
Abstract
The diagnosis and treatment of neurosarcoidosis can be very challenging for several reasons. It affects clinically 5%–10% of sarcoidosis patients, but can be found in up to 25% of autopsies. These data reveal that a high percentage of asymptomatic or misdiagnosed cases can be missed at an initial diagnostic approach. Clinical and imaging findings are often non-specific since they can be found in a large number of neurological disorders. Histopathology can also be confounding if not performed by an expert pathologist and not placed in an appropriate clinical context. In this review, we discuss clinical features, laboratory findings, imaging, and histology of neurosarcoidosis, and we report current evidence regarding drug therapy. We conclude that a correct diagnostic approach should include a multidisciplinary evaluation involving clinicians, radiologists, and pathologists and that future studies should evaluate the genetic signature of neurosarcoidosis as they could be helpful in the assessment of this uncommon disease. With head-to-head comparisons of medical treatment for neurosarcoidosis still lacking due to the rarity of the disease and an increasing number of immunomodulating therapies at hand, novel therapeutic approaches are to be expected within the next few years.
Neurosarcoidosis is a rare disorder that affects clinically 5%–10% of sarcoidosis patients, but can be found in up to 25% of autopsies, revealing that a high percentage of asymptomatic or misdiagnosed cases can be missed at an initial diagnostic approach.
A multidisciplinary evaluation is useful to achieve a correct diagnosis because clinical and imaging findings are often non-specific.
Corticosteroids are the first-line treatment for neurosarcoidosis, followed by steroid-sparing immune-modulating agents if prednisone therapy is insufficient.
Key Messages
Introduction
Sarcoidosis is a chronic idiopathic, inflammatory disorder characterized by multi-organ involvement by non-caseating granulomas. The epidemiology in general is quite complex as it shows variability due to a different racial and geographical distribution. In general, it occurs most often in countries of Northern Europe and United States and affects most frequently females.
The annual incidence is highest among African-Americans (39.1 and 29.8 cases/100,000 in females and males, respectively) followed by Caucasians (12.1 and 9.6/100,000), and peaks in patients aged 20–49 years, a decade sooner in African-Americans than in Caucasians (30–39 versus 40–49, respectively) (Citation1–3). African-Americans are also at an increased risk of mortality, tendency to multi-organ involvement, and disease chronicity (Citation3). Lungs and intrathoracic lymph nodes are the sites most frequently involved (Citation2,Citation4,Citation5), but virtually no tissue or organ is spared from sarcoidosis as it can involve organs such as heart, skin, gastrointestinal tract, liver, spleen, and joints. The involvement of central and peripheral nervous system (neurosarcoidosis) is uncommon, and the diagnosis can be very challenging if not clinically suspected, resulting in highly significant morbidity and mortality. Neurosarcoidosis manifests clinically in 5%–10% of patients with sarcoidosis, but can be identified in up to 25% of autopsies. Clinical presentation can be isolated or, more commonly, associated with other symptoms of systemic involvement such as pulmonary or eye disease (Citation6). Like the systemic form, it affects black more frequently than white people, and the disease is more common in females (Citation7). However, neurosarcoidosis seems to occur slightly later than the systemic form of the disease (mean age of onset between 33 and 41 years) (Citation8,Citation9). Children are rarely affected with neurosarcoidosis, most commonly at the age of 9–15 years (Citation10). The discordance between incidence of clinical and autopsy studies in adults reveals a great number of asymptomatic, under- or misdiagnosed cases, highlighting the need for robust criteria to recognize the disease early. In this narrative review we focus on clinical, radiological, and histopathological features of neurosarcoidosis, reviewing current diagnostic algorithms that can help toward early identification of this uncommon disorder. Current evidence on therapy and future perspectives will be also discussed.
Etiology of sarcoidosis and selective involvement of the nervous system
It is currently debated why some patients with sarcoidosis present mostly neurological rather than pulmonary manifestations of the disease. The formation of non-caseating granulomas in general seems to represent the result of an incomplete degradation of foreign or self-antigenic stimuli in genetically predisposed individuals, associated with an excessive activity of macrophages and T- and B-cells due to prolonged antigenemia (Citation11–13). Several environmental factors have been discussed as trigger agents, the most commonly accepted being those deriving from infectious disorders (e.g. mycobacteria and viruses), neoplasms, inorganic compounds (e.g. aluminium), or those derived from occupational exposure (e.g. agricultural employment or insecticides used at work) (Citation14–17).
The environmental factors, however, could not justify the onset of the disease alone, but can influence the development of sarcoidosis in genetically predisposed subjects. Genetic mutations, such as those involving the genes encoding for annexin A11 protein and butyrophilin-like 2 (BTNL2) (Citation18,Citation19), seem to influence the disease susceptibility and clinical progression (Citation20,Citation21). Some of them have been linked to certain clinical phenotypes, suggesting a predisposition to manifest a specific organ involvement (e.g. HLA-DRB1*0301 mutation and TGF-β 3 polymorphism as independent genetic risk factors for Löfgren’s syndrome and pulmonary fibrosis, respectively) (Citation22–25).
In this regard, there is increasing evidence of a specific genetic predisposition to a selective sarcoid involvement of the nervous system in some patients (Citation26–28). Caucasians present more often with manifestations deriving from peripheral nervous system involvement than do African-Americans, and patients with small fiber neuropathy have a high frequency of HLA-DQB1*0602 antigen and non-HLA polymorphic gene occurrence (Citation26).
A genetically predisposed condition of selective involvement of the nervous system can also be suggested by the evidence of a significant and specific elevation of some biomarkers in neurosarcoidosis in comparison to controls, revealing that neurosarcoidosis could be sustained partly by specific genetic profiles as found for sarcoidosis in general (Citation29–31).
Clinical manifestations of neurosarcoidosis
Neurologic involvement of sarcoidosis encompasses a variety of clinical manifestations affecting both central and peripheral nervous system. Myopathy is also described among presentation conditions of neurosarcoidosis, although it occurs rarely. The frequency of the clinical manifestations is discordant in the different case series, perhaps reflecting the non-specific or paucisymptomatic presentation in most cases (Citation9).
Central nervous system (CNS) sarcoidosis
The CNS involvement can be divided into two categories: brain and spinal cord neurosarcoidosis (BNS and SNS, respectively). BNS can present with non-specific symptoms such as fatigue, headache, cognitive dysfunction with progressive decline, fever, nausea, vomiting, and mood disorders (Citation8,Citation26,Citation32–34).
Fatigue is a core symptom of neurosarcoidosis and seems to influence the cognitive functions in the affected patients, but is not specific as it can be found in several conditions such as sleep disorders, hypothyroidism, hypogonadism, hyponatremia, hypocortisolism, and (steroid) myopathy. Clinical and laboratory findings can orient toward a specific endocrine and/or metabolic disease rather than CNS sarcoidosis. Furthermore, fatigue can also occur in association with peripheral nervous system disease (Citation35). The clinical picture of CNS sarcoidosis is most often dominated by cranial neuropathy secondary to granulomatous infiltration or to basilar, aseptic meningitis (Citation9), being described in up to 80% of cases (Citation8). In particular, most studies show that the optic nerve (II) is the most frequently affected, followed by the facial (VII) nerve. It is unknown whether these results reflect a greater likelihood of referral of patients with CN II dysfunction to centers of excellence as compared to facial neuropathy patients, who are more often managed in a community setting (Citation6,Citation8,Citation36). The damage of the II nerve from neurosarcoidosis can be associated with a poor prognosis in terms of visual recovery (Citation8). Main findings are central loss or blurred vision, retrobulbar pain, optic nerve atrophy, and papilledema resulting from local granuloma formation (Citation8,Citation37,Citation38). The VII nerve paresis is usually evident unilaterally and less frequently affects both sides of the face (Citation26,Citation35,Citation39). Rarely, the involvement of the VII nerve is associated with other specific symptoms such as Heerfordt’s syndrome, a clinical variety characterized by facial nerve palsy, fever, swelling of the parotid glands, and uveitis (Citation40). Hearing loss and vestibular dysfunction manifesting with dizziness are usually due to VIII nerve impairment (Citation6). Descriptions of trigeminal nerve (V) dysfunction, presenting, most often, with facial paresthesias and hyperesthesia and, rarely, with typical neuralgia, and of the nerves involved in eye movements (III, IV, and VI) have also been reported in the literature (Citation41–43). Other clinical findings of BNS are seizures or focal neurological deficits such as hemiparesis, often associated with brain tumor-like lesions exerting mass effect (Citation37,Citation38,Citation44,Citation45), and dysfunction of the endocrine system, which most commonly presents with diabetes insipidus, gonadotropin and TSH deficiency, and hyperprolactinemia (Citation46). Rarely, focal deficits can be secondary to the occurrence of an ischemic or hemorrhagic stroke, resulting from infiltration of vessel walls by granulomas (Citation47–50).
SNS is reported rarely, in about 14% of the cases in a recent study (Citation51). Unlike BNS, where symptoms occur within 2 years after diagnosis (Citation9), SNS manifestations are clinically evident later, and the patients have a higher age of onset (Citation52). SNS can be classified by location into intramedullary, extramedullary with leptomeningeal involvement, extradural, vertebral neurosarcoidosis, and disease of the disk space. The thoracic region is often involved (Citation53). Main presenting symptoms are paresthesias and weakness of the lower extremities (Citation51). Rarely, cases of sudden paraplegia have been described (Citation54). Bowel, bladder, erectile, and ejaculatory dysfunction have also been described as additional symptoms of spinal cord disease (Citation55).
Sarcoidosis of the peripheral nerves
Sarcoidosis of the peripheral nervous system (PNS) can affect up to 40% of patients, more often Caucasians than Afro-Americans. These data seem to be in part related to a genetic predisposition (Citation26), as discussed above. Sarcoid-related neuropathy can be mono- or multifocal associated with conduction blocks or present as polyradiculoneuropathy, sometimes in the form of Guillain–Barré-like syndrome. More often, however, it presents as symmetrical sensory motor polyneuropathy (Citation56–60). In rare cases an atypical chronic inflammatory demyelinating polyneuropathy has also been described (Citation61). Small-fiber neuropathy with or without involvement of autonomic fibers may occur and cause debilitating symptoms such as pain or restless leg syndrome (Citation62). The type and severity of symptoms can be assessed using quantitative sensory testing and related techniques (Citation63).
However, in cases of suspected neurosarcoidosis with clinical involvement of the PNS, a nerve biopsy is essential to confirm the diagnosis (Citation56).
Sarcoidosis-related myopathy
Granulomatous involvement of the muscles is often asymptomatic and found mostly at autopsy in up to 75% of all cases (Citation64); only rarely (less than 1%) does it present with acute, although non-specific, findings such as muscle atrophy, weakness, and pain (Citation26). In this regard, a useful classification includes acute, chronic, and nodular myositis. Acute myositis is characterized by non-specific symptoms such as fever and fatigue associated with muscle swelling, disabling pain, and sometimes contractures. The chronic form affects most often multiple groups of muscles, while the nodular involvement presents as multiple tumor-like masses found on muscle palpation (Citation64).
Laboratory tests
Systemic sarcoidosis is diagnosed based on the clinical picture and the histological evidence of granulomatous inflammation from cell or tissue specimens, such as lymph node biopsy or bronchoalveolar lavage (Citation4,Citation65). Accordingly, the diagnosis of definite neurosarcoidosis requires a positive CNS or nerve biopsy. Peripheral nervous system tissue is more accessible and therefore appropriate to undergo biopsy in the proper circumstances (Citation66). However, due to the broad spectrum of clinical manifestations and lack of certainty from other (e.g. imaging) diagnostic modalities, establishing the diagnosis of neurosarcoidosis is difficult. Biomarkers in serum and spinal fluid have been extensively studied for their ability to aid in the diagnosis (Citation67). Potentially, they could provide evidence of CNS involvement in known systemic sarcoidosis, or guide toward neurosarcoidosis in cases where the disease has not manifested systemically. Although certain immunological parameters are known to be affected in sarcoidosis, none of the investigated serum biomarkers have been accepted as establishing the diagnosis including angiotensin-converting enzyme (ACE) or soluble interleukin-2 receptor activity (sIL2 receptor) in the blood. The Kveim-Siltzbach test, which involves intradermal injection of tissue extracts from human sarcoidosis patients, has been used by some institutions with a reasonable specificity (Citation2), but has been abandoned by many others due to concerns regarding risk of infection and lack of standardized and commercially available test agents (Citation68).
In the original Zajicek criteria, the following laboratory tests are required for the diagnosis of at least probable neurosarcoidosis: ‘… laboratory support for CNS inflammation (elevated levels of CSF protein and/or cells, the presence of oligoclonal bands and/or MRI evidence compatible with neurosarcoidosis) and exclusion of alternative diagnoses together with evidence of systemic sarcoidosis (either through positive histology, including Kveim test, and/or at least two indirect indicators from Gallium scan, chest imaging and serum ACE)’ (Citation37). In their relatively large series of 68 patients, serum ACE was only increased in 23.5%, while calcium was normal in all, and erythrocyte sedimentation rate (ESR) positive in four. This is in line with observations from other groups: neither serum ACE nor calcium or ESR is useful to establish the diagnosis of neurosarcoidosis (Citation69,Citation70). Cerebrospinal fluid findings indicative of a chronic inflammatory disease have a much higher diagnostic yield in neurosarcoidosis (Citation71). An important part of the initial CSF examination is to rule out infectious CNS disease and malignancy.
In the work by Zajicek et al., CSF protein was elevated in 73%, leukocyte count raised in 55%, positive oligoclonal bands in CSF or both CSF and serum found in 55% (Citation37). Increased sIL2 receptor activity in the CSF might actually help to discriminate neurosarcoidosis from other chronic inflammatory CNS diseases (Citation31). Cells in the CSF (number and type of cells) as well as antibody indices (IgM, IgA, IgG) are often pathologic and may reflect disease activity (Citation72). ACE in spinal fluid, despite a low sensitivity (24%–55%), might raise the suspicion of neurosarcoidosis due to its high specificity (94%–95%) (Citation73). The CD4/CD8 T-cell ratio is typically increased in bronchoalveolar lavage of sarcoidosis patients (Citation74). In neurosarcoidosis, the diagnostic utility of this test is uncertain (Citation71,Citation75).
In summary, laboratory tests are only one component of the complex diagnostic process leading toward neurosarcoidosis. While blood biomarkers have a very limited value, CSF findings are compatible with a chronic CNS infection in the majority or patients. In a group of patients with neurosarcoidosis diagnosed at our clinic, CSF was abnormal in 12 out of 13 (Citation70).
Currently, sIL2-receptor alpha chain levels above 150 pg/mL are most specific for CNS involvement by sarcoidosis (overall accuracy of 93%) (Citation31).
Electromyography, nerve conduction, and evoked potential studies
The presence of neuropathy and myopathy can be revealed by nerve conduction studies (NCS) and electromyography (EMG). When peripheral nerves are affected, NCS can reveal anomalous sensory or motor nerve conduction consisting of absent/small potentials and reduced velocities, respectively (Citation8). Also quantitative sensory and autonomic testing can be helpful in the evaluation of small fiber neuropathy (Citation63).
EMG confirms myopathy by revealing myopathic motor unit potentials. Both NCS and EMG findings can improve after appropriate cortisone and/or immunosuppressive therapy (Citation8).
Brainstem auditory and visual evoked potentials are abnormal in up to 35% and 43% of patients, respectively, while somatosensory evoked potentials are less frequently altered. Evoked potentials can be abnormal often in asymptomatic cases, revealing a subclinical nervous system involvement in sarcoidosis, which can help in the early diagnosis of the disease (Citation76,Citation77).
The role of imaging
Imaging techniques give useful clues in the diagnosis and staging of systemic sarcoidosis. Chest X-rays and abdominal ultrasound are useful to reveal and classify the organ involvement, but procedures such as high-resolution computed tomography (HRCT), contrast-enhanced magnetic resonance imaging (CEMRI), and positron emission tomography with fluorodeoxyglucose (FDG-PET) have higher sensitivity to document the degree of the involvement and are preferred in most cases (Citation78–80). Recently, contrast-enhanced ultrasound (CEUS) has demonstrated a great potential in the diagnosis and assessment of the disease, also in the cases in which non-enhanced ultrasound is traditionally not significant (Citation81–84).
While the diagnosis of neurosarcoidosis can be easily suspected in patients with overt systemic disease with signs of neurological dysfunction, isolated neurosarcoidosis raises great diagnostic difficulty due to non-specific findings on imaging. However, the contribution of imaging (e.g. CT and MRI) is essential as it can demonstrate the central and peripheral nervous system involvement and also provide useful information to guide biopsy or follow-up of patients over time. FDG-PET can define areas of systemic hypermetabolism that can facilitate targeted tissue biopsy for diagnosis. An accurate and early recognition of nervous system involvement can allow a prompt and appropriate treatment, thus avoiding the onset of severe and/or irreversible complications that can potentially increase morbidity and mortality. Imaging findings should not be considered alone but only in an appropriate diagnostic algorithm including clinical, laboratory, radiological, and histopathological findings, to avoid misdiagnosis or classification of suspected cases (see below).
CT
CT is less sensitive than MRI for many of the imaging manifestations of CNS sarcoidosis. Granulomatous involvement can be extra- or intra-axial and can be classified according to the site affected, namely basilar, convexity, intrahemispheric, and periventricular white matter. CT can demonstrate nodular or diffuse leptomeningeal thickening after iodine contrast injection or as singular or multiple hyperdense, enhancing tumor-like lesions with edema of the adjacent white matter or mass effect to the adjacent parenchyma (Citation85). CT can sometimes reveal optic chiasma and nerve enhancement after iodine contrast administration, and can demonstrate the presence of hydrocephalus or uncommon findings such as parotid, salivary, and lacrimal gland enlargement (Citation86), calcifications, and hemorrhagic stroke (Citation85). However, CT has very low sensitivity to assess the typical small areas of ischemic injury caused by sarcoidosis-associated vasculopathy (Citation85).
MRI
MRI is the preferred diagnostic imaging technique for evaluation of neurosarcoidosis as it gives the best definition for brain and spinal disease. Leptomeningeal involvement manifests as diffuse or nodular thickening and enhancement on contrast-enhanced T1-weighted images (), with a predilection for the basilar meninges, a pattern that is often similar to that observed with tuberculosis or lymphoma (). Intraparenchymal lesions can manifest as multiple small, non-enhancing periventricular or subcortical white matter lesions giving high signal on T2-weighted images, similarly to those observed in multiple sclerosis (MS) (Citation53). Unlike MS, periventricular lesions in neurosarcoidosis do not usually resemble Dawson’s fingers. Larger lesions present as enhancing parenchymal masses involving leptomeningi and giving edema on the healthy parenchyma. These nodules can be easily misdiagnosed as primary or metastatic tumors. MRI also demonstrates well cranial nerve involvement (), although there is no close relationship between clinical and imaging findings (patients can be symptomatic with negative MRI and vice versa). When present, nerve disease can manifest as uni- or bilateral enlargement with enhancement of cranial nerves on contrast-enhanced T1-weighted images. In the case of optic nerve disease (), an infiltrative, pseudotumor-like lesion involving periorbital tissue can be observed. Similar enhancement patterns have been reported also for pituitary gland, hypothalamus, and dural involvement (Citation53). Imaging features of spinal neurosarcoidosis vary according to the site involved. Intraspinal lesions appear as hyperintense on T2- and hypointense on T1-weighted images, with patchy infiltration after gadolinium administration and fusiform enlargement of cervical and thoracic segments. Extraspinal disease, in particular leptomeningeal involvement, usually manifests as thin linear enhancement and small lesions or, rarely, as sarcoid nodules hypo- and hyperintense on T1- and T2-weighted images (). MRI also shows bone lesions well, demonstrating similar findings after gadolinium administration (Citation53,Citation87).
Figure 1. MRI pituitary, coronal T1 post-contrast image: leptomeningeal enhancement with nodularity involving both infra- and supratentorial regions, especially prominent around the suprasellar cistern and the optic chiasm (arrows). Courtesy of Dr Fang Zhu, Chicago.
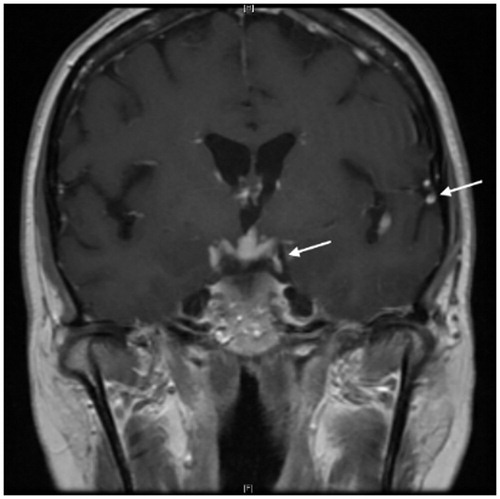
Figure 2. MRI brain, axial T1 post-contrast images: marked leptomeningeal enhancement at the base of the skull including along the midbrain (A) (arrow), surrounding the pons/medulla and along cranial nerves VII (B) (arrow). Brainstem enhancement in panel A represents involvement of pia-arachnoid with extension into Virchow–Robin spaces. Courtesy of Dr Fang Zhu, Chicago.
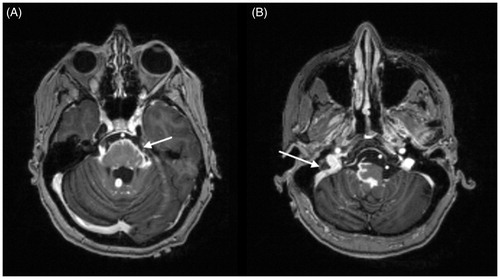
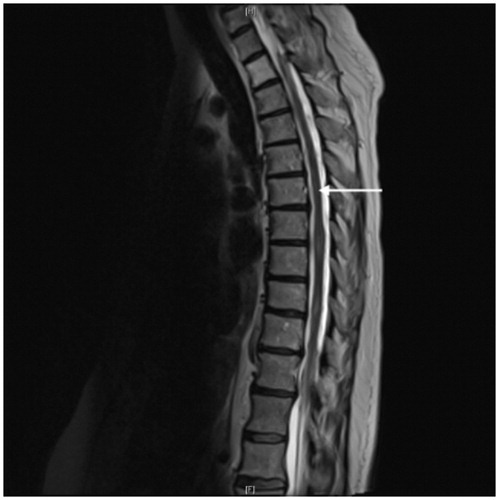
Figure 3. MRI spinal cord, sagittal T2: spinal meningeal involvement by sarcoidosis manifesting as linear leptomeningeal enhancement along the thoracic spinal cord (arrow). Courtesy of Dr Fang Zhu, Chicago.
FDG-PET
FDG-PET can be useful to assess metabolic activity of systemic sarcoidosis and has demonstrated a high efficacy in assessing stage, disease activity, and occult sites and in monitoring response to the therapy. In particular, the sensitivity and diagnostic accuracy of 18-FDG-PET is significantly higher than traditional 67Ga scintigraphy. FDG-PET can reveal areas of systemic hypermetabolism corresponding to active lesions and can be useful to reveal occult and asymptomatic sites to be examined histologically (Citation88), but there is a lack of evidence regarding its utility in neurosarcoidosis (Citation89). In general, molecular imaging has low sensitivity in CNS sarcoidosis and is not helpful to detect peripheral nerve disease (Citation53).
Histopathology
Due to the understandable reticence to undertake major invasive procedures such as biopsy to lesions suspected of representing central and peripheral nervous system involvement by sarcoidosis, the histopathologic features of neurosarcoidosis have been much less thoroughly evaluated than those of sarcoidosis involving the lungs, lymph nodes, skin, and other organs. Nonetheless, pathologic studies on biopsy and excision specimens, as well as those performed on autopsy material, where nervous system involvement is found twice as commonly as clinically suspected, have shown that neurosarcoidosis is characterized histopathologically by the same lesions as those found elsewhere in the body, i.e. the formation of epithelioid granulomas, although these tend to be located closer to vessels, be smaller, and contain giant cells less commonly (Citation90). The term granuloma, derived from the Latin granum (grain), relates to the typical gross appearance of these lesions, as tiny (2–4 mm) discrete, whitish-grey or yellowish granules, also called follicles. Neurosarcoidosis is characterized grossly and on imaging studies by either scattered, barely visible granules (miliary form), or by larger, grossly visible lesions consisting of a conglomerate of granulomas, which are characteristic of the nodular form (1–2 cm) and of the tumoral form (over 2 cm) (Citation91) of neurosarcoidosis. The use of high-resolution cross-sectional imaging has led to an increased recognition of tumoral forms of neurosarcoidosis. Such mass lesions can be mistaken for meningiomas, intraventricular tumors, and cerebellopontine angle tumors.
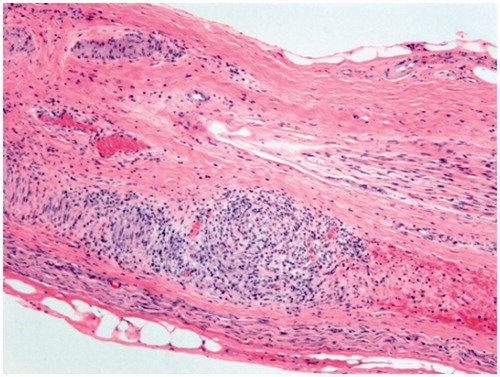
Nervous system involvement manifests most commonly as meningeal involvement, which usually occurs at the skull base and involves the leptomeninges (), but can extend from the subarachnoid space into the superficial brain parenchyma along the Virchow–Robin spaces. The basilar meningeal involvement appears as plaques or pachymeningitis () and is responsible for multiple cranial nerve and spinal root palsies. The third ventricle and choroid plexus may also be invaded by granulomas. All parts of the brain and spinal cord may be affected, but the most common sites of brain involvement are the hypothalamus and pituitary gland (), while spinal cord sites preferentially involved are the cervical or thoracic cord. Peripheral nerve involvement may also occur, as can muscle involvement, which is very common, but mostly asymptomatic. In the peripheral nerves, the granulomatous inflammation involves primarily the epineurium and perineurium and the vasa nervorum ().
Figure 4. Typical sarcoid granuloma in the subarachnoid space of the cerebellum (H&E stain, original magnification ×200): multinucleated giant cells and clusters of epithelioid histiocytes surrounded by lymphocytes expand the subarachnoid space. Underlying cerebellar parenchyma is gliotic.
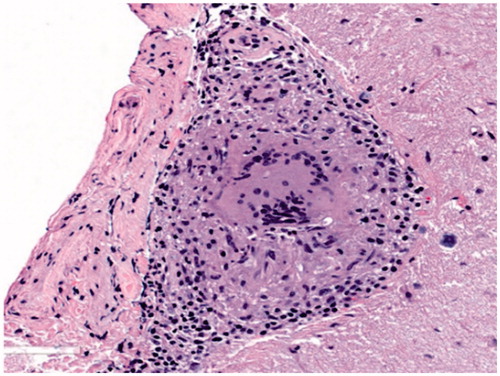
Figure 5. Neurosarcoidosis involving dura matter, H&E: compact collection of epithelioid histiocytes and multinucleated giant cells surrounded by scant lymphocytic infiltrate in densely fibrotic dura (H&E stain, original magnification ×400).
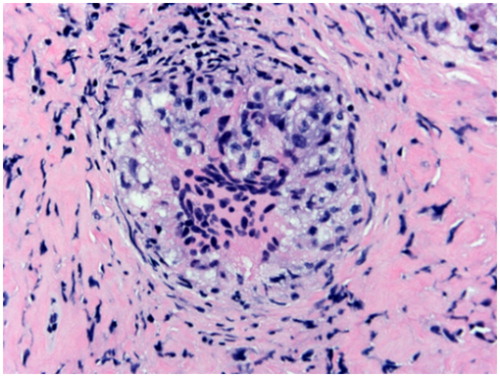
Figure 6. Sarcoidosis of pituitary gland, H&E: notice the multinucleated giant cells and presence of distinct lymphocytic cuff without significant fibrosis. Pituitary acini are present at the periphery (H&E stain, original magnification ×200).
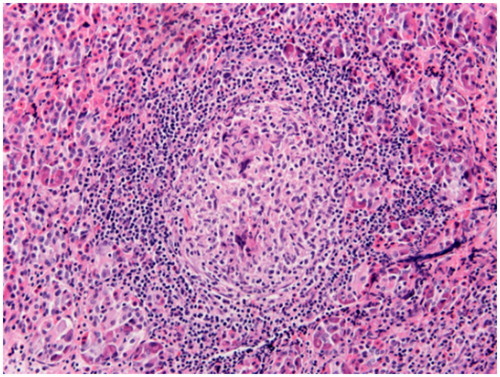
Figure 7. Non-necrotizing granuloma in the peripheral nerve (peroneal nerve biopsy): non-necrotizing granuloma is present in the epineurium (H&E stain, original magnification ×100).
Microscopically, neurosarcoidosis lesions are characterized by 150–400 μm diameter discrete rounded, compact collections of epithelioid histiocytes interspersed with lymphocytes and surrounded by a rim composed of only sparse lymphoid cells (the so-called ‘naked granulomas’), and a variable amount of fibrosis and collagen deposition, depending on the age of the granulomas. Granulomas become fibrotic starting from the periphery toward the center, showing concentric hyalinizing fibrosis with eventual replacement of the granulomas by a hyalinized nodular fibrous scar. When located within the central nervous system parenchyma, granulomas may be surrounded by dense reactive gliosis. Within the same lesion, the granulomas of neurosarcoidosis tend to be of similar size and age, although sometimes non-fibrotic granulomas may coexist with fibrotic ones. The epithelioid cells are spindle-shaped, rounded, or polygonal histiocytes with poorly defined cell borders, moderately abundant pale-staining cytoplasm, which is usually devoid of phagocytosed material or pigments, and a single ovoid, bean-shaped nucleus with fine nuclear chromatin and 1–2 small nucleoli. The presence of this abundant pale cytoplasm, imparting them an epithelial-like appearance, is responsible for the designation ‘epithelioid’.
Fusion of the epithelioid cells results in the formation of multinucleated giant cells. The multinucleated giant cells characteristic of epithelioid granulomas, also referred to as Langhans-type giant cells in honor of Theodor Langhans (1839–1915), who described them in 1868, usually have 5–20 nuclei located at the periphery of the cells and arranged in an incomplete semicircle akin to a horseshoe. Another type of multinucleated giant cell that may be encountered in epithelioid granulomas of sarcoidosis, but is not characteristic of them, is the foreign body giant cell, which is larger, and has more numerous nuclei, which are randomly distributed, but usually located in the center of the cell. The granulomas encountered in neurosarcoidosis are typically non-necrotizing, or non-caseating. The latter term refers to the lipid-rich, grossly cheese-like (‘caseous’) liquefaction necrosis characteristic of tuberculous granulomas. Microscopically, caseous necrosis appears as large geographic areas of acellular, amorphous, pale eosinophilic necrosis. This type of necrosis is never seen in sarcoid granulomas. Small foci of necrosis (granular, fibrinoid, or eosinophilic) may occasionally be present in the center of sarcoid granulomas, in a rare form of neurosarcoidosis referred to as necrotizing neurosarcoidosis. These granulomas with focal areas of coagulative necrosis are usually found side by side with classic non-necrotizing granulomas (Citation92). When such necrotic foci are present, a thorough search for infectious etiologies should be pursued before concluding that these granulomas are compatible with neurosarcoidosis. Neurosarcoidosis may also show associated vascular involvement (vasculitis) with infiltration of the adventitia and media of small arteries or veins by granulomas, giant cells, or lymphocytes, which can be responsible for foci of parenchymal necrosis.
A variety of characteristic but non-specific cytoplasmic inclusions (asteroid bodies, conchoid bodies, and Hamazaki–Wesenberg bodies) and crystals (oxalate crystals) can be found in the giant cells of sarcoid granulomas, supportive of the diagnosis of sarcoidosis. However, none of these inclusions is diagnostic of sarcoidosis, since they can be found in granulomas of other etiologies.
Immunohistochemistry
The epithelioid cells and multinucleated giant cells composing the granulomas stain for histiocytic markers (CD68, HAM-56, and CD163) and may stain weakly for CD4. CD4 also stains the T-cells within the granulomas, which may be more numerous than the epithelioid cells; the lymphocytes rimming the granulomas are usually T-cells staining for CD8, possibly admixed with rare CD20-staining B-cells and CD138-staining plasma cells, as well as rare tryptase and CD117-staining mast cells. However, immunostains are rarely needed for diagnostic purposes, since the granulomas are usually easily identified on routine (H&E) stained sections, and epithelioid granulomas of various etiologies have similar staining patterns.
Differential diagnosis
Clinical and imaging findings of neurosarcoidosis are often indistinguishable from those observed in other granulomatous disorders affecting the nervous system. Epithelioid granulomas are not specific for sarcoidosis, since they represent a non-specific reaction pattern to various etiologic agents, but can offer some precious diagnostic clues (Citation93).
Infectious disorders
The most important differential diagnosis is with tuberculosis involving the nervous system, especially in areas where tuberculosis is endemic and in patients who have lived in such areas or have a history of exposure to tuberculosis. Nervous system involvement by tuberculosis shows a similar radiological and histopathological distribution with basilar meningeal involvement and extension into the parenchyma in the form of miliary granulomas or larger tuberculomas. Although classically tuberculosis shows large necrotizing granulomas, it may occasionally show only non-necrotizing granulomas on biopsy (Citation94), and every effort should be made to rule out mycobacterial infection by acid-fast stains, auramine rhodamine stains, immunostains for mycobacteria, as well as PCR-based molecular diagnostic methods to demonstrate mycobacteria. It should be kept in mind that even after using all available methods, including cultures, special stains, and PCR-based methods, one cannot entirely rule out tuberculosis, because the sensitivities of these methods are rather low and may vary between institutions. For instance, the reported sensitivities for acid-fast stains is only 5%–25%, for culture (which may take 4–8 weeks) only 25%–85%, and for PCR-based methods around 20%–80% (Citation95). In addition, in patients living in areas endemic for fungal infections or with a history of travel to such areas, serologic testing, special stains, and molecular diagnostic methods should be employed to rule out histoplasmosis, aspergillosis, and cryptococcosis, all of which may occasionally only show non-necrotizing granulomas, especially on small biopsies.
In general, the presence of more than focal necrosis essentially excludes the diagnosis of sarcoidosis and strongly suggests an infectious process. Furthermore, all epithelioid granulomas are composed of epithelioid histiocytes, associated with variable numbers of lymphocytes, plasma cells, neutrophils, eosinophils, and mast cells. While lymphocytes are always seen in association with epithelioid granulomas, and may even outnumber the epithelioid histiocytes in sarcoidosis (Citation96), the presence of more than occasional plasma cells, neutrophils, or eosinophils within the center or periphery of granulomas argues against the diagnosis of sarcoidosis and suggests an infectious etiology. Plasma cells are prominent in the granulomas of syphilis (gumma). When syphilis is suspected, a correct approach should include also serologic testing (non-treponemal screening test such as Venereal Disease Research Laboratory (VDRL), rapid plasma reagin (RPR), or ICE syphilis recombinant antigen test) followed by treponemal test to confirm the disease (e.g. quantitative VDRL/RPR), as they have demonstrated high sensitivity and specificity (Citation97). Neutrophils can be seen in suppurative granulomas, such as those caused by Blastomyces dermatitidis, while eosinophils characterize parasitic granulomas, where exposure history, peripheral eosinophilia, and serological tests for parasite antigens can be also helpful to achieve the diagnosis (Citation98).
Polarization microscopy should be performed for the demonstration of potential polarizable foreign substances, and special stains for acid-fast organisms (Ziehl–Neelsen, Fite), spirochetes (Warthin–Starry, Dieterle), fungi (Grocott–Gomori methenamine silver), and protozoa (Giemsa) have to be performed to rule out infectious organisms.
Vasculitis
Due to the presence of vasculitis that may be occasionally associated with neurosarcoidosis, the differential diagnostic considerations also include systemic and primary central nervous system vasculitides associated with granuloma formation. Since there may be significant histopathologic overlap between these entities and neurosarcoidosis, clinical and radiological correlation is mandatory. Granulomatosis with polyangiitis (GPA), formerly known as Wegener’s granulomatosis, can involve the central nervous system as an extension from the nasal cavity, paranasal sinuses, and orbit, and can affect the optic nerve, chiasma, cranial nerves, meninges, and the pituitary gland (Citation99). Histopathologically, this disease is characterized by granulomatous inflammation and small and medium-sized vessel vasculitis with associated necrosis in patients with systemic involvement and ANCA-positive serology with antibodies to proteinase-3 (PR3) or myeloperoxidase (MPO). Primary central nervous system vasculitis (PCNSV) is characterized histologically by lymphocytic, granulomatous (50%) or necrotizing (25%) vasculitis of the leptomeningeal and cortical arteries and veins, which may be associated with thrombosis of the vessels and parenchymal hemorrhage or necrosis (Citation100). The differential diagnosis with neurosarcoidosis is difficult, and is based on the presence of necrotizing vasculitis, vasculitis in the absence of meningeal or parenchymal epithelioid granulomas, and finally the absence of systemic disease. After exclusion of infectious etiologies, the exact etiology of the non-necrotizing granulomatous inflammation cannot be determined in some patients without systemic manifestations of disease. Such ‘pathogen-free granulomatous diseases of the central nervous system’ are associated with a poor prognosis (Citation101) and may represent a vasculitis rather than neurosarcoidosis.
Neoplastic disorders
A number of neoplastic conditions are also referred to as granulomatous and/or may be accompanied by granulomas. These include lymphomatoid granulomatosis (Citation102), a B-cell lymphoma that may present with extensive infiltration of the meninges, blood vessels, and brain by at least focally atypical lymphoid cells with plasmacytoid features, causing necrosis of the surrounding tissue, and eosinophilic granuloma, a neoplastic Langerhans cell proliferation that may involve the skull and extend to the meninges and the pituitary gland. Careful histopathologic assessment of the lymphoid and histiocytic population within and outside the granulomas will disclose the cytologic atypia characteristic of these conditions and lead to the performance of confirmatory immunohistochemical stains. Pineal germinomas may be associated with sarcoid-like reactions composed of numerous small, non-necrotizing granulomas that may obscure the presence of the neoplastic germ cells, especially in small biopsies.
Other diseases
Other conditions characterized by non-epithelioid granulomas may involve the nervous system and can occasionally enter the differential diagnosis of neurosarcoidosis. Foreign body giant cell granulomas can occur as a reaction to various foreign materials, show a predominance of foreign body type giant cells, frequently found in clusters. The initiating foreign material may be demonstrated by light microscopy or polarization microscopy. Palisaded granulomas are characterized by histiocytes arranged perpendicular at the periphery of a necrobiotic center and are characteristic of rheumatoid nodules, which may occasionally involve the meninges. Lipogranulomas or xanthogranulomatous reactions show abundant foamy histiocytes sometimes associated with cholesterol clefts and a special type of multinucleated giant cell, with nuclei circularly arranged in a peripheral wreath-like arrangement, the Touton-type giant cells. Such reactions can be encountered as a reaction to the keratin debris of dermoid or epidermoid cyst (Citation103) or as part of juvenile xanthogranulomas or Erdheim–Chester disease.
Diagnostic approach
The traditional 1999 ATS/ERS/WASOG criteria defined the diagnosis of sarcoidosis with suggestive clinical picture, histopathological demonstration of non-caseating granulomas, and exclusion of other diseases able to produce similar findings (Citation104). However, these criteria have not given specific indications for each organ involvement and can sometimes result in classification bias or misdiagnosed cases in clinical practice (Citation27,Citation105,Citation106). In the specific form of neurosarcoidosis, clinical findings can manifest without systemic or respiratory symptoms and signs and can be wrongly attributed to other conditions. Furthermore, the problem sometimes becomes more complex because other diseases, such as tumors, vasculitis, or local infections, occasionally can be found in patients also having neurosarcoidosis (Citation107).
Several efforts have been made to produce instruments able to establish criteria to assess the probability of specific organ involvement by sarcoidosis. If another organ has demonstrated granulomatous inflammation previously, the WASOG Sarcoidosis Organ Assessment Instrument predicts clinical manifestations of neurosarcoidosis as:
Highly probable: a clinical syndrome consistent with granulomatous inflammation of the nervous system plus MRI findings of neurosarcoidosis or CSF examination suggestive of inflammation
At least probable: the presence of isolated VII palsy and negative MRI, clinical syndrome consistent with granulomatous inflammation of the nervous system, but MRI or CSF not suggestive for neurosarcoidosis,
Possible: the occurrence of seizures and/or cognitive decline with negative MRI
No consensus: if there was a peripheral neuropathy involving large fibers or cranial nerve palsies except for the facial nerve; negative MRI or CSF findings for neurosarcoidosis or low CSF glucose (Citation108)
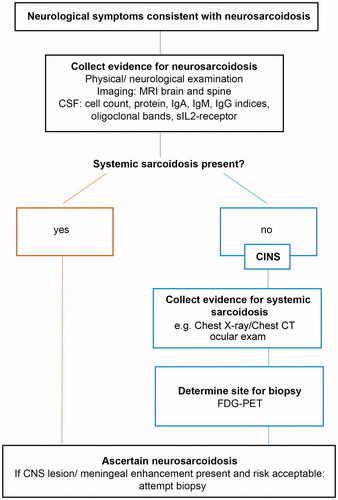
This tool has replaced the previous, outdated ACCESS instrument (Citation110) and enhances the key role of imaging, in particular MRI, to evaluate rapidly the presence of nervous system involvement (Citation108). On this basis, a useful path can indicate an early brain and spinal MRI as a mandatory step in the diagnosis of neurosarcoidosis before any invasive approach () (Citation70). Neurosarcoidosis can be suspected in patients having a history of systemic sarcoidosis, neurological symptoms, and suggestive imaging findings. However, neurological symptoms in patients with systemic disease should not be attributed to neurosarcoidosis a priori unless histopathologically confirmed (Citation67). This assumption is even more true in the case of isolated neurosarcoidosis, where the risk of misdiagnosis is high due to the lack of findings suggestive of systemic disease, making the biopsy mandatory.
Figure 8. Suggested diagnostic path for neurosarcoidosis. From Wegener, S. et al. Clinically isolated neurosarcoidosis: a recommended diagnostic path. Eur Neurol. 2015;73:71–7, with permission from S. Karger AG, Basel. CINS = clinically isolated neurosarcoidosis.
Treatment
Neurosarcoidosis is a rare condition, therefore treatment recommendations from randomized clinical trials are lacking. Whether neurosarcoidosis mandates treatment or not depends on symptom severity and course of the disease (Citation110). In systemic sarcoidosis, common causes of death are respiratory insufficiency and cardiac involvement; however, long-term immunosuppressive treatments also carry the risk of serious complications (Citation111).
There is general consensus that corticosteroids are the first-line treatment for neurosarcoidosis (Citation9,Citation26,Citation112,Citation113). If symptoms are severe, a short course of pulsed intravenous steroid treatment (such as 1 g methylprednisolone/d for 3–5 days) is usually applied. Thereafter, or if clinical symptoms are less serious, oral steroids (e.g. 40–80 mg prednisone equivalent/d) should be given for at least 1–3 months, and slowly tapered thereafter to the lowest effective dose (). Patients should be closely monitored for side effects of steroid treatment (e.g. hypertension, diabetes, gastritis). Clinical and neuroimaging examinations should be performed at short intervals to determine response to treatment. If a prednisone maintenance dose of more than 10 mg/d is required for symptom control or if clinical response is insufficient, adding a steroid-sparing immune-modulating agent is recommended (Citation26,Citation114). As second-line treatment, there is clear evidence in favor of the cytostatic methotrexate (MTX) (10–25 mg once a week with folate substitution 5 mg/week preferably taken in the morning after MTX intake). Side effects include neutropenia, as well as liver and kidney toxicity (Citation8,Citation115–117). Azathioprine with a slowly increased dose to approximately 2 mg/kg b.w. (200 mg/d at the maximum) can be used instead of MTX, with similar efficacy and steroid-sparing capacity but more infections as side effects (Citation118). Before starting azathioprine, activity of the enzyme thiopurinmethyltransferase (TPMT) should be determined to detect patients with a particular sensitivity to myelosuppressive effects of azathioprine. Leflunomide at 20 mg/d may be similarly effective to MTX with fewer side effects (Citation119). Based on promising results from neurosarcoidosis patients treated with mycophenolate mofetil (MMF) alone or in combination therapy (Citation120–122), we have increasingly used this substance at a dose of 2 g/d. It is generally well tolerated and may stabilize the disease process in steroid-refractory cases ().
Figure 9. Neurosarcoidosis treatment response to prednisone. A: MRI of a 57-year-old patient with probable neurosarcoidosis. The singular FLAIR hyperintense, contrast-enhancing CNS lesion on T1-weighted images (arrows) is completely remittent after oral prednisone therapy (B). Courtesy of Dr S. Sartoretti, Winterthur.
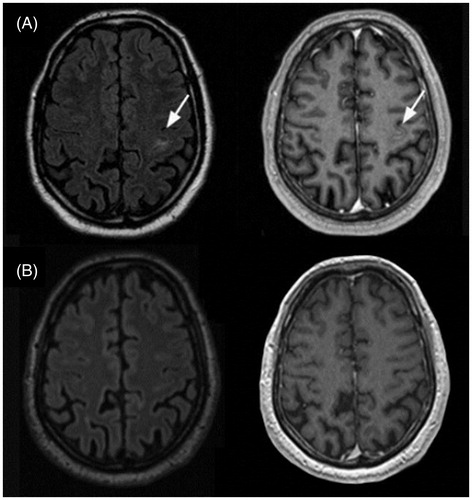
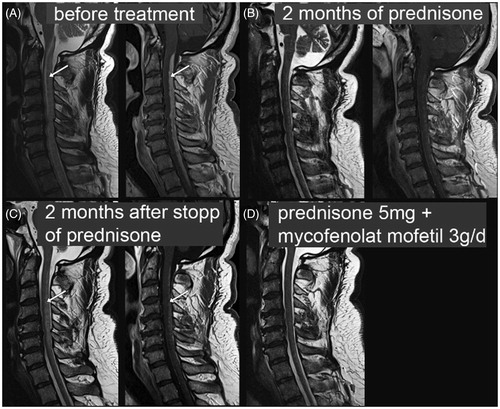
Figure 10. Spinal cord neurosarcoidosis treatment response to prednisone and mycophenolate mofetil. T2-weighted (left) and T1-weighted post-contrast images (right) of the cervical spinal cord in a 51-year-old patient with neurosarcoidosis and a tetraparesis below cervical C3 level. A: Before treatment. Arrows point to T2 hyperintensity and T1 contrast enhancement in cervical spinal cord, spreading over multiple cervical segments. B: Dramatic improvement after pulsed steroid treatment slowly tapered off. C: Relapse with clinical and imaging correlate (arrows) 2 months after complete discontinuation of prednisone. D: Clinical and imaging findings are improved under mycophenolate mofetil 3 g/d + 5 mg prednisone (no T1 post-contrast image available at that time point). Courtesy of Dr A. Rosskopf, Balgrist.
As third-line therapies, biologicals targeting tumor necrosis factor-alpha (TNF-alpha) such as infliximab and adalimumab have been recently used in patients with neurosarcoidosis (Citation123,Citation124).
Infliximab is applied intravenously at a dose of 3–5 mg/kg b.w. with a loading on weeks 0, 2, and 6 and with 4–6-weekly intervals thereafter (Citation125). Since cases with fast treatment response have been described, it may even be considered as a fast therapy inducing agent for patients in whom steroid treatment is contraindicated. Before establishing an anti-TNF-alpha therapy, a latent tuberculosis infection should be ruled out (Citation126). There are fewer data available about adalimumab; however, it is likely to be similarly effective in neurosarcoidosis to infliximab (Citation126). Adalimumab can be administered either intravenously or subcutaneously at weekly or bi-weekly intervals. Of note, anti-TNF-alpha treatment for auto-inflammatory diseases has in rare cases induced sarcoidosis, so patients should be closely monitored for this adverse treatment reaction (Citation127). There is a single case report about successful treatment of neurosarcoidosis with the monoclonal anti-CD20 antibody rituximab (Citation128). We consider this B-cell depleting, costly agent a third-line therapy in neurosarcoidosis. Due to the potential toxicity and the treatment alternatives discussed above, we recommend using cytostatic treatment with cyclophosphamide and cyclosporine only in therapy-refractive cases based on individual risk/benefit decisions (Citation26,Citation45,Citation129). When clinical deterioration progresses despite intensive immune-modulating treatment in neurosarcoidosis, a progressive multifocal leukoencephalopathy should be considered. This potentially fatal treatment complication in immunocompromised patients can be easily missed (Citation130).
With head-to-head comparisons of medical treatment for neurosarcoidosis still lacking due to the rarity of the disease and an increasing number of immunomodulating therapies at hand, novel therapeutic approaches are to be expected within the next years.
Conclusion
Neurosarcoidosis remains a diagnostic and therapeutic challenge. The difficulty of recognition and treatment of this uncommon disorder depends first on non-specific clinical and radiological findings, that can be found almost always in several other (immunological and not) neurological conditions, and then on the lack of comparative trials aimed at assessing the efficacy of the drug treatment in affected patients. Clinical algorithms could be helpful to assess nervous system involvement, but should be integrated with biopsy of affected tissues, because only histopathology can reveal the presence of non-caseating granulomas in the nervous system, the histological hallmark of neurosarcoidosis.
In view of the high genetic predisposition of some patients to manifest certain clinical pictures of neurosarcoidosis, more studies should investigate the genetic signature in these patients. The near future could see an early diagnosis of certain forms of neurosarcoidosis based on the study of patients’ genetic profile, in particular of those having risk factors for neurosarcoidosis such as long-standing sarcoidosis and symptoms as cognitive dysfunction and fatigue. At present, a thorough multidisciplinary evaluation involving clinicians, radiologists, and pathologists is essential to achieve a correct and early diagnosis. Although there are no specific recommendations, corticosteroids should be used as first-line treatment for neurosarcoidosis, followed by immunosuppressive therapy in non-responsive patients.
Declaration of interest
The authors report no conflicts of interest.
References
- Rybicki BA, Iannuzzi MC. Epidemiology of sarcoidosis: recent advances and future prospects. Semin Respir Crit Care Med. 2007;28:22–35.
- Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305:391–9.
- Rybicki BA, Major M, Popovich J Jr, Maliarik MJ, Iannuzzi MC. Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol. 1997;145:234–41.
- Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357:2153–65.
- Morgenthau AS, Iannuzzi MC. Recent advances in sarcoidosis. Chest. 2011;139:174–82.
- Pawate S, Moses H, Sriram S. Presentations and outcomes of neurosarcoidosis: a study of 54 cases. QJM. 2009;102:449–60.
- Stern BJ, Krumholz A, Johns C, Scott P, Nissim J. Sarcoidosis and its neurological manifestations. J Arch Neurol. 1985;42:909–17.
- Joseph FG, Scolding NJ. Neurosarcoidosis: a study of 30 new cases. J Neurol Neurosurg Psychiatry. 2009;80:297–304.
- Lacomis D. Neurosarcoidosis. Curr Neuropharmacol. 2011;9:429–36.
- Koné-Paut I, Portas M, Wechsler B, Girard N, Raybaud C. The pitfall of silent neurosarcoidosis. Pediatr Neurol. 1999;20:215–18.
- James DG. A clinicopathological classification of granulomatous disorders. Postgrad Med J. 2000;76:457–65.
- Tana C, Giamberardino MA, Di Gioacchino M, Mezzetti A, Schiavone C. Immunopathogenesis of sarcoidosis and risk of malignancy: a lost truth? Int J Immunopathol Pharmacol. 2013;26:305–13.
- Tana C, Tana M, Mezzetti A, Schiavone C. Sarcoidosis: old certainties and new perspectives. Ital J Med. 2012;6:186–94.
- Song Z, Marzilli L, Greenlee BM, Chen ES, Silver RF, Askin FB, et al. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic Sarcoidosis. J Exp Med. 2005;201:755–67.
- Newman LS, Rose CS, Bresnitz EA, Rossman MD, Barnard J, Frederick M, et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324–30.
- Rybicki BA, Iannuzzi MC, Frederick MM, Thompson BW, Rossman MD, Bresnitz EA, et al. Familial aggregation of sarcoidosis. A case-control etiologic study of sarcoidosis (ACCESS). Am J Respir Crit Care Med. 2001;164:2085–91.
- Tchernev G, Cardoso JC, Chokoeva AA, Verma SB, Tana C, Ananiev J, et al. The “mystery” of cutaneous sarcoidosis: facts and controversies. Int J Immunopathol Pharmacol. 2014;27:321–30.
- Hofmann S, Franke A, Fischer A, Jacobs G, Nothnagel M, Gaede KI, et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat Gene. 2008;40:1103–6.
- Rybicki BA, Walewski JL, Maliarik MJ, Kian H, Iannuzzi MC; ACCESS Research Group ACCESS Research Group. The BTNL2 gene and sarcoidosis susceptibility in African Americans and whites. Am J Hum Genet. 2005;773:491–9.
- Iannuzzi MC. Genetics of sarcoidosis. Semin Respir Crit Care Med. 2007;28:15–21.
- Dubrey S, Shah S, Hardman T, Sharma R. Sarcoidosis: the links between epidemiology and aetiology. Postgrad Med J. 2014;90:582–9.
- Spagnolo P, Sato H, Grunewald J, Brynedal B, Hillert J, Mañá J, et al. A common haplotype of the C-C chemokine receptor 2 gene and HLA-DRB1*0301 are independent genetic risk factors for Löfgren’s syndrome. J Intern Med. 2008;264:433–41.
- Pabst S, Fränken T, Schönau J, Stier S, Nickenig G, Meyer R, et al. Transforming growth factor-{beta} gene polymorphisms in different phenotypes of sarcoidosis. Eur Respir J. 2011;38:169–75.
- Hedfors E, Lindström F. HLA-B8/DR3 in sarcoidosis. Correlation to acute onset disease with arthritis. Tissue Antigens. 1983;223:200–20.
- Swider C, Schnittger L, Bogunia-Kubik K, Gerdes J, Flad H, Lange A, et al. TNF-alpha and HLA-DR genotyping as potential prognostic markers in pulmonary sarcoidosis. Eur Cytokine Netw. 1999;10:143–6.
- Hebel R, Dubaniewicz-Wybieralska M, Dubaniewicz A. Overview of neurosarcoidosis: recent advances. J Neurol. 2015;262:258–67.
- Tchernev G, Tana C, Schiavone C, Cardoso JC, Ananiev J, Wollina U. Sarcoidosis vs. sarcoid-like reactions: the two sides of the same coin? Wien Med Wochenschr. 2014;164:247–59.
- Mende D, Suchenwirth RM. Neursarcoidosis. Comparative analysis of the clinical profile based on 537 cases from the world literature up to 1963 and from 1976–1988. Fortschr Neurol Psychiatr. 1990;58:7–18.
- Linnebank M, Kesper K, Jeub M, Urbach H, Wüllner U, Klockgether T, et al. Hereditary elevation of angiotensin converting enzyme suggesting neurosarcoidosis. Neurology. 2003;61:1819–20.
- Tahmoush AJ, Amir MS, Connor WW, Farry JK, Didato S, Ulhoa-Cintra A, et al. CSF-ACE activity in probable CNS neurosarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19:191–7.
- Petereit HF, Reske D, Tumani H, Jarius S, Markus Leweke F, Woitalla D, et al. Soluble CSF interleukin 2 receptor as indicator of neurosarcoidosis. J Neurol. 2010;257:1855–63.
- Gascón-Bayarri J, Mañá J, Martínez-Yélamos S, Murillo O, Reñé R, Rubio F. Neurosarcoidosis: report of 30 cases and a literature survey. Eur J Intern Med. 2011;22:e125–32.
- Oh J, Stokes K, Tyndel F, Freedman M. Progressive cognitive decline in a patient with isolated chronic neurosarcoidosis. Neurologist. 2010;16:50–3.
- Nozaki K, Scott TF, Sohn M, Judson MA. Isolated neurosarcoidosis: case series in 2 sarcoidosis centers. Neurologist. 2012;18:373–7.
- Beste C, Kneiphof J, Woitalla D. Effects of fatigue on cognitive control in neurosarcoidosis. Eur Neuropsychopharmacol. 2015;25:522–30.
- Carlson ML, White JR Jr, Espahbodi M, Haynes DS, Driscoll CL, Aksamit AJ, et al. Cranial base manifestations of neurosarcoidosis: a review of 305 patients. Otol Neurotol. 2015;36:156–66.
- Zajicek JP, Scolding NJ, Foster O, Rovaris M, Evanson J, Moseley IF, et al. Central nervous system sarcoidosis–diagnosis and management. QJM. 1999;92:103–17.
- Delaney P. Neurologic manifestations in sarcoidosis: review of the literature, with a report of 23 cases. Ann Intern Med. 1977;87:336–45.
- Oksanen V. Neurosarcoidosis: clinical presentations and course in 50 patients. Acta Neurol Scand. 1986;73:283–90.
- Dua A, Manadan A. Heerfordt’s syndrome, or uveoparotid fever. N Engl J Med. 2013;369:458.
- Braksick S, Shah-Haque S, El-Haddad Et Al B. Neurosarcoidosis presenting as trigeminal nevralgia: a case report and review of the literature. Sarcoidosis Vasc Diffuse Lung Dis. 2013;30:153–6.
- Menezo V, Lobo A, Yeo TK, du Bois RM, Lightman S. Ocular features in neurosarcoidosis. Ocul Immunol Inflamm. 2009;17:170–8.
- Meireles J, Garrett MC, Abreu P. Isolated III cranial nerve palsy: a Hodgkin’s lymphoma? BMJ Case Rep. 2014. pii: bcr2014203999.
- Chapelon C, Ziza JM, Piette JC, Levy Y, Raguin G, Wechsler B, et al. Neurosarcoidosis: signs, course and treatment in 35 confirmed cases. Medicine (Baltimore). 1990;69:261–76.
- Lower EE, Broderick JP, Brott TG, Baughman RP. Diagnosis and management of neurological sarcoidosis. Arch Intern Med. 1997;157:1864–8.
- Langrand C, Bihan H, Raverot G, Varron L, Androdias G, Borson-Chazot F, et al. Hypothalamo-pituitary sarcoidosis: a multicenter study of 24 patients. QJM. 2012;105:981–95.
- Hodge MH, Williams RL, Fukui MB. Neurosarcoidosis presenting as acute infarction on diffusion-weighted MR imaging: summary of radiologic findings. AJNR Am J Neuroradiol. 2007;28:84–6.
- O’Dwyer JP, Al-Moyeed BA, Farrell MA, Pidgeon CN, Collins DR, Fahy A, et al. Neurosarcoidosis-related intracranial haemorrhage: three new cases and a systematic review of the literature. Eur J Neurol. 2013;20:71–8.
- Travers F, Maltête D, Morisse-Pradier H, Wallon D, Bourre B, Lefaucheur R. Intracranial hemorrhage in neurosarcoidosis. J Neurol Sci. 2014;341:185–6
- Brown MM, Thompson AJ, Wedzicha JA, Swash M. Sarcoidosis presenting with stroke. Stroke. 1989;20:400–5.
- Sohn M, Culver DA, Judson MA, Scott TF, Tavee J, Nozaki K. Spinal cord neurosarcoidosis. Am J Med Sci. 2014;347:195–8.
- Sakushima K, Yabe I, Nakano F, Yoshida K, Tajima Y, Houzen H, et al. Clinical features of spinal cord sarcoidosis: analysis of 17 neurosarcoidosis patients. J Neurol. 2011;258:2163–7.
- Smith JK, Matheus MG, Castillo M. Imaging manifestations of neurosarcoidosis. AJR Am J Roentgenol. 2004;182:289–95.
- Varron L, Broussolle C, Candessanche JP, Marignier R, Rousset H, Ninet J, et al. Spinal cord sarcoidosis: report of seven cases. Eur J Neurol. 2009;16:289–96.
- Kaiboriboon K, Olsen TJ, Hayat GR. Cauda equina and conus medullaris syndrome in sarcoidosis. Neurologist. 2005;11:179–83.
- Said G, Lacroix C, Planté-Bordeneuve V, Le Page L, Pico F, Presles O, et al. Nerve granulomas and vasculitis in sarcoid peripheral neuropathy: a clinicopathological study of 11 patients. Brain. 2002;125(Pt 2):264–75.
- Suzuki C, Tomiyama M, Baba M, Jinichi N, Ogawa M, Kurahashi K, et al. Reversible multifocal conduction block in sarcoid neuropathy. J Peripher Nerv Syst. 2006;11:93–5.
- Scott TS, Brillman J, Gross JA. Sarcoidosis of the peripheral nervous system. Neurol Res. 1993;15:389–90.
- Galassi G, Gibertoni M, Mancini A, Nemni R, Volpi G, Merelli E, et al. Sarcoidosis of the peripheral nerve: clinical, electrophysiological and histological study of two cases. Eur Neurol. 1984;23:459–65.
- Koffman B, Junck L, Elias SB, Feit HW, Levine SR. Polyradiculopathy in sarcoidosis. Muscle Nerve. 1999;22:608–13.
- Vital A, Lagueny A, Ferrer X, Louiset P, Canron MH, Vital C. Sarcoid neuropathy: clinico-pathological study of 4 new cases and review of the literature. Clin Neuropathol. 2008;27:96–105.
- Hoitsma E, Faber CG, Drent M, Sharma OP. Neurosarcoidosis: a clinical dilemma. Lancet Neurol. 2004;3:397–407.
- Mainka T, Maier C, Enax-Krumova EK. Neuropathic pain assessment: update on laboratory diagnostic tools. Curr Opin Anaesthesiol 2015;28:537–45.
- Sweiss NJ, Patterson K, Sawaqed R, Jabbar U, Korsten P, Hogarth K, et al. Rheumatologic manifestations of sarcoidosis. Semin Respir Crit Care Med. 2010;31:463–73.
- Judson MA. Advances in the diagnosis and treatment of sarcoidosis. F1000Prime Rep. 2014;6:89.
- Spiegel DR, Morris K, Rayamajhi U. Neurosarcoidosis and the complexity in its differential diagnoses: a review. Innov Clin Neurosci. 2012;9:10–16.
- Bagnato F, Stern BJ. Neurosarcoidosis: diagnosis, therapy and biomarkers. Expert Rev Neurother. 2015;15:533–48.
- Reich JM. On the nature of sarcoidosis. Eur J Intern Med. 2012;23:105–9.
- Marangoni S, Argentiero V, Tavolato B. Neurosarcoidosis. Clinical description of 7 cases with a proposal for a new diagnostic strategy. J Neurol. 2006;253:488–95.
- Wegener S, Linnebank M, Martin R, Valavanis A, Weller M. Clinically isolated neurosarcoidosis: a recommended diagnostic path. Eur Neurol. 2015;73:71–7.
- Reske D, Petereit HF, Heiss WD. Difficulties in the differentiation of chronic inflammatory diseases of the central nervous system–value of cerebrospinal fluid analysis and immunological abnormalities in the diagnosis. Acta Neurol Scand. 2005;112:207–13.
- Wengert O, Rothenfusser-Korber E, Vollrath B, Bohner G, Scheibe F, Otto C, et al. Neurosarcoidosis: correlation of cerebrospinal fluid findings with diffuse leptomeningeal gadolinium enhancement on MRI and clinical disease activity. J Neurol Sci. 2013;335:124–30.
- Khoury J, Wellik KE, Demaerschalk BM, Wingerchuk DM. Cerebrospinal fluid angiotensin-converting enzyme for diagnosis of central nervous system sarcoidosis. Neurologist. 2009;15:108–11.
- Danila E, Norkuniene J, Jurgauskiene L, Malickaite R. Diagnostic role of BAL fluid CD4/CD8 ratio in different radiographic and clinical forms of pulmonary sarcoidosis. Clin Respir J. 2009;3:214–21.
- Stern BJ, Griffin DE, Luke RA, Krumholz A, Johns CJ. Neurosarcoidosis: cerebrospinal fluid lymphocyte subpopulations. Neurology. 1987;37:878–81.
- Oksanen V, Salmi T. Visual and auditory evoked potentials in the early diagnosis and follow-up of neurosarcoidosis. Acta Neurol Scand. 1986;74:38–42.
- Gott PS, Kumar V, Kadakia J, Sharma OP. Significance of multimodality evoked potential abnormalities in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1997;14:159–64.
- Spagnolo P, Sverzellati N, Wells AU, Hansell DM. Imaging aspects of the diagnosis of sarcoidosis. Eur Radiol. 2014;24:807–16.
- Greco FG, Spagnolo P, Muri M, Paladini I, Chizzolini F, Piciucchi S, et al. The value of chest radiograph and computed tomography in pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31:108–16.
- Warshauer DM, Lee JKT. Imaging manifestations of abdominal sarcoidosis. AJR Am J Roentgenol. 2004;182:15–28.
- Tana C, Dietrich CF, Schiavone C. Hepatosplenic sarcoidosis: contrast-enhanced ultrasound findings and implications for clinical practice. Biomed Res Int. 2014;2014:926203.
- Tana C, Silingardi M, Dietrich CF. New trends in ultrasound of hepatosplenic sarcoidosis. Z Gastroenterol. 2015;53:283–4.
- Tana C, Iannetti G, Mezzetti A, Schiavone C. Splenic sarcoidosis remains a diagnostic challenge. J Clin Ultrasound. 2014;42:156.
- Tana C, Iannetti G, D’Alessandro P, Tana M, Mezzetti A, Schiavone C. Pitfalls of contrast-enhanced ultrasound (CEUS) in the diagnosis of splenic sarcoidosis J Ultrasound. 2013;16:75–80.
- Hayes WS, Sherman JL, Stern BJ, Citrin CM, Pulaski PD. MR and CT evaluation of intracranial sarcoidosis. AJR Am J Roentgenol. 1987;149:1043–9.
- Post MJ, Quencer RM, Tabei SZ. CT demonstration of sarcoidosis of the optic nerve, frontal lobes, and falx cerebri: case report and literature review. Am J Neuroradiol. 1982;3:523–6.
- Ginat DT, Dhillon G, Almast J. Magnetic resonance imaging of neurosarcoidosis. J Clin Imaging Sci. 2011;1:15.
- Treglia G, Taralli S, Giordano A. Emerging role of whole-body 18F-fluorodeoxyglucose positron emission tomography as a marker of disease activity in patients with sarcoidosis: a systematic review. Sarcoidosis Vasc Diffuse Lung Dis. 2011;28:87–94.
- Aide N, Benayoun M, Kerrou K, Khalil A, Cadranel J, Talbot JN. Impact of [18F]-fluorodeoxyglucose ([18F]-FDG) imaging in sarcoidosis: unsuspected neurosarcoidosis discovered by [18F]-FDG PET and early metabolic response to corticosteroid therapy. Br J Radiol. 2007;80:e67–71.
- van Dellen JR. Equo ne credite, Teucri. Quidquid id est, timeo Danaos et dona ferentes. World Neurosurg. 2013;80:e215–17.
- Jefferson M. The nervous system in sarcoidosis. Postgrad Med J. 1958;34:259–61.
- Tobias S, Prayson RA, Lee JH. Necrotizing neurosarcoidosis of the cranial base resembling an en plaque sphenoid wing meningioma: case report. Neurosurgery. 2002;51:1290–4; discussion 1294.
- Zajicek JP. Neurosarcoidosis. Curr Opin Neurol. 2000;13:323–25.
- Chokoeva AA, Tchernev G, Tana C, Ananiev J, Wollina U. Sarcoid-like pattern in a patient with tuberculosis. J Biol Regul Homeost Agents. 2014;28:783–8.
- Tojo K, Yazaki M, Yoshida K, Machida K, Ikeda S. Biopsy-proven tuberculous meningitis mimicking CNS sarcoidosis. Int Med. 2007;46:2001–6.
- Kosjerina Z, Zaric B, Vuckovic D, Lalosevic D, Djenadic G, Murer B. The sarcoid granuloma: ‘epithelioid’ or ‘lymphocytic-epithelioid’ granuloma? Multidiscip Respir Med. 2012;7:11.
- Calonge N; U.S. Preventive Services Task Force. Screening for syphilis infection: recommendation statement. Ann Fam Med. 2004;2:362–5.
- Garcia HH, Nash TE, Del Brutto OH. Clinical symptoms, diagnosis, and treatment of neurocysticercosis. Lancet Neurol. 2014;13:1202–15.
- Seror R, Mahr A, Ramanoelina J, Pagnoux C, Cohen P, Guillevin L. Central nervous system involvement in Wegener granulomatosis. Medicine (Baltimore). 2006;85:54–65.
- Hajj-Ali RA, Calabrese LH. Primary angiitis of the central nervous system. Autoimmun Rev. 2013;12:463–6.
- Thomas G, Murphy S, Staunton H, O’Neill S, Farrell MA, Brett FM. Pathogen-free granulomatous diseases of the central nervous system. Hum Pathol. 1998;29:110–15.
- Lucantoni C, De Bonis P, Doglietto F, Esposito G, Larocca LM, Mangiola A, et al. Primary cerebral lymphomatoid granulomatosis: report of four cases and literature review. J Neurooncol. 2009;94:235–42.
- Prayson RA, Kleinschmidt-DeMasters BK. An algorithmic approach to the brain biopsy–part II. Arch Pathol Lab Med. 2006;130:1639–48.
- Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999;160:736–55.
- Tchernev G, Chokoeva AA, Tana C, Patterson JW, Wollina U, Lotti T. Sarcoid sine sarcoidosis? A classificative, semantic and therapeutic dilemma. J Biol Regul Homeost Agents. 2015;29(1 Suppl):33–4.
- Chokoeva AA, Tchernev G, Tana M, Tana C. Exclusion criteria for sarcoidosis: a novel approach for an ancient disease? Eur J Intern Med. 2014;25:e120.
- Salvarani C, Brown RD, Christianson TJ, Huston J III, Giannini C, Miller DV, et al. Adult primary central nervous system vasculitis treatment and course: analysis of 163 patients. Arthritis Rheumatol. 2015;67:1637–45.
- Judson MA, Costabel U, Drent M, Wells A, Maier L, Koth L, et al. The WASOG sarcoidosis organ assessment instrument: an update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31:19–27.
- Judson MA, Baughman RP, Teirstein AS, Terrin ML, Yeager H Jr. Defining organ involvement in sarcoidosis: the ACCESS proposed instrument. ACCESS Research Group. A Case Control Etiologic Study of Sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16:75–86.
- Baughman RP, Nagai S, Balter M, Costabel U, Drent M, du Bois R, et al. Defining the clinical outcome status (COS) in sarcoidosis: results of WASOG Task Force. Sarcoidosis Vasc Diffuse Lung Dis. 2011;28:56–64.
- Baughman RP, Lower EE. Who dies from sarcoidosis and why? Am J Respir Crit Care Med. 2011;183:1446–7.
- Nozaki K, Judson MA. Neurosarcoidosis: clinical manifestations, diagnosis and treatment. Presse Med. 2012;41:e331–48.
- Segal BM. Neurosarcoidosis: diagnostic approaches and therapeutic strategies. Curr Opin Neurol. 2013;26:307–13.
- Lower EE, Weiss KL. Neurosarcoidosis. Clin Chest Med. 2008;29:475–92, ix.
- Whittle SL, Hughes RA. Folate supplementation and methotrexate treatment in rheumatoid arthritis: a review. Rheumatology. 2004;43:267–71.
- Schutt AC, Bullington WM, Judson MA. Pharmacotherapy for pulmonary sarcoidosis: a Delphi consensus study. Respir Med. 2010;104:717–23.
- Beegle SH, Barba K, Gobunsuy R, Judson MA. Current and emerging pharmacological treatments for sarcoidosis: a review. Drug Des Devel Ther. 2013;7:325–38.
- Vorselaars AD, Wuyts WA, Vorselaars VM, Zanen P, Deneer VH, Veltkamp M, et al. Methotrexate vs azathioprine in second-line therapy of sarcoidosis. Chest. 2013;144:805–12.
- Sahoo DH, Bandyopadhyay D, Xu M, Pearson K, Parambil JG, Lazar CA, et al. Effectiveness and safety of leflunomide for pulmonary and extrapulmonary sarcoidosis. Eur Respir J. 2011;38:1145–50.
- Chaussenot A, Bourg V, Chanalet S, Fornari JM, Lebrun C. [Neurosarcoidosis treated with mycophenolate mofetil: two cases]. Revue Neurologique. 2007;163:471–5.
- Moravan M, Segal BM. Treatment of CNS sarcoidosis with infliximab and mycophenolate mofetil. Neurology. 2009;72:337–40.
- Androdias G, Maillet D, Marignier R, Pinede L, Confavreux C, Broussolle C, et al. Mycophenolate mofetil may be effective in CNS sarcoidosis but not in sarcoid myopathy. Neurology. 2011;76:1168–72.
- Graves JE, Nunley K, Heffernan MP. Off-label uses of biologics in dermatology: rituximab, omalizumab, infliximab, etanercept, adalimumab, efalizumab, and alefacept (part 2 of 2). J Am Acad Dermatol. 2007;56:e55–79.
- Callejas-Rubio JL, Lopez-Perez L, Ortego-Centeno N. Tumor necrosis factor-alpha inhibitor treatment for sarcoidosis. Ther Clin Risk Manag 2008;4:1305–13.
- Sodhi M, Pearson K, White ES, Culver DA. Infliximab therapy rescues cyclophosphamide failure in severe central nervous system sarcoidosis. Respir Med. 2009;103:268–73.
- Redelman-Sidi G, Sepkowitz KA. IFN-gamma release assays in the diagnosis of latent tuberculosis infection among immunocompromised adults. Am J Respir Crit Care Med. 2013;188:422–31.
- Vigne C, Tebib JG, Pacheco Y, Coury F. Sarcoidosis: an underestimated and potentially severe side effect of anti-TNF-alpha therapy. Joint Bone Spine. 2013;80:104–7.
- Bomprezzi R, Pati S, Chansakul C, Vollmer T. A case of neurosarcoidosis successfully treated with rituximab. Neurology. 2010;75:568–70.
- Agbogu BN, Stern BJ, Sewell C, Yang G. Therapeutic considerations in patients with refractory neurosarcoidosis. Arch Neurol. 1995;52:875–9.
- Jamilloux Y, Neel A, Lecouffe-Desprets M, Fevre A, Kerever S, Guillon B, et al. Progressive multifocal leukoencephalopathy in patients with sarcoidosis. Neurology. 2014;82:1307–13.