
Abstract
Intracranial aneurysms, also called cerebral aneurysms, are dilatations in the arteries that supply blood to the brain. Rupture of an intracranial aneurysm leads to a subarachnoid hemorrhage, which is fatal in about 50% of the cases. Intracranial aneurysms can be repaired surgically or endovascularly, or by combining these two treatment modalities. They are relatively common with an estimated prevalence of unruptured aneurysms of 2%–6% in the adult population, and are considered a complex disease with both genetic and environmental risk factors. Known risk factors include smoking, hypertension, increasing age, and positive family history for intracranial aneurysms. Identifying the molecular mechanisms underlying the pathogenesis of intracranial aneurysms is complex. Genome-wide approaches such as DNA linkage and genetic association studies, as well as microarray-based mRNA expression studies, provide unbiased approaches to identify genetic risk factors and dissecting the molecular pathobiology of intracranial aneurysms. The ultimate goal of these studies is to use the information in clinical practice to predict an individual's risk for developing an aneurysm or monitor its growth or rupture risk. Another important goal is to design new therapies based on the information on mechanisms of disease processes to prevent the development or halt the progression of intracranial aneurysms.
Key messages
Intracranial aneurysms, with an estimated prevalence of unruptured aneurysms of 2%–6% in the adult population, are a complex disease with both genetic and environmental risk factors.
Family members of patients with intracranial aneurysms are at increased risk for developing an intracranial aneurysm, and genome-wide DNA linkage and genetic association studies have identified a large number of genetic risk factors for intracranial aneurysms.
Genome-wide microarray-based mRNA and microRNA expression studies provide unbiased information about molecular mechanisms of intracranial aneurysm and the foundation for functional studies.
An intracranial aneurysm (IA), also called a cerebral aneurysm, is a dilatation of an artery that supplies blood to the brain (Citation1). The shape of a dilatation can vary from a local sac-like pouch to long and tortuous enlargement of the diameter of a vessel, called a fusiform aneurysm. The most common form of IA is a berry, or saccular, form; consequently most research is about the saccular or berry IA (Mendelian Inheritance in Man, MIM number: 105800) unless specifically indicated otherwise. IAs form a complex disease with both genetic and environmental risk factors, and a poorly understood molecular pathology.
In this review we will summarize the current knowledge on the pathogenesis of IA with special emphasis on molecular and genetic studies in humans. We will concentrate on genome-wide DNA linkage and genetic association studies as well as microarray-based mRNA expression studies, and only discuss candidate gene studies that were of sufficient size to provide reliable data.
Intracranial aneurysm is a complex disease: clinical characteristics and risk factors
IAs are relatively common among human populations with an estimated 2%–6% prevalence of unruptured IAs in the adult population (Citation2–6). Unruptured IAs are mainly asymptomatic and are most often found either through screening high-risk patients or as purely incidental findings of magnetic resonance imaging (MRI) or computerized tomography (CT) studies for other neurological symptoms. The danger posed by IAs is the possibility of rupture leading to life-threatening subarachnoid hemorrhage (SAH) (Citation4). Many clinical studies have assessed the risk for rupture of unruptured aneurysms; however, most estimates are from retrospective studies with the concomitant biases. Retrospective series estimate the risk of rupture to be between 1% and 3% (1 to 3 per 100 patient years), whereas the prospective series estimate it to be between 0.78% and 1.4% (Citation7). The risk of rupture increases with aneurysm size, location, specifically in the posterior circulation, irregular shape, and history of previous aneurysmal SAH (Citation8,Citation9).
The most common clinical symptoms of aneurysmal SAH are a sudden onset of severe headache with stiff neck, vomiting, and photophobia. The severity of aneurysmal SAH can range in a spectrum from relatively mild and atypical, with few of the common symptoms, to sudden death. The incidence of aneurysmal SAH varies between different populations; the highest number of 22.5–32 per 100,000/y reported in Finland and Japan, while worldwide the incidence of SAH is 9.1 per 100,000/y (Citation10).
The overall mortality rate of aneurysmal SAH has not changed much during the last decades, being nearly 50% of all ruptured cases (Citation11–14). In the USA alone aneurysmal SAH is estimated to affect about 30,000 individuals each year (Citation15). Also the greatest impact of the aneurysm rupture is in the age group younger than 65 years (Citation16). The most important factor affecting the outcome is the severity of the initial aneurysm rupture. Patients with poor medical condition after aneurysmal SAH have less favorable outcome than patients with only minor neurological symptoms.
Diagnosis of SAH is primarily by CT, CT angiography (CTA), or catheter angiography. During the acute onset of symptoms CT has nearly 100% sensitivity for SAH (Citation17), and a lumbar puncture to confirm the diagnosis is seldom needed. CTA yielding high-quality images of the intracranial vascular structures is easily and rapidly available in tertiary care emergency rooms. Even though CTA is also less invasive than angiography, catheter angiography remains the standard for diagnosing aneurysm in all locations in the cranium. MRI is used only as a screening method for unruptured IAs (Citation18).
Aneurysms, whether ruptured or unruptured, can be treated either by open surgery or endovascularly, or by combining these two treatment modalities. The occlusion of the aneurysm neck prevents the risk of possible rebleeding of the aneurysm and can save the patients from secondary complications. It also enables effective prevention and treatment of possible delayed complications related to aneurysmal SAH. The treatment modality of IAs is highly dependent on the institution, some institutions favoring microsurgery and others endovascular treatment. The treatment results of IAs seem to improve with the increasing number of treated patients at the institution (Citation19,Citation20). The long-term outcomes between different treatment modalities do not differ significantly (Citation21–23). Advances in clinical management of SAH during the last two decades have substantially improved the overall outcome of aneurysmal SAH patients surviving to reach hospital even as the incidence of SAH is decreasing (Citation24).
Current smoking, hypertension, and heavy alcohol consumption are independent risk factors for aneurysmal SAH (Citation25–27). Increasing age, female sex, familial occurrence of IAs, and the use of sympathomimetic drugs are also known risk factors for aneurysmal SAH (Citation28). Patients with certain genetic disorders, such as autosomal dominant polycystic kidney disease and vascular Ehlers–Danlos syndrome (also known as type IV EDS) (Citation29), are at increased risk for aneurysmal SAH. Recently, the PHASES study found that geographical location, e.g. Finnish or Japanese origin, was also a strong risk factor for aneurysmal rupture, possibly supporting a genetic influence on rupture risk (Citation30). Of the different risk factors, only smoking and hypertension can be controlled and treated.
Family studies in intracranial aneurysms
Family history of IA is an important risk factor for IA. The largest collections of families with at least two affected members include a study by Wills et al. (Citation31) with 346 Finnish IA families and a study by the International FIA Consortium with 542 families (Citation32). A follow-up study on the Finnish IA families in which the clinical characteristics of the familial IA cases were compared to sporadic IA cases demonstrated that the familial IA group was slightly younger (46 versus 51 years in men; 50 versus 57 years in women), and had fewer females (49% versus 54%), whereas there was no difference in the number of ruptured IAs (Citation33).
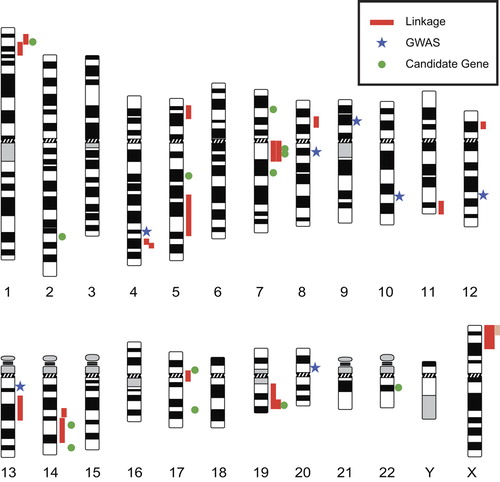
Several genome-wide DNA linkage studies have been carried out on IA (). Many of these studies concentrated on single, large families and used parametric statistical approaches (Citation34–38). The results were often restricted to the family under study and could not be extended to a larger IA population. Larger studies by three groups, the Finnish (Citation39,Citation40), the Japanese (Citation41–43), and the FIA Consortium (Citation44,Citation45), have concentrated on non-parametric statistical approaches using an affected relative pair design (). Details on the DNA linkage studies and the 13 genomic loci identified are shown in . Six of the genomic regions (1p34–36, 4q32, 7q11, 14q22, 19q13, and Xp22) were discovered in two or more independent studies, thus providing strong evidence that they represent true linkage.
Table I. DNA linkage studies on intracranial aneurysms.
The challenge for DNA linkage studies comes when fine- mapping to narrow the linkage interval and find the gene(s) harboring genetic variants contributing to the IA susceptibility. This usually requires a large number of new families. Recent technological developments in next-generation sequencing are expected to ameliorate this limitation. Interpretation of the results will remain a challenge, since incomplete penetrance and genetic heterogeneity are likely to be present.
Genetic association studies in intracranial aneurysms: from candidate gene studies to genome-wide association studies
Genome-wide linkage studies of IA families, some of which were large, have identified several susceptibility loci for IA (), but it is unknown if these same variants contribute to IA disease risk in individuals without a family history of IA. Another major approach used to identify genetic risk factors for IA is to genotype polymorphisms in unrelated (sporadic) cases and controls to determine if there is an association between the genetic marker and IA. The most widely used genetic markers in these studies are the so-called single nucleotide polymorphisms (SNPs). A genetic association study can be performed either as a candidate gene study or as a genome-wide association study (GWAS). In a candidate gene study specific, common polymorphisms are selected in candidate genes chosen for testing based on biological evidence that they may be functionally relevant to the development of IAs (i.e. functional candidate genes). Candidate genes for IA have also been selected from genetic studies on connective tissue disorders (e.g. vascular Ehlers–Danlos syndrome), known genetic diseases with IA as a phenotype (e.g. polycystic kidney disease), and gene expression studies (see below).
A recent meta-analysis of all genetic association studies (candidate gene and GWAS) of sporadic IA was conducted by Alg et al. (Citation46) and included 116,000 study participants (although there may be overlap of subjects between some studies). They identified 19 SNPs that were associated with IA in at least one genetic model. The strongest associations were from the GWAS reports (summarized in the GWAS section below), while eight SNPs from candidate gene studies were also significantly associated with IA upon a meta-analysis ().
Table II. Most significant associations with IA in candidate gene studies.
Positional candidate genes identified from IA linkage studies have been prioritized for association testing based on additional knowledge that the genes may function in the development of IA. The results suggest that the genes associated with IA are biologically relevant as they are mainly involved in vascular endothelial maintenance, integrity of the extracellular cellular matrix (ECM), and inflammation. Six SNPs associated with IA are ECM gene variants: COL1A2 (rs42524), COL3A1 (rs1800255), HSPG2 (rs3767137), SERPINA3 (rs4934), and VCAN (previously CSPG2; rs251124 and rs173686) (). Collagen types 1 and 3 are present in the adventitial and medial layers of cerebral arteries and affect vessel tensile strength (Citation47). The versican (VCAN) gene maps to 5q13-14 and also has significant association with thoracic aortic dissections (Citation48). IL6 is a proinflammatory cytokine, and rs1800796 (g.572G> C) SNP is associated with a protective effect in IA (). Vascular remodeling and inflammation contributes to the formation and progression of IAs (Citation49). The exact role of IL6 in the pathogenesis of IA is unknown as the timing of IL6 expression may directly affect the vessel wall (IL6 inhibits collagen synthesis) or expression may be due to systemic inflammation.
The 9p21 locus has been a target for IA association studies (and has subsequently been detected in GWAS) () because of the known sequence variant rs10757278, which contributes to many vascular diseases including myocardial infarction and abdominal aortic aneurysms (AAAs). While Helgadottir et al. (Citation50) were the first to report the association of the 9p21 locus (rs10757278) with IA susceptibility, rs10757278 and rs1333040 (which are in linkage disequilibrium (LD), i.e. correlated) were the SNPs with the strongest association with IA in the recent meta-analysis performed by Alg et al. (Citation46). This variant is located in a non-coding RNA called CDKN2BAS1 (also known as ANRIL). Altered CDKN2BAS1 expression is known to affect two genes involved in cell proliferation and apoptosis, CDKN2A and CDKN2B (Citation51,Citation52). SNP rs10757278 is in strong LD with rs10757272, which showed a trend toward association with IA in two GWASs (Citation53,Citation54), but not significantly associated with IA upon meta-analysis. The biological relevance of these SNPs on IA is unclear (Citation55); however, CDKN2BAS1 may affect MMP3 levels and play a role in ECM repair (Citation56).
Table III. Most significant associations from genome-wide association studies.
A recent meta-analysis (Citation57) on the ACE insertion/deletion (I/D) polymorphism, rs4646994, showed an association between the I allele and IA risk (17q23.3, odds ratio (OR) = 1.21, P = 0.003) (). Individuals with ACE I/I and I/D genotypes had significantly increased risk for IA (OR = 1.27, P = 0.03). The mechanism by which the ACE I allele contributes to IA is unknown.
Some additional candidate genes associated with IA, but not included in any meta-analysis, are noteworthy (). Genome-wide linkage studies identified a susceptibility locus for IA on chromosome 19q13 in the Finnish and Japanese populations (Citation39,Citation40,Citation42,Citation43), which includes the kallikrein gene cluster. In a follow-up study, 18 haplotype-tagging SNPs spanning the KLK gene cluster were tested for association in the Finnish and Russian populations (Citation58). Two intronic SNPs, rs1722561 and rs1701946, mapping to KLK8 and in high LD were associated with IA (). KLK8 is a serine protease that can cleave the ECM protein fibronectin. It is thus plausible to hypothesize that dysregulated kallikrein may contribute to the pathogenesis of IA (Citation58).
Another plausible candidate gene for IA is the elastin (ELN) gene. SNP rs8326 located in the 3’ untranslated region of the ELN gene (+ 659 G > C) is associated with IA and is in an at-risk haplotype for IA that includes two functional SNPs that lead to decreased transcript levels of ELN and LIMK1 () (Citation59). Elastin and LIMK1 proteins are involved in the actin depolymerization pathway and may affect the stability and synthesis of vascular walls, thus contributing to the pathogenesis of IA (Citation59). Another study (Citation60) subsequently reported an association of SNP rs6460071 in LIMK1 with increased risk of IA, but was unable to replicate the association between the ELN gene and IA.
In a follow-up study to the IA linkage region on the chromosome 17 centromere, a haplotype in the TNFRSF13B gene was found to be protective for IA in the Japanese population () (Citation61). TNFRSF13B is involved in immunity as it mediates isotype switching in B cells. This association supports a role for immunologic mechanisms in the IA pathogenesis.
An intronic SNP, rs175646, in JDP2, a positional candidate gene that maps to chromosome 14q22, was associated with IA in a Japanese cohort, and this association was replicated in a Korean IA cohort () (Citation62). JDP2 encodes Jun dimerization protein 2, a repressor of transcription activator protein 1 (AP1), which is involved in apoptosis. Hence, JDP2 may contribute to vascular remodeling in IA via apoptotic cell death.
In addition, a functional polymorphism in TCN2 p.259R> P (rs1801198; c.1025C> G; previously reported as c.776C> G), is associated with IA (Citation63). TCN2 encodes transcobalamin II, which is involved in methionine metabolism, and the protective effect in IA may be due to more effective inhibition of the nitric oxide synthase (NOS).
Unfortunately, many of the genes thought to be strong functional candidates for IA have not shown significant association with IA upon meta-analysis including: ENG, NOS3, APOE, and MMP3 (Citation46). The major limitation of genetic association studies in IA has been small sample size and failure to replicate positive associations, especially given the necessary multiple testing correction for large candidate SNP and GWAS studies (Citation64–67). Studies with fewer than 250 cases provide unreliable OR (effect) estimates (Citation68). IA association findings have been inconsistent between studies, and some candidate genes could be population-specific. Hence, in the absence of a reliable candidate gene for IA, it is premature to move this research forward to the stage of developing transgenic animal models that assess the impact of such functional candidate genes on the formation of IAs.
Genome-wide association studies
A hypothesis-free discovery approach to examine the genetic determinants of IA is GWAS by genotyping unrelated individuals (Citation69). To date there have been a total of six GWAS studies of IA, some of which have identified novel genes for IA, recently reviewed by Hussain et al (Citation70). Four of the studies included sufficient numbers of individuals to power the studies to identify associated SNPs at the genome-wide significant threshold (1 × 10−7). Here we review the most significant findings from the studies and summarize them in .
The first GWAS of IA was published in 2008 (Citation71). The discovery cohort included Finnish and Dutch cases and controls. Significant SNPs were subsequently genotyped in two pooled Japanese replication cohorts. Combination of the data sets including a total of 2196 IA cases and 8085 controls identified SNPs in three loci that passed the genome-wide significance threshold: BOLL/PLCL1 (2q33.1, OR = 1.24, P = 4.4 × 10−8, rs700651), SOX17 (8q11.23, OR = 1.36, P = 1.4 × 10−10, rs10958409), and CDKN2BAS (9p23.1, OR = 1.29, P = 1.4 × 10−10, rs1333040). In addition, rs9298506 located near SOX17, but not in LD with rs10958409, was also associated with IA (8q11.23, OR = 1.35, P = 1.8 × 10−9, rs9298506).
The same group (Citation72) added more cohorts to increase the sample population (5891 cases, 14,181 controls) and identified a total of five IA loci. Two of the loci were previously identified and had strengthened association with IA: SOX17 (8q11.23, OR = 1.28, P = 1.3 × 10−12, rs9298506) and CDKN2BAS1 (9p21, OR = 1.32, P = 1.5 × 10−22, rs1333040). In this study (Citation72), SNP rs10958409 was not associated (8q11.23, OR = 1.17, P = 9.0 × 10−7, rs10958409); however, three new associations with IA were found: CNNM2 (10q24.32, OR = 1.29, P = 1.2 × 10−9, rs12413409), STARD13 (13q13.1, OR = 1.20, P = 2.5 × 10−9, rs9315204), and RBBP8 (18q11.2, OR = 1.22, P = 1.1 × 10−12, rs11661542). It should be noted that some SNPs that reached genome-wide significance, e.g. rs700651 and rs11661542, failed to replicate and are not significant in meta-analyses.
In 2011, Yasuno et al. (Citation73) reported the results of their expanded analysis in which they genotyped 25 SNPs in 14 loci that had previously demonstrated a posterior probability of association of 0.1 to 0.5 in the discovery cohort from their 2010 study (Citation72) in Japanese replication cohorts. After combining the results from the discovery and replication cohorts they found significant association of IA with rs6841581 on chromosome 4q31.23 located 5’ of the endothelin receptor type A (EDNRA) gene (4q31.23, OR = 1.22, P = 2.2 × 10−8). They also reported trends toward association for two additional regions: FGD6 (12q22, OR = 1.16, P = 1.1 × 10−7, rs6538595) and RRBP1 (20p12.1, OR = 1.20, P = 6.9 × 10−7, rs1132274). SNPs with strong or suggestive association with IA identified in the Yasuno et al. GWAS reports (Citation72,Citation73) were also tested for association with blood pressure, an important risk factor for IA (Citation74). The suggestive IA locus at 5q23.2 (rs2287696) in PRDM6 was significantly associated with increased systolic blood pressure (5q23.2, P = 8.13 × 10−7, rs2287696). The authors (Citation74) hypothesized that variants in PRDM6 may contribute to alterations in vascular wall structure that lead to increases in systolic blood pressure and predispose an individual to IA.
Low et al. (Citation54) reported their GWAS in another Japanese cohort (1383 IA-SAH cases, 5484 controls). No SNP met genome-wide significance in the stage 1 analysis. They selected 36 unlinked SNPs with suggestive associations (P < 1 × 10−4) and seven previously reported SNPs associated with IA (nominal P < 0.05 in this study) to genotype in a replication cohort of 1048 cases and 7212 controls. They identified a genome-wide significant SNP in EDNRA (4q31.2, OR = 1.25, P = 9.6 × 10−9, rs6842241) that was more significantly associated with IA than reported in the Yasuno 2011 study (Citation73). EDNRA is an endothelin-1 receptor, which activates a G-protein(s) and their second messenger system. It is located predominantly in the vascular smooth muscle cells of the cerebrovascular system and mediates vasoconstriction and proliferation (Citation54). This SNP is located in a regulatory region of the EDNRA gene on 4q31.22. Functional promoter analysis using an electrophoretic mobility shift assay identified two alleles of rs6841581 (upstream of EDNRA, and in tight LD (r2 > 0.8) with rs6842241) that had different binding affinities to a nuclear protein(s) from human embryonic kidney cells (HEK293). Reporter assays suggested the 5’ flanking region including SNP rs6841581 might function as a transcriptional repressor and be a functional variant conferring IA susceptibility.
The most recent GWAS of IA was performed by Foroud et al. (Citation53). The study design was unique from the other GWAS of IA in that the discovery cohort included two samples: Sample 1 was 388 European cases with a strong family history of IA and 387 controls; Sample 2 was 1095 IA cases without family history of IA and 1286 controls. There were no genome-wide significant SNPs in either discovery cohort. Meta-analysis (1483 cases, 1683 controls) confirmed association with IA for two previously reported SNPs: one SNP in CDKN2BAS1 (9p21, OR = 1.36, P = 3.6 × 10−8, rs6475606) and one SNP nearby SOX17 (8q11.23, OR = 1.25, P = 8.7 × 10−5, rs1072737). In addition, they studied the effect of smoking and these two SNP genotypes on IA risk. Their results suggest that smoking acts multiplicatively with the SNP genotype, and smoking has a greater effect on risk than SNP genotype.
Gene expression studies in intracranial aneurysms
Microarray-based mRNA expression profiling provides an unbiased approach for defining the molecular signature of each organ in disease and health (Citation75). Arteries have distinct mRNA expression profiles from other tissues, and the same artery (e.g. aorta) from different individuals is more similar than other arteries from the same person, demonstrating the need for a careful selection of control tissues for expression studies (Citation75). In all published microarray-based expression studies on IA are summarized. In these nine studies (Citation76–84) genome-wide expression profiles were generated on 80 IA and 77 control tissue samples (6 intracranial control arteries, 34 superficial temporal arteries, 4 arteriovenous malformation feeder (AVMf) cell samples, 33 middle meningeal arteries), as well as on blood samples from 43 IA patients and 18 controls.
Table IV. Microarray-based mRNA and microRNA expression studies on intracranial aneurysms.
One of the studies used mRNA expression profiles as a guide to select positional candidate genes for IA and combine the expression data with DNA linkage study results (Citation76). To define the molecular signature of an intracranial artery, RNA samples isolated from both IA and non-IA intracranial arteries, and two different microarray platforms were used (Citation76). The results revealed that approximately half of the genes represented on the microarray platforms were expressed in the intracranial arteries, and the IA loci identified in DNA linkage studies harbored approximately 800 different genes expressed in intracranial arteries. Pathway analysis using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) bioinformatics tools showed enrichment of several biological pathways including the Notch and MAPK signaling pathways.
Seven of the studies listed in were designed to compare the expression profiles of either IA to non-IA tissue (Citation77–81,Citation83) or unruptured to ruptured IA (Citation82); most studies addressed both of these goals. A recent meta-analysis (Citation85) combined the gene lists reported in five of the microarray-based expression studies on IA (Citation77–81) and produced a list of 507 differentially expressed genes when comparing IA (ruptured or unruptured) to control tissue, 57 of which had altered expression levels in more than two of the five studies. Only seven genes (BCL2, COL1A2, COL3A1, COL5A2, CXCL12, TIMP4, and TNC) were found in more than three studies. It is of interest to note that all but two (BCL2 and CXCL12) of these seven genes are ECM genes.
So far only one study (Citation84) has analyzed peripheral blood samples to detect differences in gene expression between ruptured IA and non-IA controls using blood expression profiles. If successful, this systemic approach would allow assessment of mRNA expression profiles at different stages of the IA development and correlation of the molecular findings to imaging studies. A total of 57 genes were downregulated and 78 genes upregulated in the blood samples taken from patients with ruptured IA. These 135 genes were classified as genes of immune function and hematopoiesis. Quantitative reverse transcription PCR (qRT-PCR) was used to validate the findings on 16 genes in an independent set of IA patients and controls. No correlation was found between the time of blood draw related to the SAH and the gene expression. Interestingly, there was an overlap of 29 genes identified in this study and in those on stroke (Citation86) and AVM (Citation87), when analyzing gene expression profiles of blood samples.
MicroRNAs (miRNA) have been recognized as important modulators of gene expression (Citation88). The first miRNA analysis in IA revealed 18 miRNAs with decreased levels in IA tissue () (Citation83). Follow-up studies included analyzing mRNA expression profiles of IA and control samples to investigate the 681 target genes of the 18 miRNAs. Functional classification of the target genes showed enrichment in ‘migration of phagocytes’ and proliferation of many cell types.
In summary, genome-wide mRNA and miRNA expression studies provide an unbiased approach to understand IA pathophysiology. The use of GO and KEGG terms for a comprehensive functional classification of gene expression, utilizing annotated pathways rather than isolated genes, as well as the interconnectedness of the pathways, gives a more global picture of the pathology involved in IA.
Conclusions and future direction of research
The results summarized above have contributed to improved understanding of the genetics, pathophysiology, and etiology of IA. It is, however, important to understand that all studies have some limitations. One of the biggest challenges common to all human studies (DNA linkage, genetic association, and gene expression studies) is the number of samples in each study and how to re-analyze previously published data and carry out meta-analyses. In order to do this effectively, raw data from the studies should be publicly available. For example, in the case of the IA gene expression studies, six of nine studies have deposited the data to gene expression databases () thereby making a valuable contribution to science.
Another challenge, particularly in the expression studies, is the availability of appropriate control samples. In most IA gene expression studies, samples from IA were compared to samples taken from other arteries as controls. This is problematic, since it is known that arteries at different locations in the body differ from each other in structure, embryologic origin, and disease susceptibility. Using tissues originating from autopsies as controls can also be problematic, since it is possible that post-mortem changes in RNA occur leading to decreased mRNA integrity (Citation89). Nevertheless, degradation is both tissue- and time-dependent and can be minimal in the IA tissue. Yet another challenge is that IA samples in all gene expression studies were taken from IA tissue at the very late stage of the disease, when the aneurysm was large enough for surgical intervention or had already ruptured. Lack of studies from the early stages of human IA development makes it difficult to understand what is important for the initiation of vascular injury leading to IA.
The results from the genome-wide expression studies on IA have provided the foundation for many future studies. The evidence that miRNAs have an important regulatory function in vascular remodeling could be important in discovering targets for a potential therapy to stabilize or slow down IA growth. It would also be important to find the transcription factors responsible for the transcriptional control of gene expression (Citation82). Levels of mRNA and miRNA in the blood of IA patients could provide diagnostic tests by serving as biomarkers. This information could help in identifying markers predictive of rupture; furthermore, use of methods such as laser-capture microdissection to isolate specific cell populations, present in IA wall, would allow for a detailed study of their contribution to IA pathobiology.
The genetic map of IA includes risk loci on almost every human chromosome (). The results summarized in this review were derived from discoveries made using different approaches including DNA linkage (), GWAS (), and candidate gene studies (; ). It should be noted that many IA studies have limited power due to small sample size, and replication is unlikely (Citation64,Citation68). The next steps are then to investigate the molecular mechanisms by which the identified genetic risk factors contribute to the disease. As shown in and , many of the SNPs associated with IA are not in the coding regions of proteins, thus necessitating further studies to find the actual functional variants or defining the regulatory functions of the non-coding SNPs (Citation90,Citation91). These studies are also likely to include epigenetic studies such as investigating the role of DNA methylation (Citation92). Other genetic effects such as various forms of epistasis and modifier genes will also need to be considered (Citation93).
Figure 1. Genetic map of intracranial aneurysm loci. Red vertical lines adjacent to the chromosome ideograms indicate regions identified by DNA linkage studies (), pale red vertical lines indicate support for a region that arises from a family linked to two loci, blue stars indicate locations of SNPs found in genome-wide association studies (), and green round symbols indicate locations of SNPs found in candidate gene association studies (). The ideograms can be obtained from ‘Idiogram Album: Human’ (copyright© 1994 David Adler, University of Washington, Department of Pathology) at http://www.pathology.washington.edu/research/cytopages/idiograms/human/.
IA is considered a complex disease characterized by initiation, growth, and rupture, with multiple genetic and environmental risk factors that might contribute to one or more of the stages. When estimating the contribution of all the identified factors to the risk of a person to develop an IA, the growth of an IA and its rupture, the interactions of the various factors, including environmental risk factors such as smoking, should also be considered. Such analyses require large sample sizes with comprehensive data necessitating collaborative multi-center projects.
The ultimate goal of the molecular and genetic studies is to use the information to design new risk-scoring algorithms to predict an individual's risk for developing an IA or monitor its growth or rupture risk for use in clinical practice. To account for all the genetic variation contributing to IA, hundreds of genetic variants might have to be analyzed. For example, a recent meta-analysis on multiple sclerosis identified 110 non-major histocompatibility complex (MHC) susceptibility loci, which explained only about 20% of the sibling recurrence risk; this number was about 28% if including MHC loci (Citation94).
Whole exome or whole genome sequencing approaches may prove an aid in the identification of genes underlying IA linkage peaks or to identify new genetic risk variants. With the help of the high-throughput next generation sequencing (NGS) approaches it is potentially feasible to identify sequence differences between affected and unaffected individuals. NGS has so far been most useful in finding mutations in highly penetrant Mendelian disorders (Citation95). MacArthur et al. (Citation95) emphasize the difficulty of distinguishing between pathogenic and non-pathogenic variants, concluding with: ‘Objective, systematic and quantitative evaluation of the evidence for pathogenicity and sharing of these evaluations and data amongst research and clinical laboratories will maximize the chances that disease-causing genetic variants are correctly differentiated from the many rare non-pathogenic variants seen in all human genomes.’ Other groups have emphasized similar challenges and included concerns about problems with incomplete coverage and low reproducibility of variant detection (Citation96). Differentiating pathogenic variants from non-pathogenic variants under linkage peaks will remain the vexing problem it has been since the relatives in small families will share large portions (many centi-Morgans, usually many Mbp) of the chromosome identical-by-descent. For a complex disease such as IA case–control studies will require thousands of individuals. In spite of the aforementioned cautions, the ability to define substantially more variation by NGS will provide a far richer information base than we currently have.
Another important goal is to design new therapies based on the information on mechanisms of disease processes to prevent the development or halt the progression of IA. In addition, there is a potential genetic overlap among different types of aneurysm since the same sequence variant on 9p21 contributes to risk for myocardial infarction, AAA, and IA, apparently predisposing to an aberrant vascular injury response (Citation50). In addition, future studies should aim to compare the genetics of different types of aneurysms (e.g. intracranial, extracranial, thoracic, aortic), which may aid in identifying potential treatments.
Declaration of interest: The authors report no conflicts of interest.
References
- Stehbens WE. The pathology of intracranial arterial aneurysms and their complications. In: Fox JL, ed. Intracranial aneurysms. New York: Springer-Verlag; 1983. p. 272–357.
- Nakagawa T, Hashi K. The incidence and treatment of asymptomatic, unruptured cerebral aneurysms. J Neurosurg. 1994;80:217–23.
- Ronkainen A, Miettinen H, Karkola K, Papinaho S, Vanninen R, Puranen M, et al. Risk of harboring an unruptured intracranial aneurysm. Stroke. 1998;29:359–62.
- van Gijn J, Kerr RS, Rinkel GJ. Subarachnoid haemorrhage. Lancet. 2007;369:306–18.
- Vlak MH, Algra A, Brandenburg R, Rinkel GJ. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: a systematic review and meta-analysis. Lancet Neurol. 2011;10:626–36.
- Frösen J, Tulamo R, Paetau A, Laaksamo E, Korja M, Laakso A, et al. Saccular intracranial aneurysm: pathology and mechanisms. Acta Neuropathol. 2012;123:773–86.
- Morita A, Kimura T, Shojima M, Sameshima T, Nishihara T. Unruptured intracranial aneurysms: current perspectives on the origin and natural course, and quest for standards in the management strategy. Neurol Med Chir (Tokyo). 2010;50:777–87.
- Juvela S, Porras M, Poussa K. Natural history of unruptured intracranial aneurysms: probability of and risk factors for aneurysm rupture. J Neurosurg. 2000;93:379–87.
- Wiebers DO, Whisnant JP, Huston J 3rd, Meissner I, Brown RD Jr, Piepgras DG, et al. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet. 2003;362:103–10.
- de Rooij NK, Linn FH, van der Plas JA, Algra A, Rinkel GJ. Incidence of subarachnoid haemorrhage: a systematic review with emphasis on region, age, gender and time trends. J Neurol Neurosurg Psychiatry. 2007;78:1365–72.
- Ingall TJ, Whisnant JP, Wiebers DO, O’Fallon WM. Has there been a decline in subarachnoid hemorrhage mortality? Stroke. 1989;20: 718–24.
- Harmsen P, Tsipogianni A, Wilhelmsen L. Stroke incidence rates were unchanged, while fatality rates declined, during 1971–1987 in Goteborg, Sweden. Stroke. 1992;23:1410–15.
- Johnston SC, Selvin S, Gress DR. The burden, trends, and demographics of mortality from subarachnoid hemorrhage. Neurology. 1998;50: 1413–18.
- Stegmayr B, Eriksson M, Asplund K. Declining mortality from subarachnoid hemorrhage: changes in incidence and case fatality from 1985 through 2000. Stroke. 2004;35:2059–63.
- Graves EJ. Detailed diagnoses and procedures, national hospital discharge survey, 1990. Vital Health Stat 13. 1992:1–225.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain. 2001;124:249–78.
- Edlow JA. Diagnosis of subarachnoid hemorrhage. Neurocrit Care. 2005;2:99–109.
- Ronkainen A, Hernesniemi J, Puranen M, Niemitukia L, Vanninen R, Ryynänen M, et al. Familial intracranial aneurysms. Lancet. 1997; 349:380–4.
- Berman MF, Solomon RA, Mayer SA, Johnston SC, Yung PP. Impact of hospital-related factors on outcome after treatment of cerebral aneurysms. Stroke. 2003;34:2200–7.
- Cross DT 3rd, Tirschwell DL, Clark MA, Tuden D, Derdeyn CP, Moran CJ, et al. Mortality rates after subarachnoid hemorrhage: variations according to hospital case volume in 18 states. J Neurosurg. 2003;99:810–17.
- Choudhari KA, Ramachandran MS, McCarron MO, Kaliaperumal C. Aneurysms unsuitable for endovascular intervention: surgical outcome and management challenges over a 5-year period following International Subarachnoid Haemorrhage Trial (ISAT). Clin Neurol Neurosurg. 2007;109:868–75.
- Mitchell P, Kerr R, Mendelow AD, Molyneux A. Could late rebleeding overturn the superiority of cranial aneurysm coil embolization over clip ligation seen in the International Subarachnoid Aneurysm Trial? J Neurosurg. 2008;108:437–42.
- O’Kelly CJ, Kulkarni AV, Austin PC, Wallace MC, Urbach D. The impact of therapeutic modality on outcomes following repair of ruptured intracranial aneurysms: an administrative data analysis. Clinical article. J Neurosurg. 2010;113:795–801.
- Lovelock CE, Rinkel GJ, Rothwell PM. Time trends in outcome of subarachnoid hemorrhage: population-based study and systematic review. Neurology. 2010;74:1494–501.
- Knekt P, Reunanen A, Aho K, Heliovaara M, Rissanen A, Aromaa A, et al. Risk factors for subarachnoid hemorrhage in a longitudinal population study. J Clin Epidemiol. 1991;44:933–9.
- Kubota M, Yamaura A, Ono J. Prevalence of risk factors for aneurysmal subarachnoid haemorrhage: results of a Japanese multicentre case control study for stroke. Br J Neurosurg. 2001;15:474–8.
- Qureshi AI, Suri MF, Yahia AM, Suarez JI, Guterman LR, Hopkins LN, et al. Risk factors for subarachnoid hemorrhage. Neurosurgery. 2001;49:607–12; discussion 612–13.
- Kernan WN, Viscoli CM, Brass LM, Broderick JP, Brott T, Feldmann E, et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med. 2000;343:1826–32.
- Schievink WI. Genetics and aneurysm formation. Neurosurg Clin N Am. 1998;9:485–95.
- Greving JP, Wermer MJ, Brown RD Jr, Morita A, Juvela S, Yonekura M, et al. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: a pooled analysis of six prospective cohort studies. Lancet Neurol. 2014;13:59–66.
- Wills S, Ronkainen A, van der Voet M, Kuivaniemi H, Helin K, Leinonen E, et al. Familial intracranial aneurysms: an analysis of 346 multiplex Finnish families. Stroke. 2003;34:1370–4.
- Broderick JP, Brown RD Jr, Sauerbeck L, Hornung R, Huston J 3rd, Woo D, et al. Greater rupture risk for familial as compared to sporadic unruptured intracranial aneurysms. Stroke. 2009;40:1952–7.
- Huttunen T, von und zu Fraunberg M, Frosen J, Lehecka M, Tromp G, Helin K, et al. Saccular intracranial aneurysm disease: distribution of site, size, and age suggests different etiologies for aneurysm formation and rupture in 316 familial and 1454 sporadic eastern Finnish patients. Neurosurgery. 2010;66:631–8; discussion 638.
- Nahed BV, Seker A, Guclu B, Ozturk AK, Finberg K, Hawkins AA, et al. Mapping a Mendelian form of intracranial aneurysm to 1p34.3-p36.13. Am J Hum Genet. 2005;76:172–9.
- Ozturk AK, Nahed BV, Bydon M, Bilguvar K, Goksu E, Bademci G, et al. Molecular genetic analysis of two large kindreds with intracranial aneurysms demonstrates linkage to 11q24-25 and 14q23-31. Stroke. 2006;37:1021–7.
- Verlaan DJ, Dube MP, St-Onge J, Noreau A, Roussel J, Satge N, et al. A new locus for autosomal dominant intracranial aneurysm, ANIB4, maps to chromosome 5p15.2-14.3. J Med Genet. 2006;43:e31.
- Ruigrok YM, Wijmenga C, Rinkel GJ, van’t Slot R, Baas F, Wolfs M, et al. Genomewide linkage in a large Dutch family with intracranial aneurysms: replication of 2 loci for intracranial aneurysms to chromosome 1p36.11-p36.13 and Xp22.2-p22.32. Stroke. 2008;39:1096–102.
- Santiago-Sim T, Depalma SR, Ju KL, McDonough B, Seidman CE, Seidman JG, et al. Genomewide linkage in a large Caucasian family maps a new locus for intracranial aneurysms to chromosome 13q. Stroke. 2009;40:S57–60.
- Olson JM, Vongpunsawad S, Kuivaniemi H, Ronkainen A, Hernesniemi J, Ryynanen M, et al. Search for intracranial aneurysm susceptibility gene(s) using Finnish families. BMC Med Genet. 2002;3:7.
- van der Voet M, Olson JM, Kuivaniemi H, Dudek DM, Skunca M, Ronkainen A, et al. Intracranial aneurysms in Finnish families: confirmation of linkage and refinement of the interval to chromosome 19q13.3. Am J Hum Genet. 2004;74:564–71.
- Onda H, Kasuya H, Yoneyama T, Takakura K, Hori T, Takeda J, et al. Genomewide-linkage and haplotype-association studies map intracranial aneurysm to chromosome 7q11. Am J Hum Genet. 2001; 69:804–19.
- Yamada S, Utsunomiya M, Inoue K, Nozaki K, Inoue S, Takenaka K, et al. Genome-wide scan for Japanese familial intracranial aneurysms: linkage to several chromosomal regions. Circulation. 2004;110:3727–33.
- Mineharu Y, Inoue K, Inoue S, Yamada S, Nozaki K, Hashimoto N, et al. Model-based linkage analyses confirm chromosome 19q13.3 as a susceptibility locus for intracranial aneurysm. Stroke. 2007;38:1174–8.
- Foroud T, Sauerbeck L, Brown R, Anderson C, Woo D, Kleindorfer D, et al. Genome screen to detect linkage to intracranial aneurysm susceptibility genes: the Familial Intracranial Aneurysm (FIA) study. Stroke. 2008;39:1434–40.
- Foroud T, Sauerbeck L, Brown R, Anderson C, Woo D, Kleindorfer D, et al. Genome screen in familial intracranial aneurysm. BMC Med Genet. 2009;10:3.
- Alg VS, Sofat R, Houlden H, Werring DJ. Genetic risk factors for intracranial aneurysms: a meta-analysis in more than 116,000 individuals. Neurology. 2013;80:2154–65.
- Kumar VA, Caves JM, Haller CA, Dai E, Liu L, Grainger S, et al. Acellular vascular grafts generated from collagen and elastin analogs. Acta Biomater. 2013;9:8067–74.
- Breimer LH. Genetics of aneurysmal disease: the dogs that did not bark. Angiology. 2010;61:233–7.
- Penn DL, Witte SR, Komotar RJ, Sander Connolly E Jr. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J Clin Neurosci. 2014;21:28–32.
- Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–24.
- Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–12.
- Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler Thromb Vasc Biol. 2012;32:196–206.
- Foroud T, Koller DL, Lai D, Sauerbeck L, Anderson C, Ko N, et al. Genome-wide association study of intracranial aneurysms confirms role of Anril and SOX17 in disease risk. Stroke. 2012;43:2846–52.
- Low SK, Takahashi A, Cha PC, Zembutsu H, Kamatani N, Kubo M, et al. Genome-wide association study for intracranial aneurysm in the Japanese population identifies three candidate susceptible loci and a functional genetic variant at EDNRA. Hum Mol Genet. 2012;21:2102–10.
- Tromp G, Kuivaniemi H, Hinterseher I, Carey DJ. Novel genetic mechanisms for aortic aneurysms. Curr Atheroscler Rep. 2010;12:259–66.
- Olsson S, Csajbok LZ, Jood K, Nylen K, Nellgard B, Jern C. Association between genetic variation on chromosome 9p21 and aneurysmal subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2011;82:384–8.
- Chen Z, Ma J, Cen Y, Liu Y, You C. The angiotensin converting enzyme insertion/deletion polymorphism and intracranial aneurysm: a meta-analysis of case-control studies. Neurol India. 2013;61:293–9.
- Weinsheimer S, Goddard KA, Parrado AR, Lu Q, Sinha M, Lebedeva ER, et al. Association of kallikrein gene polymorphisms with intracranial aneurysms. Stroke. 2007;38:2670–6.
- Akagawa H, Tajima A, Sakamoto Y, Krischek B, Yoneyama T, Kasuya H, et al. A haplotype spanning two genes, ELN and LIMK1, decreases their transcripts and confers susceptibility to intracranial aneurysms. Hum Mol Genet. 2006;15:1722–34.
- Low SK, Zembutsu H, Takahashi A, Kamatani N, Cha PC, Hosono N, et al. Impact of LIMK1, MMP2 and TNF-alpha variations for intracranial aneurysm in Japanese population. J Hum Genet. 2011;56:211–16.
- Inoue K, Mineharu Y, Inoue S, Yamada S, Matsuda F, Nozaki K, et al. Search on chromosome 17 centromere reveals TNFRSF13B as a susceptibility gene for intracranial aneurysm: a preliminary study. Circulation. 2006;113:2002–10.
- Krischek B, Tajima A, Akagawa H, Narita A, Ruigrok Y, Rinkel G, et al. Association of the Jun dimerization protein 2 gene with intracranial aneurysms in Japanese and Korean cohorts as compared to a Dutch cohort. Neuroscience. 2010;169:339–43.
- Semmler A, Linnebank M, Krex D, Gotz A, Moskau S, Ziegler A, et al. Polymorphisms of homocysteine metabolism are associated with intracranial aneurysms. Cerebrovasc Dis. 2008;26:425–9.
- Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–9.
- Panagiotou OA, Evangelou E, Ioannidis JP. Genome-wide significant associations for variants with minor allele frequency of 5% or less—an overview: A HuGE review. Am J Epidemiol. 2010;172:869–89.
- Siontis KC, Patsopoulos NA, Ioannidis JP. Replication of past candidate loci for common diseases and phenotypes in 100 genome-wide association studies. Eur J Hum Genet. 2010;18:832–7.
- Panagiotou OA, Willer CJ, Hirschhorn JN, Ioannidis JP. The power of meta-analysis in genome-wide association studies. Annu Rev Genomics Hum Genet. 2013;14:441–65.
- Ioannidis JP, Trikalinos TA, Ntzani EE, Contopoulos-Ioannidis DG. Genetic associations in large versus small studies: an empirical assessment. Lancet. 2003;361:567–71.
- Manolio TA. Bringing genome-wide association findings into clinical use. Nat Rev Genet. 2013;14:549–58.
- Hussain I, Duffis EJ, Gandhi CD, Prestigiacomo CJ. Genome-wide association studies of intracranial aneurysms: an update. Stroke. 2013;44:2670–5.
- Bilguvar K, Yasuno K, Niemela M, Ruigrok YM, von und zu Fraunberg M, van Duijn CM, et al. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet. 2008; 40:1472–7.
- Yasuno K, Bilguvar K, Bijlenga P, Low SK, Krischek B, Auburger G, et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet. 2010;42:420–5.
- Yasuno K, Bakircioglu M, Low SK, Bilguvar K, Gaal E, Ruigrok YM, et al. Common variant near the endothelin receptor type A (EDNRA) gene is associated with intracranial aneurysm risk. Proc Natl Acad Sci U S A. 2011;108:19707–12.
- Gaal EI, Salo P, Kristiansson K, Rehnstrom K, Kettunen J, Sarin AP, et al. Intracranial aneurysm risk locus 5q23.2 is associated with elevated systolic blood pressure. PLoS Genet. 2012;8:e1002563.
- Tromp G, Kuivaniemi H. Developments in genomics to improve understanding, diagnosis and management of aneurysms and peripheral artery disease. Eur J Vasc Endovasc Surg. 2009;38:676–82.
- Weinsheimer S, Lenk GM, van der Voet M, Land S, Ronkainen A, Alafuzoff I, et al. Integration of expression profiles and genetic mapping data to identify candidate genes in intracranial aneurysm. Physiol Genomics. 2007;32:45–57.
- Krischek B, Kasuya H, Tajima A, Akagawa H, Sasaki T, Yoneyama T, et al. Network-based gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience. 2008;154:1398–407.
- Li L, Yang X, Jiang F, Dusting GJ, Wu Z. Transcriptome-wide characterization of gene expression associated with unruptured intracranial aneurysms. Eur Neurol. 2009;62:330–7.
- Shi C, Awad IA, Jafari N, Lin S, Du P, Hage ZA, et al. Genomics of human intracranial aneurysm wall. Stroke. 2009;40:1252–61.
- Marchese E, Vignati A, Albanese A, Nucci CG, Sabatino G, Tirpakova B, et al. Comparative evaluation of genome-wide gene expression profiles in ruptured and unruptured human intracranial aneurysms. J Biol Regul Homeost Agents. 2010;24:185–95.
- Pera J, Korostynski M, Krzyszkowski T, Czopek J, Slowik A, Dziedzic T, et al. Gene expression profiles in human ruptured and unruptured intracranial aneurysms: what is the role of inflammation? Stroke. 2010;41:224–31.
- Kurki MI, Hakkinen SK, Frosen J, Tulamo R, von und zu Fraunberg M, Wong G, et al. Upregulated signaling pathways in ruptured human saccular intracranial aneurysm wall: an emerging regulative role of Toll-like receptor signaling and nuclear factor-kappaB, hypoxia-inducible factor-1A, and ETS transcription factors. Neurosurgery. 2011;68: 1667–75; discussion 1675–6.
- Jiang Y, Zhang M, He H, Chen J, Zeng H, Li J, et al. MicroRNA/mRNA profiling and regulatory network of intracranial aneurysm. BMC Med Genomics. 2013;6:36.
- Pera J, Korostynski M, Golda S, Piechota M, Dzbek J, Krzyszkowski T, et al. Gene expression profiling of blood in ruptured intracranial aneurysms: in search of biomarkers. J Cereb Blood Flow Metab. 2013;33:1025–31.
- Roder C, Kasuya H, Harati A, Tatagiba M, Inoue I, Krischek B. Meta-analysis of microarray gene expression studies on intracranial aneurysms. Neuroscience. 2012;201:105–13.
- Tang Y, Xu H, Du X, Lit L, Walker W, Lu A, et al. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: a microarray study. J Cereb Blood Flow Metab. 2006;26:1089–102.
- Weinsheimer SM, Xu H, Achrol AS, Stamova B, McCulloch CE, Pawlikowska L, et al. Gene expression profiling of blood in brain arteriovenous malformation patients. Transl Stroke Res. 2011;2: 575–87.
- van Rooij E. The art of microRNA research. Circ Res. 2011;108: 219–34.
- Kuliwaba JS, Fazzalari NL, Findlay DM. Stability of RNA isolated from human trabecular bone at post-mortem and surgery. Biochim Biophys Acta. 2005;1740:1–11.
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5.
- Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–97.
- Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, et al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet. 2013;93:876–90.
- Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132:1077–130.
- Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013; 45:1353–60.
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–76.
- Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, et al. Clinical interpretation and implications of whole- genome sequencing. JAMA. 2014;311:1035–45.
- Farnham JM, Camp NJ, Neuhausen SL, Tsuruda J, Parker D, MacDonald J, et al. Confirmation of chromosome 7q11 locus for predisposition to intracranial aneurysm. Hum Genet. 2004;114:250–5.
- Kim CJ, Park SS, Lee HS, Chung HJ, Choi W, Chung JH, et al. Identification of an autosomal dominant locus for intracranial aneurysm through a model-based family collection in a geographically limited area. J Hum Genet. 2011;56:464–6.
- Wei L, Gao YJ, Wei SP, Zhang YF, Zhang WF, Jiang JX, et al. Transcriptome network-based method to identify genes associated with unruptured intracranial aneurysms. Genet Mol Res. 2013;12: 3263–73.
- Chen L, Wan JQ, Zhou JP, Fan YL, Jiang JY. Gene expression analysis of ruptured and un-ruptured saccular intracranial aneurysm. Eur Rev Med Pharmacol Sci. 2013;17:1374–81.