
Abstract
The protein brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family of growth factors involved in plasticity of neurons in several brain regions. There are numerous evidence that BDNF expression is decreased by experiencing psychological stress and that, accordingly, a lack of neurotrophic support causes major depression. Furthermore, disruption in sleep homeostatic processes results in higher stress vulnerability and is often associated with stress-related mental disorders. Recently, we reported, for the first time, a relationship between BDNF and insomnia and sleep deprivation (SD). Using a biphasic stress model as explanation approach, we discuss here the hypothesis that chronic stress might induce a deregulation of the hypothalamic-pituitary-adrenal system. In the long-term it leads to sleep disturbance and depression as well as decreased BDNF levels, whereas acute stress like SD can be used as therapeutic intervention in some insomniac or depressed patients as compensatory process to normalize BDNF levels. Indeed, partial SD (PSD) induced a fast increase in BDNF serum levels within hours after PSD which is similar to effects seen after ketamine infusion, another fast-acting antidepressant intervention, while traditional antidepressants are characterized by a major delay until treatment response as well as delayed BDNF level increase.
Brain-derived neurotrophic factor (BDNF) plays a key role in the pathophysiology of stress-related mood disorders.
The interplay of stress and sleep impacts on BDNF level.
Partial sleep deprivation (PSD) shows a fast action on BDNF level increase.
Key messages
Introduction
In our modern industrialized environment, the brain is a primary mediator and target of stress resiliency and vulnerability processes because of its capacity to adapt the behavioural and physiological responses to the social and physical stresses (Citation1,Citation2). Yet, depending on the individual genetic background and epigenetic factors, this ability is not infinite, which, once a threshold is reached, eventually increases the susceptibility to stress-related mental disorders like major depressive disorder (MDD).
Brain-derived neurotrophic factor (BDNF) belongs to the neurotrophin family and is produced by astrocytes, in addition to neurons, and the noradrenergic and serotonergic systems play a role in controlling BDNF synthesis (Citation3–5). BDNF is abundant in various brain regions implicated in mood disorders, including the hippocampus, prefrontal cortex, and amygdala, and shows a remarkable activity-dependent regulation of expression and secretion. Thus, its neurotrophic functions are connected to various physiological functions in the brain particularly relevant in neuroplasticity, memory and sleep (Citation6–9), suggesting its biological role over the entire life span.
Based on findings in brains from rodents (Citation10,Citation11) and humans (Citation12), the structure of the BDNF gene seems to be highly complex and contains numerous promoters each preceding one exon. This complexity leads through alternative splicing to the existence of distinct mRNA species that all contain exon IX, which encodes the BDNF protein (Citation10). Although the differential regulation of these transcripts is essential and may dictate subcellular BDNF mRNA targeting and translational responses following neuronal stimulation, their potential implications in neuropsychiatric disorders remain mostly unclear. Notably, only one functional mature protein is generated, irrespective of the splice variant expressed. BDNF is synthesized as a larger precursor protein known as prepro-BDNF that is cleaved into pro-BDNF, which can then be further cleaved into mature BDNF (Citation13). It has been suggested that pro-BDNF and mature BDNF activate different intracellular signaling pathways (reviewed in (Citation14)). While pro-BDNF interacts with the low-affinity neurotrophin receptor p75, mature BDNF signals through its high-affinity tropomyosin-related kinase B (TrkB) receptor, which activates several downstream effectors. Moreover, a functional Val66Met polymorphism found in the human pro-BDNF gene induced an impairment of the activity-dependent dendritic trafficking and secretion of mature BDNF protein in rat brain.
Given the complexity of BDNF system in terms of transcriptional, translational and post-translational regulatory mechanisms (Citation10), pathological alteration of BDNF may lead to defects in neuronal survival and regeneration as well as structural brain abnormalities and reduced neuronal plasticity, which in turn may impair the individual’s ability to adapt to crisis situation. Because of the role played by BDNF in regulating structural, synaptic, and morphological plasticity, BDNF has gained the most attention in the understanding of the mechanisms underlying stress-related mood disorders (). Numerous evidence from preclinical and clinical studies demonstrated that the stress-related pathophysiology of MDD is accompanied by a down-regulation of BDNF and TrkB expression, which in turn contributes to atrophy and cell loss in various brain regions, including the hippocampus (Citation15–17). It is known that major depression and inflammation are linked in many ways. Of particular interest in this context are findings showing that inflammatory events compromise neuroplasticity via reduction of BDNF and may lead in this way to the development of depression (reviewed in (Citation18)). Accordingly, several studies demonstrated that inflammation induced a significant decrease in the expression of specific BDNF variants within the rat brain (Citation19,Citation20).
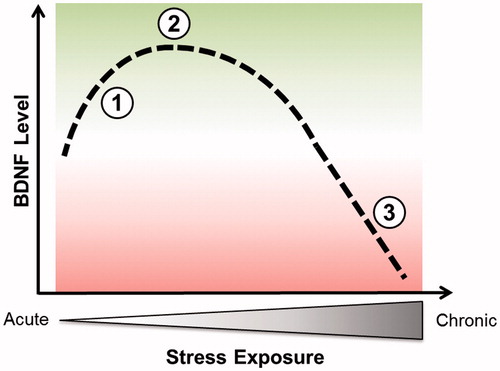
Figure 1. Conceptual representation of changes in BDNF level induced by stress: a biphasic process. Point 1 illustrates the increase of BDNF during acute stress while point 2 corresponds to a maximal benefit of short-term stress on BDNF level. Points 1 and 2 might be linked to an induced increase of synaptic transmission and plasticity. Point 3 illustrates the deleterious effects of chronic stress on BDNF content.
Concomitantly, the “neurotrophin hypothesis of depression” postulates that stress-related mood disorders result from a stress-induced decrease in BDNF expression (Citation7). The BDNF hypothesis predicts that agents that promote BDNF function might be clinically effective antidepressants. Over the years, numerous – but not the entirety of all – studies showed that antidepressant interventions were able to reverse depressive behaviours while concurrently increasing BDNF levels (Citation15,Citation21). But increases in BDNF serum levels during antidepressant treatment appear to be confined to certain antidepressants, do not always parallel clinical characteristics like depression severity and may depend on therapy response (Citation21,Citation22). Of note, lack of BDNF alone may not be sufficient to explain the pathology of stress-related mental disorders (Citation23–25), but it certainly has a prominent role in the pathophysiology of these disorders. Given that BDNF plays an important role in the pathophysiology of the stress response and in the pathogenesis of stress-associated mood disorders, and that its restoration may represent a critical mechanism underlying antidepressant therapeutic effect, many investigators have focused on serum BDNF as a “biomarker” predicting the therapy outcome in MDD, since the protein can cross the blood brain barrier and circulating BDNF was correspondingly associated with cortical BDNF concentrations (Citation16).
Sleep has a critical role in cognitive functioning including the consolidation of synaptic plasticity and long-term memory. Stress itself often disrupts sleep. Moreover, sleep disturbances such as insomnia following stressful events may further intensify the long-term changes in the brain occurring at the level of synaptic plasticity (Citation26). Conversely, stress exposure after partial or total sleep loss impairs the hypothalamic–pituitary–adrenal (HPA) response to an increase of cortisol (Citation27,Citation28). Hence, sleep disturbances may result in higher stress vulnerability, reduced environmental adaptation and cognitive impairment. Interestingly, the introduction of SD therapy (SDT) in the 1970–80s was a major step forward in the sense of its rapid and fast (within 24 h) recovery effect on depressive symptoms (Citation29,Citation30). Particularly, it has been shown that rapid eye movement (REM)-SD in MDD patients leads to a gradual improvement (Citation30), suggesting that the antidepressant effects of both selective REM-SD and total SD are based on the suppression of slow wave activity (SWA) (Citation31). Although fast-acting, SDT alone is not sufficient in most of the cases since the rate of relapse following the recovery night is rather high. However, the evidence for the rapid and robust efficacy of SDT in mood disorders is more sustainable for weeks to months when combined with antidepressant medication and/or chronotherapies (Citation32–34). Transcriptomic studies in different species through the sleep–wake cycle and after SD revealed that genes encoding proteins involved in synaptic plasticity (e.g. BDNF), specifically long-term potentiation (LTP), were up-regulated during wakefulness and down-regulated during sleep (Citation1,Citation35–37). Particularly, while short-term total SD appears to up-regulate the expression of BDNF in various regions of the brain (Citation38–42), selective SD like REM or paradoxical SD in rats showed no alterations of BDNF expression (Citation43). However, little is known regarding how the interactions between stress and sleep affect sleep-dependent plasticity and cognition, especially those explaining the rapid, even though temporary antidepressant effect of SD, since a relapse into depression occurs in most patients following the recovery night (Citation44).
Challenges and progress towards understanding the role of BDNF in stress-related mental disorders regarding the interplay of stress and the sleep homeostasis may lead to new alternative treatment strategies, providing advantages over the currently available antidepressants that are characterized by significant limitations (i.e. low response rates, treatment resistance, high incidence of relapse, and a time-lag of weeks to months for a response), highlighting a major unmet need for more efficacious and faster-acting antidepressants (Citation45).
BDNF, sleep and insomnia
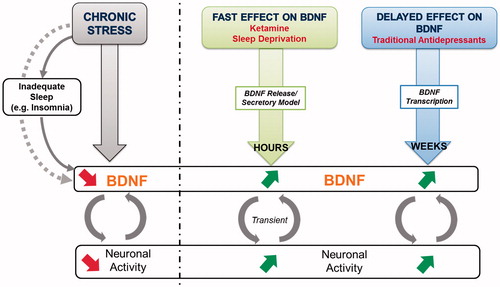
Sleep is an essential component of human cognitive performance and health. It is generally accepted that our modern society and its resulting stressors impact sleep in a negative way by altering both sleep quality and duration (Citation46). Serotonin is a major neurotransmitter candidate playing a prominent role in the regulation of both sleep–wake circadian cycles as well as in the modulation of mood states in humans (Citation47). In addition, it is known that BDNF production is stimulated by serotonin and conversely BDNF increases serotonergic signalling (Citation4,Citation48). BDNF has been shown to induce SD and spontaneous wakefulness in animals (Citation49). Moreover, a direct link with BDNF as prominent actor has been identified between the synaptic plasticity triggered by waking activities and the homeostatic sleep response (Citation50). Particularly, the degree of BDNF expression during wakefulness is directly correlated to the extent of SWA, a sensitive marker for sleep pressure and sleep need, in the subsequent period (Citation8). Support for a role of BDNF in human sleep regulation comes also from a recent study reporting impaired intensity of SWA in non-rapid eye movement (NREM) sleep in Val66Met carriers relative to Val/Val homozygotes under basal conditions and immediately following a 40 h period of waking (Citation51). Although the exact mechanism by which BDNF induces an increase in sleep SWA remains uncertain, it has been proposed that the coupling of waking-associated synaptic potentiation among connected cells may directly contribute to SWA increase (Citation8). Since the body’s stress system plays a critical role in adapting to a continuously changing and challenging environment, it is a major question how stress-related disruption of sleep may impair BDNF contents and degrade sleep function. Stress has been associated with the activation of the HPA axis. The HPA axis is a stress-responsive system that has been implicated in both mood regulation (particularly depression) and cognitive functioning. The system consists of forward-stimulating arms as well as feedback loops that together regulate the production of stress hormones including cortisol. The fact that the beginning and the end of sleep are both involved in the HPA axis activity provides a clue to estimate the effects of stress on sleep (Citation52). Chronic stress can lead to a deregulation in this biological stress system (Citation53–57), which was suggested to decrease BDNF levels in animal studies (Citation58–61) as well as in stress-related mood disorders (Citation16,Citation62,Citation63) (). Sleep problems are common features in many stress-related disorders; problems that may lead to impairment of physical and mental health since sleep loss is often followed by higher stress vulnerability (Citation64). Insomnia is considered as a state of 24-h hyperarousal rather than sleep loss and affects up to one third of the general population (Citation65). Epidemiological and clinical studies showed that a higher number of insomnia subjects also suffer from a concomitant mood disorder, mainly depression or an anxiety disorder (Citation66,Citation67). Stress-related insomnia is transient and persists for only few days. Of note, persistent insomnia increases the risk of relapse into a new MDD episode (Citation68). Although a majority of studies have concentrated on specifying the role of BDNF in depression, the mechanism underlying the effect of the sleep on the interplay between stress and BDNF was unclear until recently. Our group investigated serum BDNF levels of adults with current symptoms of insomnia and non-sleep disturbed control subjects (Citation69,Citation70). We demonstrated that subjects suffering from current symptoms of insomnia exhibited significantly decreased serum BDNF levels compared with sleep-healthy controls. In addition, serum BDNF levels were significantly correlated with severity of insomnia in all participants. Moreover, the significance of our data was further emphasized when we assessed an external control group that had recovered from occupational burnout. Again, serum BDNF levels were significantly lower in those subjects reporting symptoms of fatigue compared to sleep healthy subjects. Overall, these findings strongly support the hypothesis of increased stress vulnerability due to sleep loss, which may lead to decreased BDNF levels. A key follow-up analysis using a mediation model further suggests that sleep acts as a mediator in the interplay between stress experience and serum BDNF levels (, left panel) (Citation69), suggesting an important role of this relationship in the pathogenesis of stress-associated mental disorders. While we found reduced BDNF levels in association with insomnia, wakefulness given during SD, which can be considered as an “acute” stressor for the brain, leads to an increase in BDNF content (Citation71,Citation72). In line with a biphasic model of stress, chronic stress leads in the long-term to sleep disturbance and decrease of BDNF levels via HPA deregulation, while acute stress increases BDNF levels (). Of note, the biphasic model of stress on BDNF level is in agreement with the biphasic model of stress on the glutamate system (Citation73). The latter model argues that the acute stress-induced enhancement of glutamate transmission is dependent on activation of corticosterone receptors, while chronic stress induced opposite effects.
Figure 2. Left panel: Scheme representing the mediation model where sleep acts as key mediator in the association between stress and BDNF. Chronic stress and insomnia lead to a decrease in BDNF and synaptic activity. Right panel: fast-acting effect of ketamine and sleep deprivation versus delayed long-term effects of classical antidepressants on BDNF levels.
Major depression, sleep deprivation and circadian regulation of BDNF
Stress-related mood disorders like MDD are complex, multivariate and involve numerous neuronal substrates and brain regions. Multiple neurobiological mechanisms seem to mediate the therapeutic effects of antidepressant therapy. Some of these mechanisms, e.g. BDNF gene transcription inducing long-term potentiation, seem to play a role for neuroprotection and neurogenesis. The complexity could explain the slow onset of action in antidepressant treatments (Citation74) (). Furthermore, most currently available classical antidepressants primarily target the noradrenergic and serotonergic systems. The glutamate system has received much attention in recent years as an avenue for developing novel therapeutics since the glutamatergic system is also impaired in stress-related mood disorders (Citation75,Citation76). The use at low doses of the noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist, ketamine, has been shown to have rapid antidepressant efficacy (Citation75). In particular, patients with treatment-resistant depression benefit from sub-anaesthetic doses of ketamine with quick and persistent antidepressant effects (Citation76,Citation77). It is suggested that the NMDA receptor blockade leads to an upregulation of the AMPA receptor expression and subsequent activation of the intracellular mammalian target of rapamycin (mTOR) cascade involved in ketamine's antidepressant action (Citation78). In vitro studies also show that activation of AMPA receptors stimulates BNDF release via activation of L-type voltage-dependent calcium channels (VDCC) (Citation79). This, in turn, is followed by an increase of synaptogenesis, which might be mirrored by the neuroplasticity marker BDNF (Citation23,Citation80). In line, Haile and coworkers could demonstrate a rapid increase of plasma BDNF levels already 4 h after intravenous infusion of ketamine (Citation81). In addition to its role as an NMDA antagonist, ketamine can also alter circadian rhythms by attenuating phase-shifting responses to light. Thereby it alters the diurnal rhythms of the widespread NMDA and AMPA receptors in the suprachiasmatic nucleus and increases the expression of the core clock genes per1 and per2 via its actions on the glutamate receptors (reviewed in (Citation82)).
Some clinical studies have evaluated the changes of plasma or serum BDNF levels before and after antidepressant treatments among MDD patients and most studies report an increase of BDNF levels after antidepressant treatment (Citation83,Citation84), although very recent data indicate that the normalization of BDNF during antidepressant treatment appears to be confined to some but not all antidepressants as well as to therapy response and does not correspond with amelioration of clinical symptoms (Citation85). Nevertheless, different antidepressant strategies including antidepressant drugs and electroconvulsive therapy (ECT) were associated with an increase in BDNF levels (Citation71). Besides ketamine, manipulations of the sleep–wake cycle such as SD (total or partial) or a phase advance of the sleep period are clinically well-documented, robust, and rapid-acting methods for the treatment of severely depressed patients (Citation33). Other than SD for the whole night, partial SD (PSD) in the second half of the night has also shown comparable antidepressant effects in patients with major depression (Citation33).
Moreover, a growing body of evidence also supports a role for BDNF and its tropomyosine-related kinase receptor B (TrkB) in the modulation and mediation of circadian rhythms that appear to be disturbed in mood disorders. Alterations in rhythmic behaviours such as sleep, mood, concentration, appetite and vigilance are typical symptoms linked to depression. With regard to the link between BDNF and the HPA axis alteration in MDD, about 60% of depressed patients present a circadian deregulation of the cortisol secretion with hypercortisolemia (Citation86–88) or disturbances in the temporal pattern of secretion including a flattened circadian curve (Citation89), a reduced duration of nocturnal quiescent period (Citation90), and an earlier (Citation91) or elevated nadir (Citation92). Of note, the presence of a diurnal BDNF rhythm was also recently demonstrated in healthy humans where plasma BDNF displayed the highest concentrations in the morning, followed by a significant decrease throughout the day, and the lowest values at midnight (Citation93,Citation94). However, diurnal variation of blood BDNF was neglected until now in studies investigating MDD patients. Therefore, our group designed for the first time a study to investigate the potentially fast-acting effects of PSD on BDNF profiles in MDD patients and if a diurnal variation of serum BDNF plays a role for the therapy outcome (Citation72). Thus, post-PSD BDNF levels were investigated within hours after PSD: up to 18 h by using a 6 h interval post-PSD, specifically at 8 am, 2 pm, and 8 pm. This is in contrast to all other published studies where treatment-related BDNF changes were collected and analyzed no earlier than 1–6 weeks after antidepressive therapy with classic antidepressants.
We could confirm a similar pattern of diurnal variation of serum BDNF in patients identified as responders as it was previously reported in healthy subjects. After 6 h post-PSD, we observed an increase in BDNF level, a fast effect comparable to that of ketamine on BNDF elevation in plasma, which was detectable already after 4 h post-infusion and associated with therapeutic response. Importantly, early and rapid changes of BDNF levels after PSD and post-infusion of ketamine (Citation81) imply a rapid adaptation mechanism at the synapses, e.g. remodelling of synapses following an increase and secretion of BDNF (reviewed in (Citation6)). It can be speculated on the one hand that BDNF is secreted with a pulsatory circadian rhythm that is characterized by a progressive reduction in the amplitude of pulses throughout the day. On the other hand, it has been shown that brain BDNF is able to cross the blood brain barrier via a rapidly saturable transport system (Citation95). While BDNF peaks around midnight in rat brains and thus may exert its modulating effect on neuroplasticity and long-term potentiation (Citation96,Citation97), peripheral BDNF in the blood, namely both, of plasma and serum, peaks in the morning at 8 am and exhibits its lowest expression level at midnight, indicating that BDNF cycles in opposite phases in brain and blood. Thus, it seems legitimate to speculate that rapid changes to remodel neuronal networks involve the release of BDNF as a critical backbone, a phenomenon that might also occur in the periphery, since serum BDNF levels increase within hours ().
Furthermore and unexpectedly, not only post-PSD BDNF but also pre-PSD serum BDNF levels exhibited a diurnal pattern that was only prominent in subjects who were identified as responders. For non-responders, serum BDNF remained flat throughout the day. Accordingly, Wolkowitz and colleagues (Citation98) found that high baseline serum BDNF levels (at 10 am) before the treatment with a selective serotonin reuptake inhibitor, predicted therapy response in depressed patients. Moreover, response after treatment with duloxetine – a serotonin-norepinephrine reuptake inhibitor – in patients with MDD was also associated with a higher baseline serum BDNF concentration (Citation99). Recognition that circadian rhythm disruption might also play a key role in mood disorders has led to the development of the new antidepressant agomelatine. Recent data from various groups showed that agomelatine led to an increase in BDNF expression in treated animals, and that this effect follows a specific temporal profile (Citation100). Overall, diurnal BDNF variation seems to precede, rather than follow a favorable treatment outcome in depressed patients, suggesting that diurnal BDNF change is a prerequisite for successful therapy response independent from the specific treatment strategy, e.g. antidepressant drugs or SD.
BDNF, acute and chronic stress
In summary, our findings are in line with the hypothesis of an increased stress vulnerability due to sleep loss which may lead to a decrease in BDNF. Such a decrease might be associated with a decrease in BDNF mRNA expression in peripheral blood mononuclear cells and/or might correspond to a BDNF decline in the brain. To what extent peripheral BDNF corresponds to brain BDNF level is still not known. Nevertheless, the use of serum BDNF as potential indicator of brain alteration was justified by substantial evidence, but a time delay to monitor brain tissue alterations in the periphery should be taken into account (Citation101). While we report a reduction of BDNF levels linked to sleep disturbance reflecting chronic stress on the one side, we and others consistently showed that prolonged wakefulness caused by SD (partial or total), which can be considered as an acute stressor for the brain, leads to a rapid increase of BDNF (Citation1,Citation36). However, little is known about the differential effects of chronic and acute stress on the molecular level. Nevertheless, previous studies provide valuable leads of a biphasic modulation of hippocampal plasticity by stress, demonstrating that the same chronic stress that elicited hippocampal dendritic atrophy also reduced levels of BDNF in the rodent hippocampus while acute stress triggered opposite effects on BDNF levels () (Citation25,Citation59,Citation102). This suggests that BDNF could be a final point of convergence for the stress-induced effects in the hippocampus. BDNF-mediated signalling is involved in stress response, but the direction and nature of signalling seems to be region-specific (e.g. hippocampus versus amygdala), stress-specific and influenced by epigenetic modifications along with post-translational modifications (Citation102).
Using the biphasic model of stress on BDNF (), we hypothesise that chronic stress might induce a deregulation of the HPA system leading in the long-term to sleep disturbance and decreased BDNF levels. In this scenario, sleep is a key mediator at the connection between stress and BDNF (, left panel) and inadequate sleep, such as insomnia, can amplify the depletion in BDNF levels. In contrast, an acute stressor like SD, e.g. one night of SD, can be used as a fast-acting therapeutic intervention in some insomniac or depressed patients as a compensatory process to normalize BDNF levels.
Conclusion
We discuss here recent new insights regarding the association of sleep with BDNF, the stress-induced BDNF modifications that may be mediated by sleep and, the effect of PSD as fast-acting but transient antidepressant on serum BDNF levels ( and ).
Besides the biological link between BDNF and sleep, stress experience and subjective sleep perception interact with each other and directly affect BDNF levels, which eventually amplify the susceptibility to stress-associated mood disorders. Similar to ketamine, SD and PSD are able to induce a fast increase in serum BDNF levels within few hours. This is in contrast to the rather “delayed” effects of classical antidepressants (up to 6 weeks). Given the complexities of the pathogenesis of stress-related mood disorder, further studies including the monitoring of insomnia and sleep should be conducted. Especially the molecular mechanisms underlying the rapid response of ketamine and PSD on BDNF deserve further investigation as well as the downstream signalling pathways of BDNF and the relationship between BDNF and cortisol with regard to their role in the HPA axis deregulation.
Acknowledgements
This work was supported by the Swiss National Science Foundation (32003B_149317).
Declaration of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Gronli J, Soule J, Bramham CR. Sleep and protein synthesis-dependent synaptic plasticity: impacts of sleep loss and stress. Front Behav Neurosci. 2013;7:224.
- McEwen BS, Gianaros PJ. Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Ann N Y Acad Sci. 2010;1186:190–222.
- Juric DM, Loncar D, Carman-Krzan M. Noradrenergic stimulation of BDNF synthesis in astrocytes: mediation via alpha1- and beta1/beta2-adrenergic receptors. Neurochem Int. 2008;52:297–306.
- Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. 2008;33:73–83.
- Homberg JR, Molteni R, Calabrese F, Riva MA. The serotonin-BDNF duo: developmental implications for the vulnerability to psychopathology. Neurosci Biobehav Rev. 2014;43:35–47.
- Duman RS. Neurobiology of stress, depression, and rapid acting antidepressants: remodeling synaptic connections. Depress Anxiety. 2014;31:291–6.
- Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606.
- Faraguna U, Vyazovskiy VV, Nelson AB, Tononi G, Cirelli C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J Neurosci. 2008;28:4088–95.
- McAllister AK. Spatially restricted actions of BDNF. Neuron. 2002;36:549–50.
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–35.
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12.
- Liu QR, Walther D, Drgon T, Polesskaya O, Lesnick TG, Strain KJ, et al. Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson's Disease. Am J Med Genet B, Neuropsychiatr Genet. 2005;134B:93–103.
- Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Prog Neurobiol. 2003;69:341–74.
- Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64:238–58.
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–27.
- Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry JM. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 2002;109:143–8.
- Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16:239–49.
- Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R. Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front Cell Neurosci. 2014;8:430.
- Chapman TR, Barrientos RM, Ahrendsen JT, Hoover JM, Maier SF, Patterson SL. Aging and infection reduce expression of specific brain-derived neurotrophic factor mRNAs in hippocampus. Neurobiol Aging. 2012;33:832.e1–14.
- Thibault K, Lin WK, Rancillac A, Fan M, Snollaerts T, Sordoillet V, et al. BDNF-dependent plasticity induced by peripheral inflammation in the primary sensory and the cingulate cortex triggers cold allodynia and reveals a major role for endogenous BDNF as a tuner of the affective aspect of pain. J Neurosci. 2014;34:14739–51.
- Molendijk ML, Bus BA, Spinhoven P, Penninx BW, Kenis G, Prickaerts J, et al. Serum levels of brain-derived neurotrophic factor in major depressive disorder: state-trait issues, clinical features and pharmacological treatment. Mol Psychiatry. 2011;16:1088–95.
- Tadic A, Wagner S, Schlicht KF, Peetz D, Borysenko L, Dreimuller N, et al. The early non-increase of serum BDNF predicts failure of antidepressant treatment in patients with major depression: a pilot study. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35:415–20.
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–5.
- Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat Neurosci. 2007;10:1089–93.
- Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15(3 Pt 1):1768–77.
- Ziegenhorn AA, Schulte-Herbruggen O, Danker-Hopfe H, Malbranc M, Hartung HD, Anders D, et al. Serum neurotrophins—a study on the time course and influencing factors in a large old age sample. Neurobiol Aging. 2007;28:1436–45.
- Leproult R, Copinschi G, Buxton O, Van Cauter E. Sleep loss results in an elevation of cortisol levels the next evening. Sleep. 1997;20:865–70.
- Meerlo P, Koehl M, van der Borght K, Turek FW. Sleep restriction alters the hypothalamic-pituitary-adrenal response to stress. J Neuroendocrinol. 2002;14:397–402.
- Pflug B, Tolle R. Disturbance of the 24-hour rhythm in endogenous depression and the treatment of endogenous depression by sleep deprivation. Int Pharmacopsychiatry. 1971;6:187–96.
- Vogel GW, Vogel F, McAbee RS, Thurmond AJ. Improvement of depression by REM sleep deprivation. New findings and a theory. Arch Gen Psychiatry. 1980;37:247–53.
- Beersma DG, van den Hoofdakker RH. Can non-REM sleep be depressogenic? J Affect Disord. 1992;24:101–8.
- Bunney BG, Bunney WE. Mechanisms of rapid antidepressant effects of sleep deprivation therapy: clock genes and circadian rhythms. Biol Psychiatry. 2013;73:1164–71.
- Giedke H, Schwarzler F. Therapeutic use of sleep deprivation in depression. Sleep Med Rev. 2002;6:361–77.
- Wu JC, Kelsoe JR, Schachat C, Bunney BG, DeModena A, Golshan S, et al. Rapid and sustained antidepressant response with sleep deprivation and chronotherapy in bipolar disorder. Biol Psychiatry. 2009;66:298–301.
- Tononi G, Cirelli C. Modulation of brain gene expression during sleep and wakefulness: a review of recent findings. Neuropsychopharmacology. 2001;25:S28–35.
- Elliott AS, Huber JD, O'Callaghan JP, Rosen CL, Miller DB. A review of sleep deprivation studies evaluating the brain transcriptome. SpringerPlus. 2014;3:728.
- Porkka-Heiskanen T. Gene expression during sleep, wakefulness and sleep deprivation. Front Biosci. 2003;8:s421–37.
- Cirelli C, Tononi G. Gene expression in the brain across the sleep-waking cycle. Brain Res. 2000;885:303–21.
- Fujihara H, Sei H, Morita Y, Ueta Y, Morita K. Short-term sleep disturbance enhances brain-derived neurotrophic factor gene expression in rat hippocampus by acting as internal stressor. J Mol Neurosci. 2003;21:223–32.
- Guzman-Marin R, Ying Z, Suntsova N, Methippara M, Bashir T, Szymusiak R, et al. Suppression of hippocampal plasticity-related gene expression by sleep deprivation in rats. J Physiol. 2006;575:807–19.
- Hairston IS, Peyron C, Denning DP, Ruby NF, Flores J, Sapolsky RM, et al. Sleep deprivation effects on growth factor expression in neonatal rats: a potential role for BDNF in the mediation of delta power. J Neurophysiol. 2004;91:1586–95.
- Taishi P, Sanchez C, Wang Y, Fang J, Harding JW, Krueger JM. Conditions that affect sleep alter the expression of molecules associated with synaptic plasticity. Am J Physiol Regul Integr Compar Physiol. 2001;281:R839–45.
- Lee KS, Alvarenga TA, Guindalini C, Andersen ML, Castro RM, Tufik S. Validation of commonly used reference genes for sleep-related gene expression studies. BMC Mol Biol. 2009;10:45.
- Beck J, Hemmeter U, Brand S, Muheim F, Hatzinger M, Holsboer-Trachsler E. Modafinil reduces microsleep during partial sleep deprivation in depressed patients. J Psychiatr Res. 2010;44:853–64.
- Weizman S, Gonda X, Dome P, Faludi G. Pharmacogenetics of antidepressive drugs: a way towards personalized treatment of major depressive disorder. Neuropsychopharmacol Hungar. 2012;14:87–101.
- Brunborg GS, Mentzoni RA, Molde H, Myrseth H, Skouveroe KJ, Bjorvatn B, et al. The relationship between media use in the bedroom, sleep habits and symptoms of insomnia. J Sleep Res. 2011;20:569–75.
- Dugovic C. Role of serotonin in sleep mechanisms. Rev Neurol. 2001;157:S16–19.
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–94.
- Cirelli C. Cellular consequences of sleep deprivation in the brain. Sleep Med Rev. 2006;10:307–21.
- Huber R, Esser SK, Ferrarelli F, Massimini M, Peterson MJ, Tononi G. TMS-induced cortical potentiation during wakefulness locally increases slow wave activity during sleep. PLoS One. 2007;2:e276.
- Bachmann V, Klein C, Bodenmann S, Schafer N, Berger W, Brugger P, et al. The BDNF Val66Met polymorphism modulates sleep intensity: EEG frequency- and state-specificity. Sleep. 2012;35:335–44.
- Han KS, Kim L, Shim I. Stress and sleep disorder. Exp Neurobiol. 2012;21:141–50.
- Holsboer F, Ising M. Stress hormone regulation: biological role and translation into therapy. Annu Rev Psychol. 2010;61:81–109. C1-11.
- Miller GE, Chen E, Zhou ES. If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychol Bull. 2007;133:25–45.
- Morsink MC, Steenbergen PJ, Vos JB, Karst H, Joels M, De Kloet ER, et al. Acute activation of hippocampal glucocorticoid receptors results in different waves of gene expression throughout time. J Neuroendocrinol. 2006;18:239–52.
- Schulkin J, Gold PW, McEwen BS. Induction of corticotropin-releasing hormone gene expression by glucocorticoids: implication for understanding the states of fear and anxiety and allostatic load. Psychoneuroendocrinology. 1998;23:219–43.
- Schulte-Herbruggen O, Chourbaji S, Ridder S, Brandwein C, Gass P, Hortnagl H, et al. Stress-resistant mice overexpressing glucocorticoid receptors display enhanced BDNF in the amygdala and hippocampus with unchanged NGF and serotonergic function. Psychoneuroendocrinology. 2006;31:1266–77.
- Murakami S, Imbe H, Morikawa Y, Kubo C, Senba E. Chronic stress, as well as acute stress, reduces BDNF mRNA expression in the rat hippocampus but less robustly. Neurosci Res. 2005;53:129–39.
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–47.
- Roceri M, Cirulli F, Pessina C, Peretto P, Racagni G, Riva MA. Postnatal repeated maternal deprivation produces age-dependent changes of brain-derived neurotrophic factor expression in selected rat brain regions. Biol Psychiatry. 2004;55:708–14.
- Ueyama T, Kawai Y, Nemoto K, Sekimoto M, Tone S, Senba E. Immobilization stress reduced the expression of neurotrophins and their receptors in the rat brain. Neurosci Res. 1997;28:103–10.
- Diniz BS, Teixeira AL, Talib LL, Mendonca VA, Gattaz WF, Forlenza OV. Serum brain-derived neurotrophic factor level is reduced in antidepressant-free patients with late-life depression. World J Biol Psychiatry. 2010;11:550–5.
- Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, et al. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. 2003;54:70–5.
- Morin CM, Rodrigue S, Ivers H. Role of stress, arousal, and coping skills in primary insomnia. Psychosom Med. 2003;65:259–67.
- Basta M, Chrousos GP, Vela-Bueno A, Vgontzas AN. Chronic insomnia and stress system. Sleep Med Clin. 2007;2:279–91.
- Staner L. Sleep and anxiety disorders. Dialog Clin Neurosci. 2003;5:249–58.
- Ohayon MM, Roth T. Place of chronic insomnia in the course of depressive and anxiety disorders. J Psychiatr Res. 2003;37:9–15.
- Pigeon WR, Hegel M, Unutzer J, Fan MY, Sateia MJ, Lyness JM, et al. Is insomnia a perpetuating factor for late-life depression in the IMPACT cohort? Sleep. 2008;31:481–8.
- Giese M, Unternaehrer E, Brand S, Calabrese P, Holsboer-Trachsler E, Eckert A. The interplay of stress and sleep impacts BDNF level. PLoS One. 2013;8:e76050.
- Giese M, Unternahrer E, Huttig H, Beck J, Brand S, Calabrese P, et al. BDNF: an indicator of insomnia? Mol Psychiatry. 2014;19:151–2.
- Conti B, Maier R, Barr AM, Morale MC, Lu X, Sanna PP, et al. Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol Psychiatry. 2007;12:167–89.
- Giese M, Beck J, Brand S, Muheim F, Hemmeter U, Hatzinger M, et al. Fast BDNF serum level increase and diurnal BDNF oscillations are associated with therapeutic response after partial sleep deprivation. J Psychiatr Res. 2014;59:1–7.
- Musazzi L, Treccani G, Popoli M. Functional and structural remodeling of glutamate synapses in prefrontal and frontal cortex induced by behavioral stress. Front Psychiatry. 2015;6:60.
- Duman RS. Neural plasticity: consequences of stress and actions of antidepressant treatment. Dialog Clin Neurosci. 2004;6:157–69.
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4.
- Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64.
- Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–42.
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–64.
- Lepack AE, Fuchikami M, Dwyer JM, Banasr M, Duman RS. BDNF release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol. 2015;18:1–16 . [Epub ahead of print]. DOI: 10.1093/ijnp/pyu033.
- Duman RS, Li N. A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. Philos Trans R Soc Lond Ser B Biol Sci. 2012;367:2475–84.
- Haile CN, Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Foulkes A, et al. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int J Neuropsychopharmacol. 2014;17:331–6.
- Bunney BG, Bunney WE. Rapid-acting antidepressant strategies: mechanisms of action. Int J Neuropsychopharmacol. 2012;15:695–713.
- Brunoni AR, Valiengo L, Baccaro A, Zanao TA, de Oliveira JF, Vieira GP, et al. Sertraline vs. ELectrical Current Therapy for Treating Depression Clinical Trial—SELECT TDCS: design, rationale and objectives. Contemporary Clin Trials. 2011;32:90–8.
- Piccinni A, Marazziti D, Catena M, Domenici L, Del Debbio A, Bianchi C, et al. Plasma and serum brain-derived neurotrophic factor (BDNF) in depressed patients during 1 year of antidepressant treatments. J Affect Disord. 2008;105:279–83.
- Molendijk ML, Bus BA, Spinhoven P, Penninx BW, Prickaerts J, Oude Voshaar RC, et al. Gender specific associations of serum levels of brain-derived neurotrophic factor in anxiety. World J Biol Psychiatry. 2012;13:535–43.
- Gold PW, Loriaux DL, Roy A, Kling MA, Calabrese JR, Kellner CH, et al. Responses to corticotropin-releasing hormone in the hypercortisolism of depression and Cushing's disease. Pathophysiologic and diagnostic implications. N Engl J Med. 1986;314:1329–35.
- Murphy BE. Treatment of major depression with steroid suppressive drugs. J Steroid Biochem Mol Biol. 1991;39:239–44.
- Murphy BE. Steroids and depression. J Steroid Biochem Mol Biol. 1991;38:537–59.
- Deuschle M, Schweiger U, Weber B, Gotthardt U, Korner A, Schmider J, et al. Diurnal activity and pulsatility of the hypothalamus-pituitary-adrenal system in male depressed patients and healthy controls. J Clin Endocrinol Metab. 1997;82:234–8.
- Halbreich U, Asnis GM, Shindledecker R, Zumoff B, Nathan RS. Cortisol secretion in endogenous depression. I. Basal plasma levels. Arch Gen Psychiatry. 1985;42:904–8.
- Pfohl B, Sherman B, Schlechte J, Stone R. Pituitary–adrenal axis rhythm disturbances in psychiatric depression. Arch Gen Psychiatry. 1985;42:897–903.
- Yehuda R, Teicher MH, Trestman RL, Levengood RA, Siever LJ. Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis. Biol Psychiatry. 1996;40:79–88.
- Begliuomini S, Lenzi E, Ninni F, Casarosa E, Merlini S, Pluchino N, et al. Plasma brain-derived neurotrophic factor daily variations in men: correlation with cortisol circadian rhythm. J Endocrinol. 2008;197:429–35.
- Pluchino N, Cubeddu A, Begliuomini S, Merlini S, Giannini A, Bucci F, et al. Daily variation of brain-derived neurotrophic factor and cortisol in women with normal menstrual cycles, undergoing oral contraception and in postmenopause. Hum Reprod. 2009;24:2303–9.
- Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ. Transport of brain-derived neurotrophic factor across the blood–brain barrier. Neuropharmacology. 1998;37:1553–61.
- Liang FQ, Sohrabji F, Miranda R, Earnest B, Earnest D. Expression of brain-derived neurotrophic factor and its cognate receptor, TrkB, in the rat suprachiasmatic nucleus. Exp Neurol. 1998;151:184–93.
- Liang FQ, Walline R, Earnest DJ. Circadian rhythm of brain-derived neurotrophic factor in the rat suprachiasmatic nucleus. Neurosci Lett. 1998;242:89–92.
- Wolkowitz OM, Wolf J, Shelly W, Rosser R, Burke HM, Lerner GK, et al. Serum BDNF levels before treatment predict SSRI response in depression. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35:1623–30.
- Mikoteit T, Beck J, Eckert A, Hemmeter U, Brand S, Bischof R, et al. High baseline BDNF serum levels and early psychopathological improvement are predictive of treatment outcome in major depression. Psychopharmacology. 2014;231:2955–65.
- Soumier A, Banasr M, Lortet S, Masmejean F, Bernard N, Kerkerian-Le-Goff L, et al. Mechanisms contributing to the phase-dependent regulation of neurogenesis by the novel antidepressant, agomelatine, in the adult rat hippocampus. Neuropsychopharmacology. 2009;34:2390–403.
- Sartorius A, Hellweg R, Litzke J, Vogt M, Dormann C, Vollmayr B, et al. Correlations and discrepancies between serum and brain tissue levels of neurotrophins after electroconvulsive treatment in rats. Pharmacopsychiatry. 2009;42:270–6.
- Lakshminarasimhan H, Chattarji S. Stress leads to contrasting effects on the levels of brain derived neurotrophic factor in the hippocampus and amygdala. PLoS One. 2012;7:e30481.