mutation associated with a rare arrhythmia disorder. These mutations show substantial geographic clustering within Finland, due to an apparent founder effect. They were present in 1% of adult victims of SCD, but their role in young (< 25 years) cases with SCD remains to be explored.
Acknowledgements
Suzannah Bumpstead, Eija Hämäläinen, Kaisa Silander, Johannes Kettunen, and Antti-Pekka Sarin are acknowledged for help in the genotyping process.
Declaration of interest: The authors report no conflicts of interest. This study was supported by the Finnish Cultural Foundation (to A.M.L.); the Max Schaldach Fellowship in Cardiac Pacing and Electrophysiology (to P.A.N.); a Burroughs Wellcome Fund travel grant (to P.A.N.); Finnish Academy SALVE program ‘Pubgensense’ (#10404 to M.P.); the Wellcome Trust, the Academy of Finland (to K.K. and #129494 and #139635 to V.S.); the Finnish Foundation for Cardiovascular Research (to K.K. and V.S.); the Sigrid Juselius Foundation (to K.K. and V.S.); the National Institutes of Health (HL080025, HL098283 to C.N.-C.); the Doris Duke Charitable Foundation (to C.N.-C.); and the Burroughs Wellcome Fund (to C.N.-C.).
References
- Adabag AS, Luepker RV, Roger VL, Gersh BJ. Sudden cardiac death: epidemiology and risk factors. Nat Rev Cardiol. 2010;7:216–25.
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–220.
- Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al Samara M, et al. Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis. 2008;51:213–28.
- Jouven X, Desnos M, Guerot C, Ducimetière P. Predicting sudden death in the population: the Paris Prospective Study I. Circulation. 1999; 99:1978–83.
- Friedlander Y, Siscovick DS, Arbogast P, Psaty BM, Weinmann S, Lemaitre RN, et al. Sudden death and myocardial infarction in first degree relatives as predictors of primary cardiac arrest. Atherosclerosis. 2002;162:211–6.
- Tan HL, Hofman N, van Langen IM, van der Wal AC, Wilde AA. Sudden unexplained death: heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation. 2005;112:207–13.
- Behr ER, Dalageorgou C, Christiansen M, Syrris P, Hughes S, Tome Esteban MT, et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 2008;29:1670–80.
- Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J. 1975;89:378–90.
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–33.
- Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–9.
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–80.
- Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358:169–76.
- Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2011;9:223–33.
- Swan H, Piippo K, Viitasalo M, Heikkilä P, Paavonen T, Kainulainen K, et al. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035–42.
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200.
- Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–90.
- Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–84.
- Peltonen L, Jalanko A, Varilo T. Molecular genetics of the Finnish disease heritage. Hum Mol Genet. 1999;8:1913–23.
- Fodstad H, Swan H, Laitinen P, Piippo K, Paavonen K, Viitasalo M, et al. Four potassium channel mutations account for 73% of the genetic spectrum underlying long-QT syndrome (LQTS) and provide evidence for a strong founder effect in Finland. Ann Med. 2004;36(Suppl 1):53–63.
- Lahtinen AM, Lehtonen A, Kaartinen M, Toivonen L, Swan H, Widén E, et al. Plakophilin-2 missense mutations in arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2008;126:92–100.
- Lahtinen AM, Lehtonen E, Marjamaa A, Kaartinen M, Heliö T, Porthan K, et al. Population-prevalent desmosomal mutations predisposing to arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2011;8:1214–21.
- Marjamaa A, Laitinen-Forsblom P, Wronska A, Toivonen L, Kontula K, Swan H. Ryanodine receptor (RyR2) mutations in sudden cardiac death: studies in extended pedigrees and phenotypic characterization in vitro. Int J Cardiol. 2011;147:246–52.
- Marjamaa A, Salomaa V, Newton-Cheh C, Porthan K, Reunanen A, Karanko H, et al. High prevalence of four long QT syndrome founder mutations in the Finnish population. Ann Med. 2009;41:234–40.
- Straus SM, Kors JA, De Bruin ML, van der Hooft CS, Hofman A, Heeringa J, et al. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J Am Coll Cardiol. 2006;47: 362–7.
- Noseworthy PA, Havulinna AS, Porthan K, Lahtinen AM, Jula A, Karhunen PJ, et al. Common genetic variants, QT interval, and sudden cardiac death in a Finnish population-based study. Circ Cardiovasc Genet. 2011;4:305–11.
- Nieminen T, Lehtimäki T, Viik J, Lehtinen R, Nikus K, Kööbi T, et al. T-wave alternans predicts mortality in a population undergoing a clinically indicated exercise test. Eur Heart J. 2007;28:2332–7.
- Slawnych MP, Nieminen T, Kähönen M, Kavanagh KM, Lehtimäki T, Ramadan D, et al. Post-exercise assessment of cardiac repolarization alternans in patients with coronary artery disease using the modified moving average method. J Am Coll Cardiol. 2009;53:1130–7.
- Tobin MD, Kähönen M, Braund P, Nieminen T, Hajat C, Tomaszewski M, et al. Gender and effects of a common genetic variant in the NOS1 regulator NOS1AP on cardiac repolarization in 3761 individuals from two independent populations. Int J Epidemiol. 2008;37:1132–41.
- Marjamaa A, Newton-Cheh C, Porthan K, Reunanen A, Lahermo P, Väänänen H, et al. Common candidate gene variants are associated with QT interval duration in the general population. J Intern Med. 2009;265:448–58.
- Raitakari OT, Blom-Nyholm J, Koskinen TA, Kähönen M, Viikari JS, Lehtimäki T. Common variation in NOS1AP and KCNH2 genes and QT interval duration in young adults. The Cardiovascular Risk in Young Finns Study. Ann Med. 2009;41:144–51.
- Koskela J, Kähönen M, Fan M, Nieminen T, Lehtinen R, Viik J, et al. Effect of common KCNE1 and SCN5A ion channel gene variants on T-wave alternans, a marker of cardiac repolarization, during clinical exercise stress test: the Finnish Cardiovascular Study. Transl Res. 2008;152:49–58.
- Koskela J, Kähönen M, Nieminen T, Lehtinen R, Viik J, Nikus K, et al. Allelic variant of NOS1AP effects on cardiac alternans of repolarization during exercise testing. Scand J Clin Lab Invest. 2012;72:100–7.
- Lahtinen AM, Noseworthy PA, Havulinna AS, Jula A, Karhunen PJ, Kettunen J, et al. Common genetic variants associated with sudden cardiac death: the FinSCDgen study. PLoS One. 2012;7:e41675.
- Vartiainen E, Laatikainen T, Peltonen M, Juolevi A, Männistö S, Sundvall J, et al. Thirty-five-year trends in cardiovascular risk factors in Finland. Int J Epidemiol. 2010;39:504–18.
- Heistaro S. Methodology report, Health 2000 Survey. Helsinki: Publications of the National Public Health Institute, KTL B 26; 2008.
- Kattainen A, Salomaa V, Härkänen T, Jula A, Kaaja R, Kesäniemi YA, et al. Coronary heart disease: from a disease of middle-aged men in the late 1970s to a disease of elderly women in the 2000s. Eur Heart J. 2006;27:296–301.
- Tyynelä P, Goebeler S, Ilveskoski E, Mikkelsson J, Perola M, Löytönen M, et al. Birthplace predicts risk for prehospital sudden cardiac death in middle-aged men who migrated to metropolitan area: The Helsinki Sudden Death Study. Ann Med. 2009;41:57–65.
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease- related lesions begins in middle age. Ann Neurol. 2009;65:650–7.
- Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc. 2003;78:1479–87.
- Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7.
- Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, Fasoli G, et al. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Mol Genet. 1994;3:959–62.
- Peters S, Tr mmel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2004;97:499–501.
- van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–8.
- Watkins DA, Hendricks N, Shaboodien G, Mbele M, Parker M, Vezi BZ, et al. Clinical features, survival experience, and profile of plakophylin-2 gene mutations in participants of the arrhythmogenic right ventricular cardiomyopathy registry of South Africa. Heart Rhythm. 2009;6:S10–S17.
- van der Zwaag PA, Cox MG, van der Werf C, Wiesfeld AC, Jongbloed JD, Dooijes D, et al. Recurrent and founder mutations in the Netherlands : Plakophilin-2 p.Arg79X mutation causing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Neth Heart J. 2010;18:583–91.
- Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. Homozygosity for a HERG potassium channel mutation causes a severe form of long QT syndrome: identification of an apparent founder mutation in the Finns. J Am Coll Cardiol. 2000;35:1919–25.
- Piippo K, Swan H, Pasternack M, Chapman H, Paavonen K, Viitasalo M, et al. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: implications for estimation of disease prevalence and molecular diagnostics. J Am Coll Cardiol. 2001;37:562–8.
- Fodstad H, Bendahhou S, Rougier JS, Laitinen-Forsblom PJ, Barhanin J, Abriel H, et al. Molecular characterization of two founder mutations causing long QT syndrome and identification of compound heterozygous patients. Ann Med. 2006;38:294–304.
- Hall C, Li S, Li H, Creason V, Wahl JK 3rd. Arrhythmogenic right ventricular cardiomyopathy plakophilin-2 mutations disrupt desmosome assembly and stability. Cell Commun Adhes. 2009;16:15–27.
- Fishman GI, Chugh SS, Dimarco JP, Albert CM, Anderson ME, Bonow RO, et al. Sudden cardiac death prediction and prevention: report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation. 2010;122:2335–48.
- Chugh SS, Senashova O, Watts A, Tran PT, Zhou Z, Gong Q, et al. Postmortem molecular screening in unexplained sudden death. J Am Coll Cardiol. 2004;43:1625–9.
- Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol. 2007;49:240–6.
- Albert CM, Nam EG, Rimm EB, Jin HW, Hajjar RJ, Hunter DJ, et al. Cardiac sodium channel gene variants and sudden cardiac death in women. Circulation. 2008;117:16–23.
- Adabag AS, Peterson G, Apple FS, Titus J, King R, Luepker RV. Etiology of sudden death in the community: results of anatomical, metabolic, and genetic evaluation. Am Heart J. 2010;159:33–9.
- Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner's cases. Mayo Clin Proc. 2004;79:1380–4.
- Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600–7.