
Abstract
Homozygous familial hypercholesterolemia (HoFH) represents the most severe lipoprotein disorder, generally attributable to mutation(s) of the low-density lipoprotein receptor (LDL-R), i.e. autosomal dominant hypercholesterolemia type 1 (ADH1). Much lower percentages are due to alterations of apolipoprotein B (ADH2), or gain-of-function mutations of proprotein convertase subtilisin/kexin type 9 (PCSK9) (ADH3). In certain geographical areas a significant number of patients may be affected by an autosomal recessive hypercholesterolemia (ARH). Mutations may be also combined (two mutations of the same gene, compound heterozygosity), or two in different genes (double heterozygosity). Among the most innovative therapeutic approaches made available recently, inhibitors of the microsomal transfer protein (MTP) system have shown a high clinical potential. MTP plays a critical role in the assembly/secretion of very-low-density lipoproteins (VLDL), and its absence leads to apo B deficiency. MTP antagonists dramatically lower LDL-cholesterol (LDL-C) in animals, although a reported increase of liver fat delayed their clinical development. Lomitapide, the best-studied MTP inhibitor, reduces LDL-C by 50% or more in HoFH patients, with modest, reversible, liver steatosis. Recent US approval has confirmed an acceptable tolerability, provided patients adhere to a strictly low-fat regimen. There are no clinical data on atherosclerosis reduction/regression, but animal models provide encouraging results.
Key messages
Homozygous familial hypercholesterolemia (HoFH) is among the most severe forms of hyperlipidemia, in the majority of cases consequent to LDL-R mutation(s).
Mutations in the LDLRAP1 gene, encoding the adaptor protein for the LDL-R (i.e. autosomal recessive hypercholesterolemia) or, more rarely, mutations in other genes causing altered apolipoprotein B (apo B) structure or gain-of-function of PCSK9 may also lead to a severe homozygous form of hypercholesterolemia.
HoFH patients very frequently undergo treatment with LDL-apheretic procedures. Availability of the inhibitors of the microsomal transfer protein (MTP) system can provide an effective treatment option. Lomitapide, the first marketed MTP inhibitor, is a very powerful cholesterol-lowering agent, with acceptable safety.
Introduction: the therapeutic challenge of homozygous hypercholesterolemia
Homozygous familial hypercholesterolemia (HoFH) is one of the most severe lipid disorders found in the clinic. It is generally caused by a deficiency of LDL-receptor (LDL-R) activity (autosomal dominant hypercholesterolemia type 1, ADH1). The same mutation of the LDLR gene may occur in both alleles (true Ho) or different mutations may be present in each allele (compound heterozygote, CH), in both cases leading to extreme hypercholesterolemia (Citation1). However, more rarely, other causes have been found, resulting in a similar phenotype (Citation2), such as the combination of one mutation in LDL-R with a binding defective apolipoprotein B (apo B) (Citation3) or with a gain-of-function mutation of proprotein convertase subtilisin/kexin type 9 (PCSK9) (Citation4). Moreover, while it was generally believed that HoFH would occur in about one per million individuals, in some specific regions of the world the prevalence of HoFH is higher for a founder effect in isolated populations () (Citation1,Citation5–9). Recently, a very high prevalence of HoFH (up to 1:40,000) was predicted in the Danish population, based on the estimated frequency (1:200) of heterozygous FH (HeFH) subjects defined by clinical criteria (Citation10).
Table I. Frequency of HoFH, autosomal dominant form due to LDL-R mutations (ADH1).
Whatever the real number, it is apparent that probably thousands of patients, also taking advantage of the newer biotech methods to identify mutation carriers, will be diagnosed as affected by HoFH. This diagnosis will have a significant impact on the patient's life expectancy. These patients, whatever the diagnosis, will need in fact intensive treatment with available lipid-lowering drugs, to which they may respond to a minimal degree, eventually being moved to more demanding procedures such as LDL-apheresis (Citation11).
As indicated, although HoFH is due to LDL-R mutation(s) (ADH1) in about 95% of the cases, the disease may be also associated to a number of different genotypes, not directly linked to the LDL-R expression. The best known are:
autosomal recessive hypercholesterolemia (ARH), linked to a defective adaptor protein (low-density lipoprotein receptor adapter protein 1, LDLRAP1) moving the LDL-R to the cell surface (Citation12). ARH is the most frequent abnormality leading to HoFH, e.g. in Sardinia, where the prevalence of ARH may be as high as ∼1:34,000 (Citation13,Citation14)
defective apo B, with sequence changes impairing the ability to bind the LDL-R (ADH2) (Citation15)
increased activity of PCSK9 gain-of-function mutants, which increase the catabolic activity on the LDL-R and may lead to severe hypercholesterolemia (ADH3) (Citation16)
Two of these, i.e. defective apo B and enhanced PCSK9 activity, are seldom seen. In contrast, ARH is frequent, with a significant number of carriers particularly in Southern Europe and in the Mediterranean area (Citation13). This leads to a phenotype not markedly different from that of the classical LDL-R deficiency. The experience of one of the authors of this review indicates that, not infrequently, an ARH carrier presents with an as difficult or more difficult-to-treat disease than the classical LDL-R deficiency (Citation17). Phenotypic comparisons would, however, appear to indicate a less severe clinical presentation versus the classical ADH1 (Citation14), and therapeutic responses may, at times, be more satisfactory (Citation18).
One further reason for the increased frequency of HoFH is that pure homozygosity for a single LDL-R mutation (or for an ARH mutation) is nowadays more rarely seen. In contrast, CH, i.e. carriers of two different mutations of the LDL-R are becoming more frequent, and sometimes double heterozygotes (DH) for a mutation in both LDLR and LDLRAP1 genes or other combinations have been observed (Citation19), possibly as a result of a reduction of consanguineous marriages and of an increased mobility of people in various parts of the world. The phenotypes of CHs and of some DHs are quite similar to those due to homozygosity for LDL-R mutations, with similar therapeutic challenges (Citation4). Finally, just restricting attention to the LDL-R mutations, five different genotypes in cultured skin fibroblasts have been originally described (Citation20) that present with differential responses to lipid-lowering treatments. More recently, in vitro assays in cultured fibroblasts have led to a classification of either receptor-negative (< 2% residual activity of the LDL receptors) or receptor-defective (2%–25% residual activity). This latter classification is more widely used, although in many patients both genotype and receptor activity may be available.
Drug treatment of HoFH of different origin still encounters extreme difficulties. Statins, by inhibiting cholesterol biosynthesis, may improve the expression of LDL-R on the cell surface, but have a limited efficacy in patients who do not have any LDL-R (HoFH carrying receptor-negative mutations) (Citation21). Other drug treatments, e.g. fibrates, nicotinic acid, resins, or others, may exert some effect, but of a minimal degree, thus not allowing these patients to be handled successfully (Citation22,Citation23). Recent data on PCSK9 antagonists indicate an overall modest activity in HoFH with residual receptor activity (HoFH carrying receptor-defective mutations), while receptor-negative HoFHs appear not to respond at all (Citation24). The only remaining option, i.e. LDL apheresis, is curtailed by not infrequent problems, such as the need of prolonged venous access, not suitable for children of a few years of age, considerable cost, and difficult organization (Citation11).
In order to improve drug treatment of severe hypercholesterolemia, particularly of the homozygous type, a novel drug approach is being brought to the medical community, making use of a drug target that does not involve LDL-R activation. The microsomal transfer protein (MTP) inhibitory treatment provides in fact a highly active cholesterol-lowering medication for these patients, who are ‘orphans’ regarding effective pharmacotherapeutic strategies. The biochemical basis and the clinical effectiveness of this new drug approach are the objects of this review article.
Microsomal transfer protein (MTP): what is it and how is it regulated?
MTP plays a critical role in the assembly/secretion of very- low-density lipoproteins (VLDL) in the liver and of chylomicrons in the intestine. MTP is found in the microsomal fraction of liver and intestine and is involved in the transfer of triglycerides (TG), cholesterol esters (CE), and phospholipids (PL) to newly synthesized apo B (Citation25). The absence of functional MTP was first detected in patients with abetalipoproteinemia (ABL), a rare genetic disorder presenting with a defect in the assembly/secretion of plasma apo B-containing lipoproteins and, in addition, with increase of fat in the liver (steatosis) (Citation26). After the initial observation that MTP was absent in ABL individuals, a biochemical characterization followed (Citation27).
MTP is a heterodimer composed of the multifunctional protein disulfide isomerase (PDI; 58 kDa) and a unique large M-subunit of about 97 kDa, that can be isolated as a soluble protein from the lumen of the microsomal fraction of liver and intestine () (Citation28). This large subunit was indeed not detectable in ABL patients (Citation27). As an ancillary finding in the clinical evaluation, it was detected that, in these patients, not only were VLDL absent, but also chylomicrons were markedly reduced; in addition there appeared to be some deficiency of lipid-soluble vitamins and other nutritional factors that are transported by apo B-containing lipoproteins (Citation28).
Figure 1. Biochemical mechanisms of the activity of MTP of the inhibition of this assembly. A: (a) Synthesis of apo B occurs on the rough endoplasmic reticulum. (b) MTP consists of two subunits: the P subunit is the ubiquitous ER-resident enzyme protein disulfide isomerase (PDI), known to facilitate disulfide bond formation during nascent protein biosynthesis. The M subunit contains binding sites for apo B and lipids. (c) By interacting with nascent apo B, MTP facilitates its translocation, lipidation, and the prevention of degradation. (d) The apo B-containing lipoprotein is assembled. B: (e) MTP inhibition by mutation of the apo B binding site or following inhibitor use (f) leads apo B to proteasomal degradation via heat shock protein 70 (hsp70) and ubiquitination.
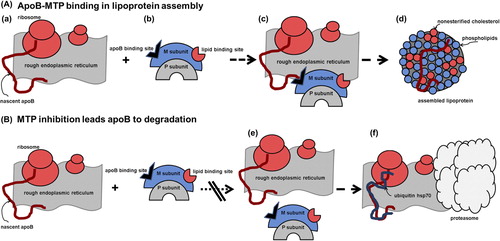
The facilitated transfer of lipid to nascent apo B by MTP occurs while the protein is being co-translationally translocated across the endoplasmic reticulum (ER) membrane. This aids in the assembly of primordial lipoprotein particles preventing presecretory proteasomal degradation of apo B (Citation29). Kinetic studies predict that MTP has two lipid binding sites (Citation30): the fast site is implicated in TG and PL transfer, whereas the slow site is implicated in PL transfer only. PL transfer activity of MTP appears, however, to be sufficient to support secretion of apo B-containing lipoproteins in vitro and in vivo (Citation28). Very recently this same group of authors characterized the effects of various MTP mutations in ABL patients, in particular on functional properties, by analyzing PL and TG transfer activities (Citation31). All mutants co-localized with calnexin and interacted with PDI. Some of these, known to be deficient in TG transfer activity, also lacked PL transfer activity. Such appears to be the case particularly of novel mutants. ABL is thus associated with the absence of both TG and PL transfer activities in MTP, and the pathogenesis of ABL should be attributed to the absence of both.
Liver MTP is involved in the maturation of pre-VLDL (synthesized in the ER even in the presence of very low levels of MTP) into VLDL by inducing the folding and lipidation of apo B during translocation in the ER. In mice with MTP deletion (MTPΔ/Δ) and ldlr-/- the hepatocytes secrete dense apo B100-containing lipoprotein particles; on the contrary in mice MTPΔ/Δ and ldlr+/+ the secretion of these particles is abolished, since the presence of a normal LDL-R targets pre-VLDL to intracellular degradation, mainly in the ER or in the cytoplasm by a ubiquitin-dependent proteasomal process (Citation32). These authors noted that MTP gene transcription is negatively regulated by the sterol regulatory element binding proteins (SREBPs) through their binding to the MTP promoter (region from –124 to –109 bp containing the GCAGCCCAC SRE-binding sequence). In particular, both SREBP2 and SREBP1 are activated under sterol-depleted conditions, explaining the down-regulation of the MTP gene in these conditions. The SREBPs, in particular SREBP2, are activators of the transcription of LDLR, hydroxymethylglutaryl-coenzyme A (HMG-CoA) synthase and reductase genes. Interestingly, both wild-type SREBP2 (amino acids 1–481) and its truncated form (amino acids 31–481, lacking the transactivation domain) inhibit MTP transcription by about 40%. On the contrary the LDLR transcription up-regulation requires full-length SREBP2, thus pointing out that different mechanisms are involved in the transcriptional regulation of the MTP and LDLR genes in sterol-depleted conditions (Citation33–35). Kinetic studies using endogenous labeling of apo B with deuterated leucine have documented that HoFH patients carrying LDL-R-negative mutations have an increased production rate of VLDL2 and intermediate-density lipoprotein (IDL), whereas Ho/CH-FH carrying LDL-R-defective mutations do not show significant differences from controls in the production rate of these lipoproteins (Citation36,Citation37). These findings suggest an inverse correlation between residual LDL-R activity and MTP expression and activity and, possibly, an inverse correlation between LDL-R activity and effectiveness of a pharmacological inhibition of MTP.
Studies on the in vivo regulation of MTP (Citation25) have detected, among others, that MTP expression is up-regulated by the dietary fat content. In an early study Lin et al. (Citation38) evaluated hamsters fed high-fat versus high-sucrose diets. They reported a 55% increase over controls of the liver MTP large subunit mRNA levels and 126% of the intestinal (duodenum and jejunum) mRNA levels in the high-fat versus control group. The high-sucrose diet also raised liver MTP mRNA by 55%, but did not affect the intestinal MTP mRNA levels. A dose dependency of the increase of liver MTP mRNA levels in hamsters was reported by Bennett et al. with increasing dietary fat content (Citation39).
The identity of fatty acids more significantly involved in MTP regulation has received limited attention. Dorfman et al. (Citation40) showed that in hamsters on a high-fat diet MTP mRNA abundance was markedly raised after monounsaturated (canola oil) and polyunsaturated fatty acid (soybean oil) intakes over butter and stick margarine intakes. Other possible regulatory factors of MTP expression, investigated in HepG2 cells, e.g. glucagon, glucose, and fatty acid supplements, have displayed generally minimal or no effects on the MTP large subunit mRNA levels (Citation41). Insulin treatment appears instead to reduce gene expression to 50% of control levels (Citation42).
The clinical observations and the accompanying biochemical findings stimulated interest in a further evaluation of the lipid transport properties of MTP and on the possible development of MTP antagonism as a mode of treatment of hyperlipoproteinemias.
Early studies on MTP antagonism as a therapeutic target
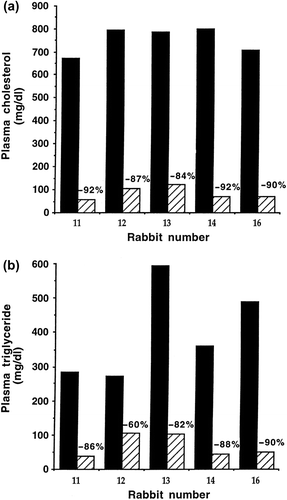
The clinical observation of dramatic plasma lipid reductions in ABL patients and the ancillary findings in the different evaluated animal models, as well as in the in vitro studies, strongly suggested that antagonism to MTP might be a valuable approach to hyperlipoproteinemias. This encouraged, at Bristol Myers Squibb, a high-throughput screening of a large chemical library to identify inhibitors of MTP-mediated TG transfer. The initial product (BMS-200150) inhibited MTP-mediated transport of TG between membranes in vitro, also inhibiting secretion from HepG2 cells, a human liver-derived cell line. The product, however, proved not to be active in animal models. This encouraged further chemical modifications, until reaching compound 9, an MTP inhibitor possessing subnanomolar potency in both the lipid transfer and HepG2 apo B secretion assays, as well as inhibiting lipoprotein production in fasted rats (Citation43). Compound 9 (BMS-201038) was also tested on plasma lipid levels in Watanabe heritable hyperlipidemic (WHHL) rabbits, an animal model with a hepatic LDL-R activity < 5% of normal, resulting in dramatically reduced plasma lipids ().
Figure 2. Cholesterol and triglyceride reductions in WHHL rabbits treated orally for 14 days with lomitapide (10 mg/kg). Total cholesterol and triglyceride levels were measured 18 hours after the last dose. Solid bars represent basal concentrations; diagonally hatched bars represent concentrations at the end of treatment. From ref. (Citation43) with permission.
The exciting information on BMS-201038 stimulated activity in different settings, leading to the development from Bayer Yakuhin of implitapide (Citation44), that reduced plasma lipids and atheroma in WHHL rabbits, and from Pfizer of CP-346086 (Citation45). This latter too was effective in animal models, as well as in healthy volunteers, receiving 30 mg/day of the agent for two weeks: reductions of total cholesterol, low-density lipoprotein cholesterol (LDL-C), and TG by 47%, 72%, and 75% relative to the individual baselines were reported, with few changes in high-density lipoprotein cholesterol (HDL-C). A very powerful activity in a guinea pig model (Citation46) was also displayed by JTT-130, an MTP inhibitor with an activity (per dose) at least 10-fold higher versus atorvastatin and with little, if any, liver lipid accumulation.
Enthusiasm on the potential development of MTP inhibitors was, however, hampered by the observation both of steatorrhea and of fat-soluble vitamin malabsorption, in addition to liver steatosis in animal models, with the possible exception of JTT-130. These last observations are an obvious consequence of MTP inhibition at the liver level.
Interest in the clinical use of the BMS-201038 was, however, still alive at Harvard and at the University of Pennsylvania School of Medicine. In the former institution a woman with severe chylomicronemia and repeated bouts of pancreatitis, unresponsive to any dietary or drug treatment has been, in fact, given for 12 years the drug with significant success, i.e. prevention of pancreatitis, in spite of the difficulty in keeping the drug constantly available. A very recent report shows that this patient's fatty liver, present before treatment, progressed to steatohepatitis and fibrosis after 12 to 13 years (Citation47).The authors suggest that the repeated bouts of pancreatitis are likely to have been responsible for the progression of liver disease.
A major breakthrough in the development of MTP inhibitors was the clinical evaluation by Cuchel et al. (Citation48) of BMS-201038 at four different doses (0.03, 0.1, 0.3, and 1.0 mg/kg) each for 4 weeks in six HoFH patients. All patients tolerated titration to the highest dose. At this dose LDL-C was reduced by 50.9% and apo B by 55.6% from baseline. Kinetic studies showed a markedly reduced apo B production. The most serious events were nausea and steatorrhea. There were elevations of liver aminotransferase levels with accumulation of hepatic fat which, at the highest dose, ranged from less than 10% to more than 40%. After this study BMS donated the compound to the University of Pennsylvania School of Medicine. This then licensed the drug candidate to Aegerion Therapeutics. Aegerion received an initial grant from the US Federal Government in connection with the government's qualifying therapeutic discovery project grant program. In December 2007 Aegerion initiated a phase III open-label trial in patients with well-characterized HoFH, to determine the safety and effectiveness of long-term use of the drug, now called lomitapide () (Citation49), the maximum tolerated dose being up to 60 mg/day. At this moment lomitapide is the only MTP inhibitor having reached drug status and, currently, in clinical use.
Figure 3. Chemical structure of lomitapide.
Lomitapide. Pharmacological and clinical profile
Studies on lomitapide by BMS and later by Aegerion have ranged from investigations on kinetics, nutrient/drug interactions, to the effects of associated lipid-lowering treatments.
Pharmacokinetics and drug interactions
Pharmacokinetics (PK) and the absorption, distribution, metabolism, and excretion (ADME) profiles have been the object of a number of studies involving bioavailability, food effects, as well as kinetics in conditions of liver and kidney impairment.
Lomitapide, in earlier studies, was found to be absorbed by at least 33% after oral administration, but more detailed recent studies indicated that bioavailability is actually around 7%, thus suggesting a high first-pass metabolism (Citation50). The unchanged compound is highly protein-bound (99.5%), and the elimination half-life is 34.4 hours, 33% being excreted in the urine (all as metabolites) and 52.9% in feces. Lomitapide undergoes extensive metabolism by cytochrome P450 (CYP)3A4, and the major metabolites (M1–M3) are pharmacologically inactive with respect to MTP inhibition (Citation50).
An open-label study compared patients with mild to moderate hepatic impairment to matching subjects with normal liver function. The major findings were indicative of some delay of t1/2β (half-life after reaching complete distribution) in conditions of liver impairment. In mild impairment Cmax and AUCINF values were 4% and 47% higher versus matched controls, with a mean t1/2β about 12 hours longer. In conditions of moderate liver impairment mean Cmax and AUCINF were 164% and 361% higher versus matched control values, whereas mean t1/2β was not significantly longer versus controls and mildly impaired patients, respectively (Citation50).
In contrast, comparison of PK of a single 60 mg dose between patients with end-stage renal disease (ESRD) on hemodialysis and matched healthy subjects indicated only moderate increases of Cmax and AUC0-72 in ESRD. ESRD was also associated with higher levels of the M1 but not of the M3 metabolite (Citation50). These data indicate that no adjustment in dosage regimen is needed for patients with minor, moderate, or severe renal impairment.
Food interactions
In view of the apparent frequency of gastrointestinal (GI) side effects during therapy with lomitapide, a phase I randomized open-label study was carried out in 25 healthy volunteers (Citation50). Lomitapide absorption was evaluated by determining plasma levels following either an overnight fast, or 5 minutes after a low-fat or a high-fat breakfast. Significant increases in Cmax and AUC (up to + 77% and + 58%, respectively) were noted after the high-fat breakfast compared with fasting. No food effect on Tmax was detected. GI complaints were more common following dosing with the high-fat breakfast.
Drug interactions
Lomitapide is a substrate mainly of CYP3A4, not of 2D6. In view of the frequent concomitant use of other lipid-lowering medications, most of these were tested in combination with lomitapide.
Three statins were tested (Citation51). Lomitapide 10 mg/daily increased the simvastatin Cmax and AUC by 35% and 39%, respectively, and simvastatin lactone Cmax and AUC both by approximately 65%. At 60 mg/daily the Cmax and AUCINF of simvastatin lactone were raised 1.6-fold and 1.7-fold, respectively. The atorvastatin 20 mg Cmax was raised to a minimal extent, i.e. + 12%, with lomitapide 10 mg and 60 mg. The rise was + 38% for the sum of active atorvastatin moieties. The effects on the AUC were less significant. No significant effects of lomitapide 10 mg daily on rosuvastatin 10 mg exposure were detected, but the 60 mg dose increased rosuvastatin AUCINF by 32%.
No significant effects of lomitapide 10 mg were noted on ezetimibe PK, whereas the same dose raised the AUCINF of the N-methyl nicotinamide metabolite by 36% with no other changes of Cmax or AUCINF of niacin metabolites (Citation51). Interestingly, lomitapide 10 mg/daily reduced the Cmax and AUCINF of fenofibric acid by –30% and –10%, respectively (Citation51).
The possibility of an interaction with the PK of oral contraceptives was also assessed. Lomitapide 50 mg/daily was tested in combination with ethinylestradiol (EE) and norgestimate. On both of these lomitapide exerted a raising effect (+ 92% for Cmax and AUC0-t of EE and + 102% for Cmax and + 106% for AUC0-t for the norgestimate 17-deacetyl metabolite).
Finally the effects on warfarin PK by lomitapide 60 mg were of a minimal extent: the INRmax increased by 22% and the AUCINR by only 7%, thus probably not adding a significant variant to the INR goals in patients on chronic anti-thrombotic treatment (Citation50).
Lomitapide is sensitive to CYP3A4 inhibition, since co- administration of ketoconazole raised Tmax and AUCINF of the agent approximately 15-fold and 27-fold. The half-life was prolonged from 39.0 hours to 63.7 hours. There was an apparent increase of GI adverse events upon co-administration of ketoconazole (Citation50). Recently the European authority (Committee for Medicinal Products for Human Use, CHMP, 26 July 2013) recommended withdrawal of oral ketoconazole from the market, based on liver toxicity and high risk of potentially dangerous interactions.
Clinical development of lomitapide and evaluation of lipid-lowering activity
Therapeutic trials in hyperlipidemias—phase II trials
Phase II trials involved 460 patients with dyslipidemias receiving monotherapy or combinations with other lipid-lowering agents, i.e. atorvastatin, ezetimibe, and fenofibrate (Citation52). Lomitapide was administered orally in daily doses ranging from 2.5 to 10 mg. Lomitapide reduced LDL-C levels by a mean of –35% from baseline as monotherapy and up to –66% from baseline when administered in combination with atorvastatin. A reduction of TG levels up to –50% and a weight loss up to –3% after 12 weeks of therapy were also recorded. Average liver fat levels after 12 weeks of exposure across doses of lomitapide were + 7%, no treatment arm exceeding + 10%.
Particular attention was given to combinations of lomitapide with ezetimibe (Citation52), since ezetimibe is the lipid-lowering agent with the least interaction with lomitapide. In a prospective double-blind 12-week trial, 84 patients were randomized to ezetimibe 10 mg/day; lomitapide 5 mg/day for the first 4 weeks; 7.5 mg/day for the second 4 weeks and 10 mg/day for the last 4 weeks; ezetimibe 10 mg/day + lomitapide administered with the same titration. This study clearly indicated that lomitapide lowers LDL-C from –19% at 5 mg to –30% at 10 mg/day (). Combined therapy produced similar but larger dose-dependent decreases, i.e. –35%, –38%, and –46%, respectively. A small number of patients discontinued drug treatment for adverse events, i.e. five with ezetimibe alone, nine with lomitapide alone, and four with the combined treatment. Subjective side effects were mainly gastrointestinal of a minimal degree, and drug interruptions were essentially due to a small number of transaminase elevations.
Table II. Evaluation of lomitapide in moderate hypercholesterolemic patients with and without ezetimibe (Citation52).
Phase III trial in HoFH
The major advance in the treatment with MTP inhibitors came with the outcome of a single-blind, open-label phase III study of lomitapide in patients with HoFH (Citation53). A total of 29 patients were recruited in four countries, i.e. US (7 subjects), South Africa (11 subjects), Italy (6 subjects), and Canada (5 subjects). All patients received a full genetic characterization by established methodologies. Of these 29 patients, one was not a carrier of an LDL-R mutation, being homozygote for an insert in the ARH gene in exon 4. All the others were carriers of previously diagnosed, well-defined LDL-R mutations, with well-known LDL-R residual activity. Nine of the 29 were HoFH, whereas the other 20 were CH. Current lipid-lowering therapy (including LDL apheresis) was maintained from 6 weeks before baseline through at least week 26. Lomitapide dose was escalated on the basis of safety and tolerability from 5 mg to a maximum of 60 mg/day. The primary end-point was mean percent change in levels of LDL-C from baseline to week 26. Every patient remaining on lomitapide through week 78 was the object of a detailed safety assessment. At present, some of the patients have exceeded 5 years of treatment.
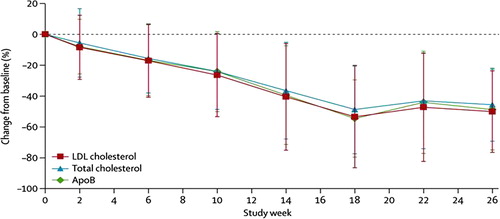
Twenty-eight patients (one patient dropped out at week 2) completed at least 6 weeks of treatment, and 23 of the 28 completed both the efficacy phase (26 weeks) and the full study (78 weeks). Interestingly, it was noted that patients reaching 26 weeks (or 6 months) of treatment tended to remain steadily on the drug without any drop-out up to this time. The mean final dose of lomitapide was 40 mg/day. LDL-C was reduced by a mean of 50% (95% CI, from –62% to –39%) from baseline (mean 8.7 ± 2.9 mmol/L) to week 26 (4.3 ± 2.5 mmol/L). LDL-C levels were lower than 2.6 mmol/L in eight patients at 26 weeks. At week 26, the reductions of apo B and TG were 49% and 45%, respectively, and remained significantly reduced, by –48% and –31%, at the end of the study. Concentrations of LDL-C remained reduced by –44% at week 56 and –38% at week 78 () (Citation53). It should be noted that in some patients the baseline levels were likely to be elevated by the interruption or significant reductions in the frequency of LDL apheresis.
Figure 4. Mean percent changes in total, LDL-C, and apo B levels during the phase III study on lomitapide in HoFHs. From ref. (Citation53) with permission.
We divided the patients of the study led by Cuchel et al. (Citation53) into subgroups () according to their LDL-R residual activity and to their LDL-R mutation classes. We considered only those for whom full data were available up to at least 6 weeks, in order to achieve an acceptable activity of the drug. Therefore, out of the 28 patients, we excluded the following from our analysis: 3 patients for discontinuation due to drug intolerance and 1 for non-compliance, and 1 patient was analyzed separately (see below).
Table III. LDL-Cholesterol responses to lomitapide of homozygous FH patients according to residual LDL-R activity and to predicted LDL-R classes of mutation.
The majority of patients belonged to mutation classes 1–2, i.e. those with the most severe receptor alterations (). A small number were in classes 3–5, where defects in intracellular LDL-R transport or altered intracellular cycling are mainly responsible for the disease. The majority of included patients were CH (n = 18), and, interestingly, out of these 18 CH, three could be ranked as class 1, two as class 2A, eleven as class 2B, two as class 3, one as class 4, and four as class 5. The vast majority of patients thus belonged to the most severe types of LDL-R mutation classes (Citation20).
By considering patients as distributed into the different LDL-R mutation classes, it was apparent () that the highest effects, in terms of absolute reductions, occurred in patients belonging to classes 1, 2A, and 2B. A lower effect was noted in the three patients belonging to class 3 and 4 and in the four patients in class 5. The change per dose (mmol/L of LDL-C reduction × mg/day of drug) was far higher in patients belonging to classes 1, 2A, and 2B. In particular, it is noticeable that patients belonging to classes 1 and 2B had the highest response, as mmol/L absolute reduction per mg of drug. Finally, just considering the final dose (also keeping in mind that it may reflect tolerability, physician's decision, and others), this did not vary significantly in the groups of patients belonging to the different genotypes. By considering the residual LDL-R receptor activity in our patients treated with lomitapide, it was clear that subjects with receptor-negative mutations (i.e. classes 1 and 2A) respond to treatment better than subjects with receptor-defective mutations (classes 3, 4, and 5). The differences among the three groups are particularly evident in terms of absolute LDL-C decrease and of absolute LDL-C decrease per mg of lomitapide (P = 0.018 and P = 0.048, respectively).
The different response of patients belonging to the different LDL-R classes of mutations may also help in shedding light on the issue of VLDL–IDL production rate in HoFH. Two previous studies (Citation36,Citation37) concluded that production rates of VLDL and IDL was raised only in receptor-negative individuals, i.e. belonging to classes 1 or 2A, and some to class 2B. In receptor-defective patients, belonging to different phenotypic classes, production rates did not differ from those of controls. Kinetic studies using endogenous labeling of apo B with deuterated leucine documented that HoFH patients carrying an LDL-R-negative mutation (Promoter + Ex1 del, C660X) had, with respect to healthy individuals, an increased production rate of VLDL2 (0.52 ± 0.20 versus 0.31 ± 0.04 mmol/kg/day) and IDL (0.59 ± 2.06 versus 0.35 ± 0.04 mmol/kg/day). On the contrary, Ho/CH-FH carrying LDL-R-defective mutations (c.-137C> T + E387K, S156L + S156L, C249Y + C255W) did not show significant differences from controls in the production rates of VLDL2 and IDL (Citation36,Citation37). Goldstein et al. (Citation54), indeed, showed that in fibroblasts from HoFHs carrying receptor-negative mutations (null alleles) the HMG-CoA reductase activity is dramatically raised, i.e. with an about 60 times greater number of enzyme molecules versus controls. This activity is instead suppressed in fibroblasts from HoFH patients carrying receptor-defective mutations (i.e. the residual activity of LDL-Rs seems to be sufficient to suppress cholesterol synthesis, as in normal cells). In the presence of increased concentrations of lipoprotein-assembling components, it appears that MTP activity can enhance the production rate of lipoproteins. MTP inhibition should thus result in a larger reduction of VLDL and IDL synthesis and in a more effective LDL-lowering. In the presence of residual LDL-R activity, as observed in patients carrying LDL-R defective mutations, intracellular degradation of apo B and partial reuptake of the newly secreted VLDL–IDL may be similar or close to those observed in subjects with normal LDL-R activity. Some exceptions are, however, possible, since the attributed classes do not always reflect the functioning of residual LDL-Rs as inducers of apo B degradation.
A possible confirmation of the hypothesis of a link between high liver LDL production and response to lomitapide is provided by the case of patient 22-004 (who was analyzed separately), homozygous for W66G (atypical defective class 3), a French-Canadian mutation considered as receptor-defective belonging to class 3 (defective LDL binding) or to class 5 (defective LDL-R recycling) (Citation55). The majority of homozygous and heterozygous carriers show a mild clinical phenotype, less severe than that in patients with receptor-negative mutations (Citation55,Citation56). However, unexpectedly, some homozygotes may present with a severe phenotype (Citation57). Patient 22-004, with high LDL-C level (14.6 mmol/L) and a poor response to treatment with atorvastatin 80 and ezetimibe 10 mg/day, was a high responder to lomitapide treatment (–79.6% LDL-C from baseline) thus suggesting an atypical class 3, probably associated to an increased liver VLDL production. This patient presented with severe obesity, hepatic steatosis, and microalbuminuria, clearly indicative of insulin resistance, known to increase the expression of MTP and apo B, to reduce the presecretory degradation of apo B, and to increase lipoprotein production by the liver (Citation58,Citation59).
Other clinical findings in lomitapide-treated patients
Reduction of LDL-apheretic schedules occurred in the majority of patients who were following this therapeutic procedure. This will be the object of a separate publication (unpublished observation, Stefanutti C, University of Rome ‘La Sapienza’, 2014).
No specific side effects or other ancillary changes, such as weight loss or others, were noted to be associated to any specific class.
The most common adverse events were gastrointestinal (27 out of 29 patients). Four patients showed an increase in hepatic transaminase (ALT > 5 × ULN), which returned to normal on reducing the dose or stopping/restarting the study drug (Citation53). Occurrence of liver steatosis, a feared complication of lomitapide therapy, similar to other agents affecting apo B synthesis, such as the anti-sense nucleotide mipomersen (Citation56), proved to be less serious than expected. Liver fat content (as assessed by an NMR technology) increased in the first 26 weeks, from 1.0% to 8.6%, remaining stable during the rest of the study, without further increases. There was no apparent connection between subjective GI side effects, liver enzyme changes, and rises of liver fat.
The evaluation of this series of patients with LDL-R mutations, albeit unplanned, appears to indicate that the MTP inhibitory treatment is dramatically more effective in patients belonging to LDL-R mutation classes characterized by clear deficiency. This type of evaluation allows the comparison of the effects of lomitapide with that of an inhibitor of PCSK9. A study with this latter agent was, in fact, recently reported: of the eight selected HoFH patients some were totally refractory to treatment, whereas others showed a modest response (Citation24). In particular, the two patients listed as LDL-R-negative (CH classes 2A/2A and 1/2A, respectively) showed no response, whereas modest to no responses (from + 2% to –16%) were noted in the three patients listed as LDL-R-defective (two homozygous for class 2B mutations, one CH for class 2A/2B mutations). Satisfactory responses (–38 to –48%) were finally recorded in the remaining three patients, also listed as LDL-R-defective (one homozygous for a class 2B mutation and two CH for two different class 2B mutations). Notably, two receptor-defective patients who were homozygous for the same class 2B mutation showed a strikingly different response: one was a good responder and the other a very poor responder, in spite of comparable suppression of plasma PCSK9 levels (Citation24). The comparison of lomitapide with the PCSK9 inhibitor, although indirect, would suggest a stable, satisfactory lipid response in essentially all HoFH patients treated with lomitapide, versus a response restricted only to some LDL-R-defective patients with the PCSK9 inhibitor.
The phase III study with lomitapide eventually led to the US FDA approval for HoFH as well as the approval by the European Medicine Agency (EMA)/CHMP and by the Mexican COFEPRIS, and it has led to a considerable rise in the number of patients treated with lomitapide. This last finding is also indicative of the fact that most likely a larger number of HoFH is found in the general population, i.e. far above the frequently described 1 per million. Overall, the clinical experience has been non-eventful, i.e. with only a small number of patients (less than 10%) not tolerating therapy. It should be underlined that in the phase III study no patient on active therapy for over 6 months did eventually stop treatment. This indicates that probably dietary compliance is a key factor in maintaining drug compliance. This is now the object of intensive investigation.
MTP inhibition and arterial disease
In the phase III study, only one patient undergoing continued treatment suffered a major cardiovascular event (acute coronary syndrome and angina, unlikely related to lomitapide). While of course this does in no way prove that MTP inhibition provides an effective preventive management of atherosclerosis, nonetheless it fits well with the lipid findings reported in the WHHL rabbit (Citation43). An interesting, infrequently quoted experimental study on MTP inhibition was provided by Lieu et al. (Citation57) on the REVERSA mouse model. This has overexpression of apo B and defective LDL-R, being homozygous for a conditional allele of MTP. Expression of MTP can be antagonized by Cre in the liver, thus rapidly reducing cholesterolemia from 13.6 to 2.6 mmol/L in fat-fed mice. While aortas from the REVERSA mice on a high-fat diet for 20 weeks show extreme disease, the induction of Cre (by pI-pC) at 4 weeks of age totally prevents atheroma formation. Very interestingly, in animals on a chow diet, Cre induction reduces cholesterolemia from 6.0 to 0.96 mmol/L. In these animals, moreover, MTP inhibition does not induce liver steatosis, thus supporting the concept that, in the presence of a low fat intake, liver steatosis will be prevented, most likely also in humans.
Most recently, by employing an MTP inhibitor chemically different from lomitapide, a very rapid regression of atherosclerosis was shown in a rodent model (Citation60). LDL-R-deficient (ldlr−/−) mice fed a Western diet for 16 weeks were switched to chow diet or chow diet plus MTP inhibitor for just 2 weeks. The rapid reduction in plasma lipids and atheromas by the MTP inhibitor was accompanied by a significant decrease in lipid content and monocyte-derived (CD68+) cells in aortic plaques compared to chow diet controls. MTP inhibitor-treated mice had an increased arterial collagen content, a marker associated with increased stability of plaques.
Conclusions
In conclusion, evaluation of a long-term open-label study of lomitapide in HoFH proved that this approach, besides an acceptable tolerability, provides a very effective treatment option for patients, up to now really pharmacotherapeutic ‘orphans’. These patients, particularly those belonging to the most severe LDL-R mutation classes, have excellent responses, not only in terms of lipid reduction, but also in the associated therapeutic interventions, i.e. LDL apheresis, leading to either total abandonment or considerable reduction in the procedures.
The present-day status of lomitapide appears quite favorable, and entrance into the daily practice of lipid and non-lipid specialists has shown, up to now, no significant toxic events and satisfactory compliance by patients. The future of the drug appears to be that of an extension of indications to pediatric patients, also in view of the clear arterial benefit of starting statin treatment before the age of 8 in severely hypercholesterolemic, not homozygous, FH patients (Citation61). In addition, the clinical report of an excellent response to the drug of a chylomicronemic patient also indicates this as a possible further indication for lomitapide (Citation47). A careful follow-up of a possible arterial benefit of the agent will be crucial also in the understanding of the potential for wider clinical indications. Whether these or similar agents will ever enter a wider preventive field, e.g. that of statins, should be also considered. Clinical practice shows that, at times, statin tolerability is not as satisfactory as generally believed (Citation62). A better understanding of the complete mechanism of action of lomitapide and of similar agents, also considering a potentially very effective anti-atherogenic activity, will be of value in a possible future extension of therapeutic indications.
Declaration of interest: The authors report no conflicts of interest.
References
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–8.
- Marais AD, Blom DJ. Recent advances in the treatment of homozygous familial hypercholesterolaemia. Curr Opin Lipidol. 2013;24:288–94.
- Rubinsztein DC, Raal FJ, Seftel HC, Pilcher G, Coetzee GA, van der Westhuyzen DR. Characterization of six patients who are double heterozygotes for familial hypercholesterolemia and familial defective apo B-100. Arterioscler Thromb. 1993;13:1076–81.
- Pisciotta L, Priore Oliva C, Cefalu AB, Noto D, Bellocchio A, Fresa R, et al. Additive effect of mutations in LDLR and PCSK9 genes on the phenotype of familial hypercholesterolemia. Atherosclerosis. 2006; 186:433–40.
- Sjouke B, Kusters DM, Kindt I, Besseling J, Defesche JC, Sijbrands EJ, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J. 2014 Feb 28. [Epub ahead of print]
- Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Tada H, et al. Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis. 2011;214:404–7.
- Fahed AC, Safa RM, Haddad FF, Bitar FF, Andary RR, Arabi MT, et al. Homozygous familial hypercholesterolemia in Lebanon: a genotype/phenotype correlation. Mol Genet Metab. 2011;102:181–8.
- Moorjani S, Roy M, Gagne C, Davignon J, Brun D, Toussaint M, et al. Homozygous familial hypercholesterolemia among French Canadians in Quebec Province. Arteriosclerosis. 1989;9:211–16.
- Jelassi A, Slimani A, Jguirim I, Najah M, Maatouk F, Varret M, et al. Effect of a splice site mutation in LDLR gene and two variations in PCSK9 gene in Tunisian families with familial hypercholesterolaemia. Ann Clin Biochem. 2011;48(Pt 1):83–6.
- Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012;97:3956–64.
- Thompson GR, Catapano A, Saheb S, Atassi-Dumont M, Barbir M, Eriksson M, et al. Severe hypercholesterolaemia: therapeutic goals and eligibility criteria for LDL apheresis in Europe. Curr Opin Lipidol. 2010;21:492–8.
- Cohen JC, Kimmel M, Polanski A, Hobbs HH. Molecular mechanisms of autosomal recessive hypercholesterolemia. Curr Opin Lipidol. 2003;14:121–7.
- Arca M, Zuliani G, Wilund K, Campagna F, Fellin R, Bertolini S, et al. Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: a clinical and molecular genetic analysis. Lancet. 2002;359:841–7.
- Pisciotta L, Priore Oliva C, Pes GM, Di Scala L, Bellocchio A, Fresa R, et al. Autosomal recessive hypercholesterolemia (ARH) and homozygous familial hypercholesterolemia (FH): a phenotypic comparison. Atherosclerosis. 2006;188:398–405.
- Innerarity TL, Mahley RW, Weisgraber KH, Bersot TP, Krauss RM, Vega GL, et al. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J Lipid Res. 1990;31:1337–49.
- Abifadel M, Guerin M, Benjannet S, Rabes JP, Le Goff W, Julia Z, et al. Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis. 2012;223:394–400.
- Sirtori CR, Catapano AL, Franceschini G, Corsini A, Noseda G, Fragiacomo C, et al. Aortic and coronary atheromatosis in a woman with severe hypercholesterolaemia without LDL receptor alterations. Eur Heart J. 1991;12:818–24.
- Naoumova RP, Neuwirth C, Lee P, Miller JP, Taylor KG, Soutar AK. Autosomal recessive hypercholesterolaemia: long-term follow up and response to treatment. Atherosclerosis. 2004;174:165–72.
- Soufi M, Rust S, Walter M, Schaefer JR. A combined LDL receptor/LDL receptor adaptor protein 1 mutation as the cause for severe familial hypercholesterolemia. Gene. 2013;521:200–3.
- Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24:133–70.
- Miltiadous G, Xenophontos S, Bairaktari E, Ganotakis M, Cariolou M, Elisaf M. Genetic and environmental factors affecting the response to statin therapy in patients with molecularly defined familial hypercholesterolaemia. Pharmacogenet Genomics. 2005;15:219–25.
- Raper A, Kolansky DM, Cuchel M. Treatment of familial hypercholesterolemia: is there a need beyond statin therapy?Curr Atheroscler Rep. 2012;14:11–16.
- Marais AD, Firth JC, Blom DJ. Homozygous familial hypercholesterolemia and its management. Semin Vasc Med. 2004;4:43–50.
- Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–20.
- Wetterau JR, Lin MC, Jamil H. Microsomal triglyceride transfer protein. Biochim Biophys Acta. 1997;1345:136–50.
- Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258: 999–1001.
- Sharp D, Blinderman L, Combs KA, Kienzle B, Ricci B, Wager-Smith K, et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature. 1993;365: 65–9.
- Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond). 2012; 9:14.
- Wang S, McLeod RS, Gordon DA, Yao Z. The microsomal triglyceride transfer protein facilitates assembly and secretion of apolipoprotein B-containing lipoproteins and decreases cotranslational degradation of apolipoprotein B in transfected COS-7 cells. J Biol Chem. 1996; 271:14124–33.
- Atzel A, Wetterau JR. Identification of two classes of lipid molecule binding sites on the microsomal triglyceride transfer protein. Biochemistry. 1994;33:15382–8.
- Khatun I, Walsh MT, Hussain MM. Loss of both phospholipid and triglyceride transfer activities of microsomal triglyceride transfer protein in abetalipoproteinemia. J Lipid Res. 2013;54:1541–9.
- Larsson SL, Skogsberg J, Bjorkegren J. The low density lipoprotein receptor prevents secretion of dense apoB100-containing lipoproteins from the liver. J Biol Chem. 2004;279:831–6.
- Shimano H, Shimomura I, Hammer RE, Herz J, Goldstein JL, Brown MS, et al. Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J Clin Invest. 1997;100:2115–24.
- Sato R, Miyamoto W, Inoue J, Terada T, Imanaka T, Maeda M. Sterol regulatory element-binding protein negatively regulates microsomal triglyceride transfer protein gene transcription. J Biol Chem. 1999;274:24714–20.
- Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998;101:2331–9.
- Tremblay AJ, Lamarche B, Ruel IL, Hogue JC, Bergeron J, Gagne C, et al. Increased production of VLDL apoB-100 in subjects with familial hypercholesterolemia carrying the same null LDL receptor gene mutation. J Lipid Res. 2004;45:866–72.
- Millar JS, Maugeais C, Ikewaki K, Kolansky DM, Barrett PH, Budreck EC, et al. Complete deficiency of the low-density lipoprotein receptor is associated with increased apolipoprotein B-100 production. Arterioscler Thromb Vasc Biol. 2005;25:560–5.
- Lin MC, Arbeeny C, Bergquist K, Kienzle B, Gordon DA, Wetterau JR. Cloning and regulation of hamster microsomal triglyceride transfer protein. The regulation is independent from that of other hepatic and intestinal proteins which participate in the transport of fatty acids and triglycerides. J Biol Chem. 1994;269: 29138–45.
- Bennett AJ, Billett MA, Salter AM, White DA. Regulation of hamster hepatic microsomal triglyceride transfer protein mRNA levels by dietary fats. Biochem Biophys Res Commun. 1995;212:473–8.
- Dorfman SE, Wang S, Vega-Lopez S, Jauhiainen M, Lichtenstein AH. Dietary fatty acids and cholesterol differentially modulate HDL cholesterol metabolism in Golden-Syrian hamsters. J Nutr. 2005;135: 492–8.
- Hagan DL, Kienzle B, Jamil H, Hariharan N. Transcriptional regulation of human and hamster microsomal triglyceride transfer protein genes. Cell type-specific expression and response to metabolic regulators. J Biol Chem. 1994;269:28737–44.
- Lin MC, Gordon D, Wetterau JR. Microsomal triglyceride transfer protein (MTP) regulation in HepG2 cells: insulin negatively regulates MTP gene expression. J Lipid Res. 1995;36:1073–81.
- Wetterau JR, Gregg RE, Harrity TW, Arbeeny C, Cap M, Connolly F, et al. An MTP inhibitor that normalizes atherogenic lipoprotein levels in WHHL rabbits. Science. 1998;282:751–4.
- Shiomi M, Ito T. MTP inhibitor decreases plasma cholesterol levels in LDL receptor-deficient WHHL rabbits by lowering the VLDL secretion. Eur J Pharmacol. 2001;431:127–31.
- Chandler CE, Wilder DE, Pettini JL, Savoy YE, Petras SF, Chang G, et al. CP-346086: an MTP inhibitor that lowers plasma cholesterol and triglycerides in experimental animals and in humans. J Lipid Res. 2003;44:1887–901.
- Aggarwal D, West KL, Zern TL, Shrestha S, Vergara-Jimenez M, Fernandez ML. JTT-130, a microsomal triglyceride transfer protein (MTP) inhibitor lowers plasma triglycerides and LDL cholesterol concentrations without increasing hepatic triglycerides in guinea pigs. BMC Cardiovasc Disord. 2005;5:30.
- Sacks FM, Stanesa M, Hegele RA. Severe hypertriglyceridemia with pancreatitis: thirteen years’ treatment with lomitapide. JAMA internal medicine. 2014;174:443–7.
- Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–56.
- Lomitapide. Am J Cardiovasc Drugs. 2011;11:347–52.
- Perry CM. Lomitapide: a review of its use in adults with homozygous familial hypercholesterolemia. Am J Cardiovasc Drugs. 2013;13: 285–96.
- Tuteja S DD, Dunbar RL, Movva R, Gadi R, Bloedon LT, et al. Pharmacokinetic interactions of the microsomal triglyceride transfer protein inhibitor, lomitapide, with drugs commonly used in the management of hypercholesterolemia. Pharmacotherapy. 2014;34: 227–39.
- Samaha FF, McKenney J, Bloedon LT, Sasiela WJ, Rader DJ. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2008;5:497–505.
- Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013; 381:40–6.
- Goldstein JL, Dana SE, Brunschede GY, Brown MS. Genetic heterogeneity in familial hypercholesterolemia: evidence for two different mutations affecting functions of low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 1975;72:1092–6.
- Leitersdorf E, Tobin EJ, Davignon J, Hobbs HH. Common low-density lipoprotein receptor mutations in the French Canadian population. J Clin Invest. 1990;85:1014–23.
- Visser ME, Akdim F, Tribble DL, Nederveen AJ, Kwoh TJ, Kastelein JJ, et al. Effect of apolipoprotein-B synthesis inhibition on liver triglyceride content in patients with familial hypercholesterolemia. J Lipid Res. 2010;51:1057–62.
- Lieu HD, Withycombe SK, Walker Q, Rong JX, Walzem RL, Wong JS, et al. Eliminating atherogenesis in mice by switching off hepatic lipoprotein secretion. Circulation. 2003;107:1315–21.
- Verges B. Abnormal hepatic apolipoprotein B metabolism in type 2 diabetes. Atherosclerosis. 2010;211:353–60.
- Costandi J, Melone M, Zhao A, Rashid S. Human resistin stimulates hepatic overproduction of atherogenic ApoB-containing lipoprotein particles by enhancing ApoB stability and impairing intracellular insulin signaling. Circ Res. 2011;108:727–42.
- Hewing B, Parathath S, Mai CK, Fiel MI, Guo L, Fisher EA. Rapid regression of atherosclerosis with MTP inhibitor treatment. Atherosclerosis. 2013;227:125–9.
- Rodenburg J, Vissers MN, Wiegman A, van Trotsenburg AS, van der Graaf A, de Groot E, et al. Statin treatment in children with familial hypercholesterolemia: the younger, the better. Circulation. 2007;116:664–8.
- Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Therapy. 2005;19:403–14.