
Abstract
Background. Finnish type of hereditary gelsolin amyloidosis (FGA) is one of the most common diseases of Finnish disease heritage. Existing FGA knowledge is based only on smaller patient series, so our aim was to elucidate the natural course of the disease in a comprehensive sample of patients and to build up a national FGA patient registry.
Methods. An inquiry about the known and suspected signs of FGA, sent to the members of Finnish Amyloidosis Association, telephone contacts, and hospital records were utilized to create the registry.
Results. A total of 227 patients were entered to the database. The first symptom was ophthalmological for 167 patients (73.6%) at the mean age of 39 years. Corneal lattice dystrophy (CLD) was reported at the mean age of 43 years. Impaired vision, polyneuropathy, facial nerve paresis, and cutis laxa appeared on average between 52 and 57 years. Carpal tunnel syndrome (CTS) was reported by 86 patients (37.9%). Nine patients (4.0%) had a pacemaker, and 12 (6.1%) had cardiomyopathy.
Conclusions. The first symptom was ophthalmological in most cases. Except for CLD no prominent difference in the age of appearance was found between the major symptoms. CTS, cardiac pacemakers, and cardiomyopathy were remarkably more common compared to the general population.
In the clinical course of Finnish gelsolin amyloidosis eye problems appear at first, followed later on by impaired vision, polyneuropathy, facial nerve paresis, and cutis laxa which develop quite simultaneously.
When it comes to possible treatment options in the future it is important to remember that a significant risk of carpal tunnel syndrome and also cardiological manifestations are associated with this disease.
The National Finnish Gelsolin Amyloidosis Patient Registry, FIN-GAR, was established for this study and other upcoming studies in the future, considering also possible specific treatment options.
Introduction
Finnish type of hereditary gelsolin amyloidosis (FGA), also known as Meretoja syndrome (Citation1) or AGel amyloidosis, is one of the most common diseases of Finnish disease heritage (Citation2). Estimations from 400 patients to 1,000 gene defect carriers in Finland have been presented (Citation2). Moreover, the same disease caused by the same mutation at the same site has been reported from many other European countries and also from North America as well as from Asia, Mexico, and India (Citation2–4). It is inherited autosomally dominantly and has a penetrance of 100% (Citation2). FGA is caused by a mutation in the gene coding for gelsolin located on chromosome 9 (Citation5), where guanine is replaced by adenosine (640G→A), earlier known as c.G654G> A (Citation6,Citation7). The point mutation causes defective folding of the gelsolin protein, with its accumulation as gelsolin amyloid in various tissues. Another, less common, point mutation in the same place, guanine being replaced by thymine, has been reported from Denmark, Czechoslovakia, France, and Brazil (Citation2). Also, newly discovered gelsolin mutations (580G→A and 633C→A) have been described to cause renal amyloidosis (Citation8,Citation9). All molecular genetically characterized FGA patients in Finland have had the same major gelsolin gene mutation.
The signs and symptoms of FGA are multifaceted and are thought to depend on the tissue in which the gelsolin amyloid accumulates. Furthermore, the severity of the symptoms varies remarkably between patients for unknown reasons. Symptoms commonly begin to appear by the fortieth birthday. The most common symptoms are known to emerge in eyes, peripheral nerves, and skin. The first symptoms are typically eye dryness, irritability, and sensitivity to light. The first finding is normally corneal lattice dystrophy (CLD) (Citation2) diagnosed by an ophthalmologist. The diagnosis of CLD can be made without any preceding symptoms. With increasing age, CLD and eye ulceration can lead in impaired vision (Citation10).
Skin problems are often first seen as drooping eyelids. With advancing disease the loosening of facial skin, a phenomenon called cutis laxa, leads in some cases to a mask-like face with diminished expression (Citation11). The advancing paresis of facial nerves, a form of peripheral neuropathy either on one or later on both sides (Citation12), also contributes to facial problems. Other peripheral nerve problems are polyneuropathy and carpal tunnel syndrome (CTS). Polyneuropathy is predominantly reported as sensory and causes numbness and paresthesia in extremities that can later cause ataxia-like symptoms in form of walking difficulties and clumsiness in hands. Skin dryness and thinness are reported to increase with advancing age, causing abrasions easily and scarring. FGA patients are possibly also more prone to get bruises.
Cardiological findings including arrhythmias and atrioventricular blocks of different grades have been described. Proteinuria and serious renal problems in homozygote patients have even been reported to lead to the requirement of renal transplantation. However, these features are not fully characterized. So far it is thought that FGA has only a minor effect on life span (Citation13). Moreover, because of the wide variety of symptoms and signs, it has a remarkable effect on the quality of life.
Earlier publications have either included smaller series of patients or case reports, making it difficult to make solid conclusions about the clinical course of the disease. Moreover, there are still open questions regarding the order and age of appearance of symptoms. Therefore, we established a national registry for FGA, namely FIN-GAR (The Finnish Gelsolin Amyloidosis Patient Registry). Our aim was to collect comprehensive information of the symptoms and the course of the disease from as many patients as possible. To our knowledge, this is the largest reported sample of FGA patients so far. With detailed information from the patients, the natural course of FGA can be described more accurately.
Patients and methods
This study was approved by the local Ethics Committee of Helsinki University Hospital (HUCH), and permission to establish the FIN-GAR and collect data from patient records was granted by HUCH. The FIN-GAR was created at a protected net site at HUCH, to which only the authors have access. The initial questionnaire was sent to all FGA patients who were members of the Finnish Amyloidosis Association (SAMY, with 228 members in 2013; www.suomenamyloidoosiyhdistys.fi). SAMY took care of the initial correspondence. The questionnaire was designed to capture known and also suspected features of FGA and the year of their appearance. Other medical conditions, operations, and medications were also asked about. The patients signed an informed consent and replied to the questions of the questionnaire. These index patients were also asked to contact their relatives known to have FGA but who were not members of SAMY and ask for permission to contact them in order to recruit them for the study. In addition to this, 16 deceased patients were included in the registry, as they had been part of earlier FGA studies (Citation2). Furthermore, our aim in the future is to characterize the late phases of this disease.
In case of any remaining open questions, the patients were called and asked for more details. A total of 184 out of 211 living patients (87%) required a phone call to resolve open issues. When applicable, also hospital patient records were used to complement the data. The hospital patient records were available from all deceased patients. After all patient information had been entered to the registry it was cleaned and coded.
Statistical methods
Distributions of the continuous variables were studied and tested for normality. Univariate analyses were performed with a non-parametric Mann–Whitney U test. Dichotomous variables were compared with Fisher's exact test. Then the cumulative prevalence of the symptoms according to the patient's age at the time of symptom development () was outlined. These figures were constructed based on the data from the patients with the symptom of interest. IBM SPSS 19.0 (SPSS, Inc., an IBM company, New York, USA) was used for the analyses.
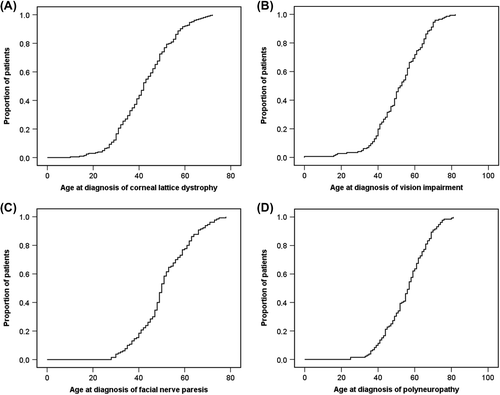
Figure 1. The cumulative prevalence of symptom according to the patient's age at the time of symptom development. A: Corneal lattice dystrophy. B: Vision impairment. C: Facial nerve paresis. D: Polyneuropathy.
Results
Between October 2013 and August 2014, a total of 227 patients with FGA were entered into the FIN-GAR, and their mean age was 61.5 years at the data lock. The baseline information is given in . The mean age of death for 16 deceased patients included in the database was 77.4 years. Their cause of death, however, was not ascertained. In the entire series the original diagnosis was based on ophthalmological examination (186 patients, 81.9%), genetic testing (32 patients, 14.1%), and skin biopsy (4 patients, 1.8%); 5 patients reported that their diagnosis was made using some other method. A total of 47 patients had had the diagnosis before any symptoms, and these patients were diagnosed either by an ophthalmologist or a gene test. Nine of them were diagnosed by the age of 25 years (out of which one at the age of 10), 27 between 26 and 39 years, and 11 were over 40 years old. The diagnosis of a 10-year-old child was based on CLD, and the course of her disease has otherwise been mild; she is now 39 years old. FGA being a hereditary disease, 224 patients reported to have relatives with FGA. Only two patients, both of whom were adopted, did not know any relatives with FGA, and the data were missing for one patient.
Table I. Baseline information.
As CLD is considered practically prerequisite and with positive family history sufficient for the diagnosis of FGA, the initial diagnosis was made by an ophthalmologist in the majority (186 patients), including 47 individuals who were diagnosed prior to their first symptoms. Genetic testing, including the previously mentioned 32 patients with original DNA diagnosis, had been performed in altogether 123 (54.2%) patients. To date, all known genetically tested FGA patients have carried the same 640G→A mutation in the gene coding for gelsolin (2, Sari Kiuru-Enari personal communication). In the skin biopsies of patients with histologically verified FGA (n = 4) positive identification of gelsolin amyloid was performed by Congo red staining combined with immunohistochemistry with an antibody raised against variant gelsolin amyloid subunits (Citation14).
By the age of 39 years 50% of the patients had developed their first symptom. The mean age at the first symptom was 38 years in women and 42 years in men. The first symptom was ophthalmological, typically dry or irritable eyes, in 124 (81.6%) women and 43 (61.4%) men. The other first symptoms were neurological for 17 females (11.2%) and 11 men (15.7%) and dermatological for 10 females (6.6%) and 15 men (21.4%) ().
Table II. First symptoms.
The prevalences of the different symptoms are given in . CLD was diagnosed at the mean age of 42.6 years and reported in 210 (94.2%) patients. Half of the patients with CLD were diagnosed within 42 years of age (). This condition was not reported in 13 patients, and information was not available for 4 patients. Corneal ulcer (one or more) was reported in 107 (51.4%) and impaired vision in 161 (72.2%) patients. Dry eyes and drooping eyelids were very common symptoms as 207 (95.4%) and 186 (84.5%) patients suffered from them, respectively. Operations were performed frequently due to ophthalmological manifestations. Half of the patients with cataracts were diagnosed by the age of 67 years. Eyelid operations (one or more) were performed for 115 patients (68.5%). A total of 177 patients (83.9%) reported visiting an ophthalmologist annually.
Table III. The prevalence of individual symptoms and signs.
Table IV. The age of appearance of signs and symptoms.
Peripheral somatic nerve symptoms (e.g. numbness/tingling or weakness/clumsiness of feet or hands) and facial nerve weakness were the most common neurological symptoms (69.5% and 69.6%, respectively) assessed by the questionnaire. The type of polyneuropathy, based on data reported by patients or in some cases available from hospital medical records, was known in 150 patients. According to these, polyneuropathy was sensory for 92 (41.3%), sensori-motor for 53 (23.8%), and motor for 5 (2.2%) patients. Myokymias were reported in 113 (50.2%) patients. Operations for carpal tunnel syndrome (CTS) (one or more) were performed for 55 patients (64% of those with diagnosed CTS).
Cutis laxa, a phenomenon of loose, hanging skin seen especially on the face, was reported in 170 patients (76.9%). Facial surgery (one or many) was performed for 96 patients (44.9%). Other dominating skin symptoms were abnormal bruising reported in 151 (67.4%) and dry skin reported in 157 (83.1%) patients.
Arrhythmias were seen in 74 patients (32.7%). Cardiomyopathy was reported in 12 patients (6.1%) and a pacemaker in 9 patients (3.9%). Proteinuria was seen in 32 patients (14.3%), and renal transplantation surgery was performed for 5 patients (2.2%). Sleep apnea was reported in 15 patients (6.7%).
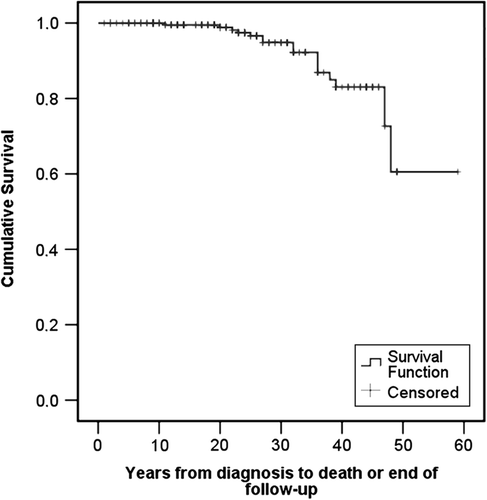
The main symptoms (CLD, impairment of vision, polyneuropathy, and facial paresis) seem to develop at the same rate, as seen in . The median and interquartile ranges for each of the main symptoms are listed in . The overall survival from diagnosis to death or end of follow-up is illustrated in .
Figure 2. The overall survival curve from diagnosis to death or end of follow-up.
Discussion
In this study, we improve the existing knowledge of the course of Finnish gelsolin amyloidosis (FGA). The data are based on a nationwide survey analysis of 227 patients. FGA is one of the known hereditary amyloid diseases. The most common disease in this group is transthyretin (TTR) amyloidosis. Contrary to FGA, in which the hypothesis is that it does not strongly affect life span, TTR amyloidosis leads to death, the typical time between onset and death being 5 to 10 years (Citation15). There has been extensive research in TTR amyloidosis which has led to new treatment options (Citation16). There are also new possible treatment options for FGA upcoming (Citation17,Citation18), which is encouraging and speaks for the necessity of a patient registry such as FIN-GAR.
The number of FGA patients in Finland is unknown, but estimations from 400 symptomatic patients to 1,000 gene defect carriers have been presented (Citation2). Guided by the earlier estimations, we here approximate that our present sample represents on average more than one-third of the total number of patients, giving a sufficient sample size for drawing conclusions. Two-thirds (68.7%) of the patients in the database were females. It is known that women are more interested in participating in medical trials, probably explaining this finding. The mean age of diagnosis in our study was 41 years. For most of the patients the diagnosis could have been made much earlier since CLD, considered a prerequisite for the clinical diagnosis, starts to form in the third decade (Citation13,Citation19). However, the benefit of a presymptomatic diagnosis is small since only symptomatic treatment options are available at present. It is unlikely that all 47 patients diagnosed before symptoms really did not have any symptoms, as the disease manifests typically during the fourth decade. Probably the patients diagnosed by the age of 25 were asymptomatic and maybe also some of those diagnosed between 26 and 39 years. It is likely that the older patients just did not remember when the first symptoms appeared, in particular as they typically develop insidiously.
Corneal ulcers were a common finding in this study (51.4%), and the prevalence seems to be higher than in a previous study where one-third suffered from them (Citation11). Repetitive corneal erosions together with subepithelial amyloid accumulation in the cornea lead to loss of vision and possibly even to corneal blindness, often requiring corneal transplantation (Citation20). Although many patients (72.2%) reported impaired vision, it is noteworthy that the mean age of the patients was 61.5 years so there are obviously confounding factors influencing this finding. In one smaller series impaired vision appeared from the age of 60 years (Citation2). Glaucoma was quite usual (21.2%) among the patients, possibly caused by the accumulation of amyloid (Citation21,Citation22). In this study, the prevalence of glaucoma was slightly lower than in a previous report in which it was 26% among the patients over 40 years of age (Citation21). Cataracts were found in 42.7% of the patients, which was more common than expected. It has been suggested to be associated with FGA (Citation2,Citation10), but there has been no estimate of its frequency. The prevalence of cataracts in the Finnish population over 65 years is approximately 33%. Since in this study the mean age was 61.5 years, cataracts seem to be more common among FGA patients than in the general population. There has been a suggestion that accumulation of amyloid in the ciliary body or alterations in the aqueous humor may have an effect (Citation10).
According to the nature of the disease a majority (95.4%) reported suffering from dry eyes, which has been noted to be very common in FGA (Citation2,Citation10). In this study the prevalence of blepharochalasis, or lid drooping, was 84.5%. In other studies it has been reported to appear in 90% (Citation11) or 94.6% (Citation21). However, taking into account the age range of this study and the fact that this condition gets worse with advancing age, the result complies fairly well with other studies.
Polyneuropathy was one of the main symptoms, as it was reported for almost 70% of the patients. It has been related to gelsolin amyloid deposition in nerve tissues of FGA patients (Citation23). It has been described to appear during the fifth decade of life (Citation2,Citation11), and this could also be seen in this study. As many as 69.6% of the patients suffered from facial nerve paresis, which is one of the major features in FGA and is common from the fifth decade (Citation19). However, it is noteworthy that facial nerve weakness can be difficult to notice at an early stage, and probably minor findings could be detected at a younger age with careful examination. Although almost one-fourth of the patients reported impaired hearing, further studies were necessary to estimate the role of FGA in this finding. Obviously aging or other confounding factors make any conclusions impossible in a survey study. Hearing problems have been noticed with FGA (Citation2,Citation11).
This study showed that dysarthria is not very common but noteworthy, however, as about one-fourth of the patients suffered from it. Dysarthria reflects facial and hypoglossal nerve paresis in FGA (Citation11). About one-third of the patients reported balance problems and/or clumsiness, which may indicate sensory ataxia observed earlier in FGA (Citation24). However, no exact conclusions can be made without clinical examinations, especially when regarding the frequency of sensory neuropathy among FGA patients. Therefore no exact conclusions of its prevalence can be made without clinical examination, especially regarding the frequency of sensory neuropathy among FGA patients. CTS prevalence was surprisingly high as nearly 40% suffered from it. Although electrophysiological findings compatible with CTS have been reported to be as common as 60%, the clinical findings have been scanty (Citation25). CTS was reported in only 1 out of 30 patients in a previous study (Citation11). According to our findings and in contrast to the previous study, it seems that CTS is one of the most common symptoms of FGA and more common than previously assumed.
The prevalence of cutis laxa (76.9%) was in line with earlier reports (Citation26). It is one of the major signs of the disease (Citation26), requiring usually plastic surgery that often needs to be repeated, as the results do not last for a long time (Citation12). It is also a significant cosmetic handicap, giving a picture of early facial aging. Drooping eyelids may impinge on vision by making the visual field smaller. Cutis laxa can also cause or, with facial and hypoglossal neuropathy, aggravate problems with drooling, speech, or eating. As half of the facial surgeries were performed by the age of 55, it seems that cutis laxa becomes clinically significant during and after the fifth decade, which supports other estimations (Citation1,Citation2). Abnormal bruising was also a very common symptom (67.4%), as reported also previously in the literature (Citation11). This could be due to amyloid angiopathy (Citation27) or altered platelet function (Citation28).
Proteinuria remains a quite rare symptom (reported for 32 patients, 14.3%) among heterozygotes, and there was only one known homozygote patient in this study. Despite the rarity of renal problems, a renal transplantation was performed for five patients, among them the one patient who was homozygous for the gelsolin 640G→A mutation. This gives more accurate new and important information about this serious risk. One-third of patients reported swelling which could also be due to kidney problems.
Arrhythmias were reported for 33% of patients, therefore being quite prominent in our material and complying well with a previous finding of quite high prevalence of arrhythmias in FGA patients (Citation29). Arrhythmias are naturally a very common phenomenon in the general population. However, nine patients (4.0%) had a cardiac pacemaker, whereas the prevalence of pacemakers in the adult population was shown to be 0.47% recently (Citation30). This could be a sign of a gelsolin-related pathological background for at least some of the arrhythmias. A total of 6.1% of patients reported cardiomyopathy. However, the type of cardiomyopathy was not asked. Their mean age was 66 years, ranging from 48 to 86 years. A heart transplant surgery has been performed for one Finnish FGA patient due to cardiomyopathy (Citation31). In Finland it is estimated that the prevalence of dilating and hypertrophic cardiomyopathies is 0.04% and 0.2%, respectively. Therefore cardiomyopathy seems to be more prevalent among the patients with FGA.
As we analyzed the order of appearance of the major symptoms, it seemed that many of them appeared slowly and simultaneously (, ). Many symptoms progressed so slowly that for a patient it may be difficult to notice and remember the exact time of the first appearance of an individual symptom. Excluding CLD, which appeared earlier, there was no significant difference in the order of appearance of other symptoms. This could be due to the fact that amyloid accumulates systemically at a constant rate, consequently giving rise to different symptoms quite simultaneously. Also the pre-amyloid fibrils formed during amyloidogenesis (Citation32) as well as the variant gelsolin protein could have pathogenic role (Citation33).
This study is the largest FGA patient series so far. Even though there might be some uncertainties due to the fact that the information was derived from the patient questionnaire and telephone contacts, the study gives an extensive description of the course of this disease. More accurate information was received by calling most of the patients to resolve open questions. We also used medical records, which were available for about 20% of the patients. CLD was not reported for 13 patients. The most probable reason for this was that the particular term of CLD and also the term ‘eye degeneration’ was unknown to them. All four patients with missing data on CLD had a genetic verification of the disease. Previous studies have been focusing more on describing particular symptoms and pathophysiology behind them with markedly smaller patient numbers. The larger patient sample, considering that FGA is a rare disease, gives more reliable results about the prevalence of different symptoms.
In this study we have focused on current clinical data in FIN-GAR. However, our aim in the future is to complement the overall picture through a study focusing on the late phases of FGA. As there are possible upcoming treatment options for FGA in the future (Citation17,Citation18), it is necessary to have knowledge of the natural history of FGA, and the patient registry could be utilized in a clinical trial later. However, in expectance of specific future therapeutic options, it is important also to search for reliable biomarkers in order to plan better and more individualized follow-up for the patients.
Acknowledgements
We thank the Finnish Amyloidosis Association SAMY for their cooperation.
Funding: Financial support was received from Helsinki University Hospital Research Foundation (TYH 2013315 and 2014111) and Finska Läkaresällskapet.
Declaration of interest: The authors report no conflicts of interest.
References
- Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1:314–24.
- Kiuru-Enari S, Haltia M. Hereditary gelsolin amyloidosis. In: Said G, Krarup C, editors. Peripheral nerve disorders, volume 115: Handbook of clinical neurology. Amsterdam: Elsevier; 2013. p. 659–81.
- Maramattom BV, Chickabasaviah YT. A new Indian family affected by gelsolin amyloidosis. Neurol India. 2013;61:673–5.
- Gonzalez-Rodriguez J, Ramirez-Miranda A, Hernandez-Da Mota SE, Zenteno JC. TGFBI, CHST6, and GSN gene analysis in Mexican patients with stromal corneal dystrophies. Graefes Arch Clin Exp Ophthalmol. 2014;252:1267–72.
- Kwiatkowski DJ, Westbrook CA, Burns GAP, Morton C. Localization of gelsolin proximal to ABL on chromosome 9. Am J Hum Genet. 1988;42:565–72.
- Haltia M, Levy E, Meretoja J, Fernandez-Madrid I, Koivunen O, Frangione B. Gelsolin gene mutation – at codon 187 – in familial amyloidosis, Finnish: DNA-diagnostic assay. Am J Med Genet. 1992;42:357–9.
- Maury CPJ, Kere J, Tolvanen R, de la Chapelle A. Finnish hereditary amyloidosis is caused by a single nucleotide substitution in the gelsolin gene. FEBS Lett. 1990;276:75–7.
- Efebera YA, Sturm A, Baack EC, Hofmeister CC, Satoskar A, Nadasdy T, et al. Novel gelsolin variant as the cause of nephrotic syndrome and renal amyloidosis in a large kindred. Amyloid. 2014;21:110–12.
- Sethi S, Theis JD, Quint P, Maierhofer W, Kurtin PJ, Dogan A, et al. Renal amyloidosis associated with a novel sequence variant of gelsolin. Am J Kidney Dis. 2013;61:161–6.
- Carrwik C, Stenevi U. Lattice corneal dystrophy, gelsolin type (Meretoja's syndrome). Acta Opthalmol (Oxf). 2009;87:813–19.
- Kiuru S. Familial amyloidosis of the Finnish type (FAF): a clinical study of 30 patients. Acta Neurol Scand. 1992;86:346–53.
- Pihlamaa T, Rautio J, Kiuru-Enari S, Suominen S. Gelsolin amyloidosis as a cause of early aging and progressive bilateral facial paralysis. Plast Reconstr Surg. 2011;127:2342–51.
- Meretoja J. Genetic aspects of familial amyloidosis with corneal lattice dystrophy and cranial neuropathy. Clin Genet. 1973;4:173–85.
- Haltia M, Ghiso J, Prelli F, Gallo G, Kiuru S, Somer H, et al. Amyloid in familial amyloidosis, Finnish type, is antigenically and structurally related to gelsolin. Am J Pathol. 1990;136:1223–8.
- Liepnieks J, Zhang L, Benson M. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75:324–7.
- Hund E. Familial amyloidotic polyneuropathy: current and emerging treatment options for transthyretin-mediated amyloidosis. Appl Clin Genet. 2012;5:37–41.
- Van Overbeke W, Verhelle A, Everaert I, Zwaenepoel O, Vandekerckhove J, Cuvelier C, et al. Chaperone nanobodies protect gelsolin against MT1-MMP degradation and alleviate amyloid burden in the gelsolin amyloidosis mouse model. Mol Ther. 2014;22:1768–78.
- Van Overbeke W, Wongsantichon J, Everaert I, Verhelle A, Zwaenepoel O, Loonchanta A, et al. An ER-directed gelsolin nanobody targets the first step in amyloid formation in a gelsolin amyloidosis mouse model. Hum Mol Genet. 2015;24:2492–507.
- Meretoja J. Inherited systemic amyloidosis with lattice corneal dystrophy [dissertation]. Helsinki: University of Helsinki; 1973.
- Mattila J, Krootila K, Kivela T, Holopainen J. Penetrating keratoplasty for corneal amyloidosis in familial amyloidosis, Finnish type. Ophthalmology. 2015;122:457–63.
- Meretoja J. Comparative histopathological and clinical findings in eyes with lattice corneal dystrophy of two different types. Ophtalmologica. 1972;165:15–37.
- Kivela T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja's syndrome. Invest Ophthalmol Vis Sci. 1994;35:3759–69.
- Kiuru-Enari S, Somer H, Seppäläinen A-M, Notkola I-L, Haltia M. Neuromuscular pathology in hereditary gelsolin amyloidosis. J Neuropathol Exp Neurol. 2002;61:565–71.
- Tanskanen M, Paetau A, Salonen O, Salmi T, Lamminen A, Lindsberg P, et al. Severe ataxia with neuropathy in hereditary gelsolin amyloidosis: a case report. Amyloid. 2007;14:89–95.
- Kiuru S, Seppäläinen A-M. Neuropathy in familial amyloidosis, Finnish type (FAF): electrophysiological studies. Muscle Nerve. 1994;17: 299–304.
- Kiuru-Enari S, Keski-Oja J, Haltia M. Cutis laxa in hereditary gelsolin amyloidosis. Br J Dermatol. 2005;152:250–7.
- Kiuru S, Salonen O, Haltia M. Gelsolin-related spinal and cerebral amyloid angiopathy. Ann Neurol. 1999;45:305–11.
- Kiuru S, Javela K, Somer H, Kekomaki R. Altered platelet shape change in hereditary gelsolin Asp187Asn-related amyloidosis. Thromb Haemost. 2000;83:491–5.
- Chastan N, Baert-Desurmont S, Saugier-Veber P, Derumeaux G, Cabot A, Frebourg T, et al. Cardiac conduction alterations in a French family with amyloidosis of the Finnish type with the p.Asp187Tyr mutation in the GSN gene. Muscle Nerve. 2006;33:113–19.
- Bradshaw P, Stobie P, Knuiman MW, Briffa TG, Hobbs MST. Trends in the incidence and prevalence of cardiac pacemaker insertions in the ageing population. Open Heart. 2014;1:e000177.
- Fernandez A, Herreros J, Monzonis A, Panizo A. Heart transplantation for Finnish type familial systemic amyloidosis. Scand Cardiovasc J. 1997;31:357–9.
- Solomon JP, Page LJ, Balch WE, Kelly JW. Gelsolin amyloidosis: genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit Rev Biochem Mol Biol. 2012;47:282–96.
- Weeds AG, Gooch J, McLaughlin P, Maury CP. Variant plasma gelsolin responsible for familial amyloidosis (Finnish type) has defective actin severing activity. FEBS Lett. 1993;335:119–23.