
Abstract
Mast cell (MC) disease has long been thought to be just the rare disease of mastocytosis (in various forms, principally cutaneous and systemic), with aberrant MC mediator release at symptomatic levels due to neoplastic MC proliferation. Recent discoveries now show a new view is in order, with mastocytosis capping a metaphorical iceberg now called “MC activation disease” (MCAD, i.e. disease principally manifesting inappropriate MC activation), with the bulk of the iceberg being the recently recognized “MC activation syndrome” (MCAS), featuring inappropriate MC activation to symptomatic levels with little to no inappropriate MC proliferation. Given increasing appreciation of a great menagerie of mutations in MC regulatory elements in mastocytosis and MCAS, the great heterogeneity of MCAD’s clinical presentation is unsurprising. Most MCAD patients present with decades of chronic multisystem polymorbidity generally of an inflammatory ± allergic theme. Preliminary epidemiologic investigation suggests MCAD, while often misrecognized, may be substantially prevalent, making it increasingly important that practitioners of all stripes learn how to recognize its more common forms such as MCAS. We review the diagnostically challenging presentation of MCAD (with an emphasis on MCAS) and current thoughts regarding its biology, epidemiology, natural history, diagnostic evaluation, and treatment.
Recent discoveries show a new view of the realm of mast cell disease is in order, with mastocytosis capping a metaphorical iceberg now termed “MC activation disease” (MCAD, i.e. disease principally manifesting inappropriate MC activation) and with the bulk of the iceberg being comprised of the recently recognized “MC activation syndrome” (MCAS), featuring inappropriate MC activation to symptomatic levels with little to no inappropriate MC proliferation.
Most MCAD patients present with decades of chronic multisystem polymorbidity generally of an inflammatory ± allergic theme, but diagnostic recognition is challenged by great heterogeneity of clinical presentation which may be due to great underlying mutational heterogeneity in assorted mast cell regulatory elements.
Although few biomarkers predictive of helpful therapy are yet available, most MCAD patients are able to eventually identify significantly helpful therapy by persistently and methodically stepping through trials of the many treatments shown helpful across this patient population.
Key messages
Introduction
Mast cell (MC) disease has long been thought to be just the one rare disease of mastocytosis, hallmarked by MC neoplasia and divided principally into cutaneous mastocytosis (CM) and systemic mastocytosis (SM, with several subtypes, Supplementary Table 1) (Citation1). Most SM harbors constitutively activating mutations in codon 816 of transmembrane tyrosine kinase KIT, the dominant MC regulator (Citation2), driving classic SM features (MC proliferation, spindling, aggregation, tryptase overexpression, and CD25 expression) (Citation3–5). Most mastocytosis symptoms result from dysregulated production/release of MC mediators (e.g. histamine, tryptase, chymase, heparin, interleukins, eicosanoids) and correlate poorly with tumor load. Suspicion of disease of primary aberrant MC activation (MCA), sans proliferation, arose a quarter century ago (Citation6). In 2007, the first such cases (now termed “MCA syndrome” (MCAS)) were reported (Citation7,Citation8), followed by discovery of many KIT mutations in MCAS (Citation9,Citation10) (exclusive of codon 816 but nevertheless seemingly constitutively activating). Similar mutational menageries in KIT and other MC regulatory elements have been found in mastocytosis (Citation11), suggesting such biology is operant in MCAS and engendering a term of “MC activation disease” (MCAD) encompassing mastocytosis and MCAS. MCAS seems prevalent and may underlie much chronic inflammatory disease. We review the presentation and other aspects of MCAD.
Presentation
MCs soon leave their marrow birthplace, briefly circulate, and ultimately site (sparsely) in all tissues, preferentially at environmental interfaces and lymphovascular tissue, optimally positioned to serve as sentinels of adverse environmental change (Citation2). Although the MC’s mediator repertoire varies with siting, collectively MCs express more than 200 mediators, each with many effects (Citation12). In MCAD, constitutive MC mediator production/release and inappropriate MC reactivity directly and indirectly effect diverse local and distant reactions in all tissues/systems. Due to MCs’ wide distribution, enormous range in normal biological activities, and great heterogeneity in mediator expression patterns, diagnostically challenging extreme heterogeneity in clinical presentation of MCAD is the norm.
Many MC mediators produce inflammatory effects, creating MCAD’s nebulous clinical signature of multisystem polymorbidity of generally (but not necessarily) inflammatory nature (). Classic flushing and anaphylaxis often spur consideration of MCAD, especially SM, but less distinctive symptoms often are the only signs, explaining typically great latency in diagnosis of SM (Citation13) and MCAS (Citation14). Symptoms course chronically or episodically, waxing and waning with varying frequencies and amplitudes. Opposite effects appear in different patients (e.g. polycythemia (Citation15) versus red cell aplasia (Citation16)), at different times (e.g. alternating diarrhea and constipation (e.g. Citation17) as in irritable bowel syndrome (IBS, increasingly linked to MCA (e.g. Citation18,Citation19))), and in different sites (e.g. co-existing (heparin-induced) osteoporosis and (histamine-induced) osteosclerosis (e.g. Citation17,Citation20)).
Table 1. Symptoms and findings in mast cell (MC) activation disease (Citation73).
Aberrant reactivities – dietary, medication, and/or environmental – may be numerous. Medication reactivities often are directed against excipients. Some intolerances (e.g. acetaminophen, iodine, airplanes) may seem odd until recognition of excipient (e.g. povidone in iodine–povidone sterilization solution) or physical triggers (e.g. bathwater’s heat, barometric changes during air travel).
Likely prevalent, MCAD (especially MCAS) is reasonable to suspect in poorly explained chronic multisystem polymorbidity (particularly if inflammatory). MCAD patients often acquire many idiopathic diagnoses (Supplemental Table 4). MCAD may be present if definitively diagnosed illness (e.g. low-stage lymphoma) accounts poorly for certain findings (e.g. recurrent presyncope). Of note, MCAD both may be comorbid with definitively diagnosed illness and can cause such illness.
Biology
MCs putatively arose in multicellular eukaryotes 500 million years ago (Citation21). Their evolved arrays of sensory and response mechanisms engender diverse havoc when MC dysfunction emerges.
Immature MCs arise in marrow from myeloid progenitors and complete maturation peripherally. White adipose tissue, too, is a MC progenitor reservoir (Citation22). MCs secrete pre-stored mediators and synthesize de novo mediators in response to allergic, microbial, and non-immune triggers. MCs are immune effectors and regulators, central in innate and adaptive immunity (Citation23). MCs are sensitive to numerous stimuli, activating various intracellular pathways which intersect to modulate character and magnitude of response. The best characterized MCA mechanism is antigenic cross-linking of IgE bound to MC-surface FcɛRI. IgE-independent MCA (and inhibition) occurs via many other receptors, too. MCs exhibit tissue-specific regulation and mediator expression. For example, connective tissue MCs express tryptase and chymase (MCTCs), contain more heparin, and respond to neuropeptides in contrast to mucosal MCs, which express tryptase but not chymase (MCTs), contain less heparin, and do not respond to neuropeptides. Other MC phenotypes exist, and MCs can transform their phenotypes (Citation24).
Apart from critical involvement in allergy, MC mediators critically guide development, and maintain integrity/function, in all tissues (Citation12,Citation25). MCs regulate host defense by acting as innate immune cells, interacting with the specific immune system, inducing/regulating inflammation, and recruiting other immune cells. MCs orchestrate microbial, toxic, and physical environmental defense through classic non-selective degranulation, selective mediator release (“piecemeal” or “differential” degranulation), and transgranulation (e.g. Citation26,Citation27). MCs also aid wound healing, tissue remodeling, and degrading certain endogenous toxins (e.g. endothelin-1 or neurotensin) in bacterial infection. The fine regulation of these mechanisms infers potential for diverse havoc from dysregulated MCs.
Human MCs express KIT 10-fold more brightly than other cells (Citation2,Citation28). Binding of stem cell factor to homodimeric KIT conformationally alters KIT’s intracellular domains, autoinhibiting KIT at the juxtamembrane domain and activating KIT’s two kinase domains. Downstream pathways promote MC survival, mediator production/release, and other functions. Various mutational patterns, then, cause various clinical consequences. KITD816V is found in most (>90%) SM (Citation29) and may drive previously noted hallmark features. MCAS patients harbor multiple KIT mutations (Citation9,Citation10), but KITD816V appears rare and no other recurrent patterns are yet apparent. This mutational heterogeneity in KIT in MCAS (other genes not yet investigated), together with similar mutational heterogeneity in KIT and other MC regulatory elements in mastocytosis (Citation11), broadly align with clinical heterogeneity observed in MCAS and mastocytosis.
Mastocytosis, like other neoplasms, usually arises somatically (e.g. Citation11,Citation29,Citation30). Primary MCAS appears similar (Citation11,Citation31). Yet, MCAD has a familial predisposition (Citation32). Complexity multiplies further in that different affected members of a kindred usually present differently and bear different MC mutational profiles. Inheritable epigenetic mutations may create genetic fragility such that various stressors may induce progenitor mutations principally expressed in MCs (e.g. Citation11,Citation31,Citation33). Lifestyle-associated factors (diet, intestinal microbiome, alcohol) may influence MCAD phenotype (Citation34,Citation35).
Classification
Mastocytosis is classified by the World Health Organization (WHO) scheme (Supplementary Table 1), but recent finding of KITD816V in marrow in 100% of a cohort of adult-onset CM patients (Citation36) suggests mastocytosis is inherently systemic. Some argue MCAD should be divided into primary (i.e. proven clonal), secondary (reactive), and idiopathic categories (Citation37); others counter MCAD’s clinical behavior is sufficiently variable, and clonality testing available in clinical laboratories is sufficiently limited, to preclude meaningful classification until full genotyping is routinely available (Citation31). Distinction of proven monoclonal MCAS (MMCAS) from “non-clonal” MCAS (Citation37–39) is of unclear value given their seemingly identical prognosis and therapeutic approach and given limitations in clonality testing (usually just PCR probes for KITD816V and flow cytometry for the CD117 + CD25+ or CD117 + CD2+ pathognomonic signatures seen in a minority of mastocytosis patients and rarely in MCAS) (Citation40). Routine comprehensive genotyping may clarify diagnosis and expedite identification of effective personalized therapies.
Epidemiology
The rarity of mastocytosis precludes accurate identification of incidence and prevalence. Only preliminary data have been reported for MCAS. For mastocytosis, in western Europe incidence is ∼5–10/1,000,000 (e.g. Citation13,Citation41), prevalence ∼5–13/100,000 (e.g. Citation41,Citation42). Cases of CM outnumber SM by ∼10:1; e.g. prevalence of SM is ∼1:364,000, or 2.7/1,000,000 (Citation31). Most CM manifests in childhood (two-thirds before age 2); most SM is diagnosed in middle age or later. In Germany, ∼5–10% of the general population may harbor MCAD (Citation31,Citation32), unsurprising given suggestions MCAS underlies some common idiopathic inflammatory diseases (e.g. fibromyalgia (e.g. Citation43,Citation44), IBS (e.g. Citation19)).
Mastocytosis occurs roughly equally in males and females (Citation45). In MCAS, a 3:1 female preponderance is observed (Citation31). Underdiagnosis is certain given recent recognition of, and difficulty diagnosing, MCAS. Given the erratic, nebulous, often non-life-threatening symptoms of MCAD, cultural issues may abet underdiagnosis in males. No racial predilection has been reported.
Natural history, prognosis, and familial predisposition
Usually recognized in retrospect, symptoms typically initially manifest in adolescence or even earlier. Younger age at initial clinical manifestation may predict less successful therapy, possibly due to epigenetic anticipation (Citation33). Childhood CM often regresses by late adolescence, but later emergence, in many such remitters, of illnesses likely attributable to MCAS questions whether spontaneous cure truly occurs.
In MCAD, variable periods of relatively stable symptoms (generally waxing and waning, but with acute flares occasionally in some and frequently in others) are punctuated by step-wise sustained escalations in baseline symptoms (possibly from subclonal evolution) soon following major stressors predictably physiologic (e.g. puberty) and unpredictably pathologic (e.g. physical or psychological trauma).
Clinical presentation is very diverse. Symptoms can occur in any organs/tissues/systems (), often temporally staggered (i.e. different symptoms at different times), fluctuating over years to decades. Symptom-free intervals eventually shorten. Finally, symptoms become chronic, still with fluctuating intensity but overall steadily increasing.
Most mastocytic neoplasia (CM and indolent SM (ISM)) behaves indolently, which does not preclude severe mediator-induced symptoms. In spite of symptom burdens, CM/ISM patients enjoy normal lifespan (Citation45,Citation46). SM with eosinophilia and other advanced mastocytosis (SM with associated hematologic malignancy, aggressive SM (ASM), and MC leukemia) confer worse prognoses (median survival 6 months with MC leukemia) (Citation45–47), although investigational therapies appear promising. Transformation of CM/ISM to advanced mastocytosis is rare; transformation of MCAS to SM has not yet been studied or reported, but the authors have never observed such.
ASM (defined by the so-called B- and C-findings, Supplementary Table 2) often rapidly progresses, causing organ dysfunction (Citation45,Citation46). Adverse prognosticators (Supplementary Table 3) include huge osteolyses, weight loss, malabsorption, hepatomegaly with portal hypertension, splenomegaly with hypersplenism, and > 5% MCs in marrow smears (Citation46,Citation48).
Many MCAD patients wonder whether ill family, or desired offspring, might bear the disease. While most MCAD-associated mutations appear somatic (Citation31), familial MCAD is common and may be epigenetically rooted (Citation32,Citation33). Irrespective of MCAD variant and gender, ∼75% of index patients have at least one first-degree affected relative (Citation32). Approximately 50% of children of MCAD patients appear affected (e.g. Citation32), but without predictors of likelihood for developing MCAD, no advice can be given regarding reproduction.
Diagnostic evaluation
All MCAD features inappropriate MCA. A validated symptom questionnaire is available to help identify patients with a mast cell mediator release syndrome (Citation40). Mastocytosis additionally features substantial MC proliferation, typically suggested by suspicious skin lesions () and/or serum tryptase persistently >20 ng/ml. Following this diagnostic fork, testing for mastocytosis focuses on finding characteristic cytoproliferation (usually, MC clusters in skin lesion or marrow biopsies), while testing for MCAS seeks biochemical evidence of activation, i.e. elevated levels of relatively specific MC mediators including tryptase, histamine or its metabolite N-methylhistamine, prostaglandin (PG) D2 or its metabolite 11-β-PGF2α, heparin, chromogranin A (in appropriate settings), or certain leukotrienes (e.g. leukotriene E4) (Citation40). Many factors routinely confound accurate testing (Citation40). Negative initial testing does not refute a presentation suggestive of MCAD, and repeat testing (ideally when particularly symptomatic) often proves diagnostic. Even absent frank neoplasia, decreased apoptosis alone sometimes drives increased density of diffusely scattered MCs in biopsies (e.g. gastrointestinal tract).
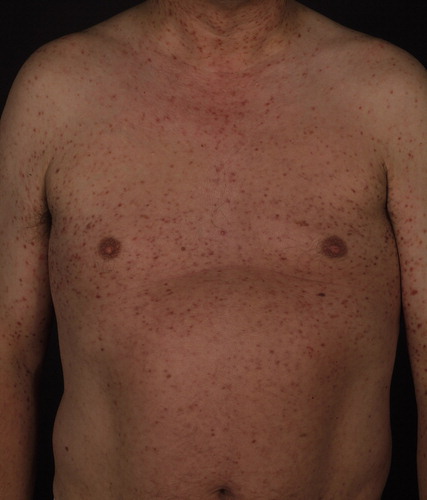
Figure 1. Urticaria pigmentosa in a patient with KITD816V-positive systemic mastocytosis.
shows a diagnostic algorithm (Citation40,Citation49). shows criteria for classifying MCAD. Although there is increasing concern that one of the minor diagnostic criteria for SM (KIT codon 816 mutation) might drive other (major and minor) criteria, at present, diagnosis of mastocytosis follows the WHO criteria. In practice, the scheme in usually diagnoses MCAS by finding chronic MCA symptoms and elevated MC mediators absent mastocytosis. Other schemes (e.g. (Citation49)) use more constrained sets of symptoms/mediators and may limit diagnosis. Of note, ∼20% of ISM patients lack MC clusters in marrow and ∼30% exhibit serum tryptase <20 ng/ml (e.g. Citation50). Hence, although an initial serum tryptase <20 ng/ml (“low-tryptase”) makes SM much less likely, initial misdiagnosis of uncommon low-tryptase SM as MCAS is possible – but may not be clinically consequential. Marrow examination in suspected SM is standard of care. However, prognosis and therapy of ISM and MCAS are presently indistinguishable, and since low-tryptase SM virtually always is indolent, indications for marrow examination when MCAS is more suspected than SM (by tryptase <20 ng/ml) remain unclear. SM due to KIT codon 816 mutations is associated with myeloid neoplasms (and, less frequently, B-cell neoplasms) frequently enough to warrant routine marrow examination when SM is suspected (e.g. tryptase >20 ng/ml or frequent unprovoked anaphylaxis). Patchiness of MC infiltration in most SM renders one-sixth of unilateral biopsies non-diagnostic (Citation51), suggesting bilateral biopsies should be standard. The frequency of associated hematologic neoplasms in marrow biopsies at time of diagnosis of MCAS has not yet been studied but in our experience appears very low. Thus, at present, marrow examination in suspected MCAS is optional.
Figure 2. Diagnostic approach to MCAD (from (Citation40)).

Table 2. Criteria proposed to define mast cell activation disease (for references, see text) when all other diagnoses that could better explain the full range and chronicity of the findings in the case have been excluded.
In the future, routinely available MC mutational assessment may simplify the process for diagnosing primary MCAD, but exclusion of differential diagnoses will remain essential in diagnosing all types of MCAD. Given the MC’s complex essential biology, which guarantees extraordinarily diverse clinical presentations across the MCAD population, it can become too easy to attribute mysterious illness to MCAD. By the time of their evaluation for MCAD, most patients have already undergone extensive prior evaluations ruling out many diagnoses, but it still behooves the clinician, prior to diagnosing any form of MCAD, to again carefully consider whether MCAD indeed explains the identified symptoms and findings better than alternative diagnoses (e.g. infection, porphyria, hereditary angioedema, etc.). For example, though anaphylaxis typically is dominantly a MC activation-driven event, a patient whose presentation is limited to anaphylaxis (idiopathic or not) would be served best by a diagnosis of anaphylaxis and by treatments targeted at controlling anaphylaxis, whereas the patient suffering not only anaphylaxis but also a bevy of other inflammatory ± allergic comorbidities, and with laboratory evidence of MC activation but no other evident unifying diagnosis such as mastocytosis, would be better served by a diagnosis of MCAS. Furthermore, diagnosis of MCAD does not constrain treatment to solely MC-targeted interventions; as an extreme example, a myocardial infarction (MI) driven by MCAD (i.e. Kounis syndrome) may attain better outcome with addition of MC-targeted treatment, but without standard MI treatment, outcome can be expected to be poor.
Treatment
Unless clearly secondary or an isolated tumor, MCAD presently is incurable. As the primary feature of MCAD is inappropriate MCA (Citation39,Citation52,Citation53), treatment invariably involves trigger identification/avoidance plus control of MC mediator production/action (Supplementary Figure 1).
Participation in therapeutic clinical trials is best for complicated MCAD patients. However, few such trials in mastocytosis have been conducted (Supplementary Figure 1), and there have been no therapeutic trials in MCAS yet. Patients with mild disease can be treated conventionally.
Although no biomarkers reflecting symptom type or severity, or predicting therapeutic response, are yet validated, the tolerability and the efficacy of most MCAD therapies become evident within 1–2 months. Therapies not significantly helpful should be halted to mitigate polypharmacy. With no response predictors yet identified, cost-based sequencing of therapeutic trials seems reasonable. Successful regimens appear highly personalized, possibly reflecting mutational heterogeneity.
Multiple simultaneous regimen changes are discouraged lest cause-effect determination be confounded. Poorly tolerated agents should prompt consideration of excipient-directed reactivity, though some drugs (e.g. cromolyn) sometimes cause initial symptom flares which usually soon abate. Temporary abstinence from gluten-, yeast-, and cow’s milk protein-containing foods during the initial month of drug therapy can improve response rate (e.g. (Citation54)), possibly more from reducing trigger exposure than truly improving medication efficacy. Immune system activators may dramatically worsen the disease.
Blockade of elevated mediators may help. NSAIDs help some MCAD patients but trigger others; aspirin desensitization may be useful. Although typically not first-line, acute and chronic immunosuppressants can be considered (Supplementary Figure 1). Glucocorticoids sometimes help control flares, but chronic toxicities can become unacceptable. Because MC mediator release can result in local and remote effects, inhibition of local MCs (e.g. with poorly absorbed cromolyn) may yield remote benefits. The humanized anti-IgE murine monoclonal antibody omalizumab has been described in multiple case reports as safe and effective in MCAD (e.g. Citation55), although a definitive trial has yet to be conducted. Tyrosine kinase inhibitors (TKIs), on-market and investigational (Supplementary Figure 1), have yielded highly variable responses in SM. Responses have been reported with TKIs in MCAS, too (Citation15,Citation16,Citation56). Low TKI doses often are effective in MCAD. Symptoms in MCAD may be due to mediator release from normal cells (e.g. MCs, basophils, other granulocytes, lymphocytes, etc.) secondarily activated by MCs (e.g. Citation57), helping explain why symptom intensity and pattern does not correlate with MC burden (e.g. Citation47). Distinction in MC pathways which promote MC accumulation versus mediator production/release may explain why TKIs reduce MC burdens and symptoms to different degrees (e.g. Citation58).
In the mastocytosis patient with significant MC burden and aggressive clinical course, cytoreductive drugs are prescribed with anti-mediator drugs (Citation45,Citation59). Effective cytoreductive therapies are few and offer modest response rates, qualities, and durations. Cytoreductive options include interferon-α (IFN) and cladribine (Supplementary Figure 1), commonly with glucocorticoids. IFN can ameliorate mastocytosis-related organopathy but often incurs considerable adverse effects (e.g. flu-like symptoms, myelo suppression, depression, hypothyroidism), limiting use in MCAD (e.g. Citation60). PEGylated interferon-α has been shown as efficacious as, and less toxic than the non-PEGylated form in some myeloproliferative neoplasms, but its use has not been reported in MCAD. Cladribine is generally reserved for last-choice treatment of patients with IFN-refractory/intolerant ASM; potential toxicities include significant, prolonged myelosuppression and lymphopenia and opportunistic infections. Polychemotherapy (as in acute leukemia) is investigational. Allogeneic stem cell transplantation sometimes yields remissions in mastocytosis (Citation61).
If anaphylaxis is provoked by a known allergen, especially hymenoptera venom, immunotherapy should be considered with recognition of potential risks (e.g. Citation62). In patients with repeated life-threatening anaphylactoid episodes, self-administration of epinephrine on demand is recommended, with cautions to first call for assistance and recline before injection. The use of ß-adrenoceptor blockers is discouraged in MCAD for reasons including potential inhibition of epinephrine effect, but glucagon can be an effective alternative.
Even absent anaphylaxis, symptomatic flares can require urgent/emergent evaluation to exclude mimicking phenomena (e.g. myocardial infarction) with highly effective therapies and to access MCA-controlling treatments not readily available otherwise. For example, intravenous histamine H1 and H2 receptor blocking, and sometimes also benzodiazepines and/or glucocorticoids, commonly help settle MCA flares quickly.
In most MCAD patients, some improvement is usually attainable, although patient and providers must exercise patience; sometimes many lines (and formulations) of therapy must be tried. In 32 SM patients, after 2 years of therapy, 10% showed complete response to anti-mediator therapy, 44% showed major response (>50% of symptoms disappeared and/or frequency of severe events decreased), 41% showed partial response (10–50% of symptoms disappeared), and only 6% showed no response (Citation63). In another retrospective dataset of anti-mediator response in 135 MCAS patients, 51% reported significant improvement (decrease of mean symptom intensity on visual analog scale), 11% remained unchanged and 38% reported worsening (GJM, unpublished data).
Fertility and reproduction
MCs critically assist in governing fertility and reproduction (e.g. Citation64). In the few studies of SM in pregnancy, no consistent effects were seen (e.g. Citation65,Citation66). In one study, one-third of pregnant women with SM suffered mild exacerbation of SM-related symptoms during pregnancy, but no predictors were identified (Citation66). No studies of pregnancy in MCAS have been performed. In spite of MC heparin content, MCAD-induced coagulopathy (Citation67) may spur placental microthrombi, preventing nidation or disrupting embryonic blood supply and threatening miscarriage (G. J. Molderings & H.-J. Hertfelder, unpublished observations). The use of low molecular weight heparins should be considered, especially after prior idiopathic miscarriage, though given the propensity of MCAD to affect the immune system including induction of autoimmunity, idiopathic miscarriage in MCAD should prompt testing for anti-phospholipid antibody syndrome, too. Ample reports describe histamine H1 and H2 receptor blockers as safe in pregnancy (e.g. (Citation68)), but there have been no drug safety studies specifically in the gravid MCAD population.
Monitoring
The time intervals, types, and extents of serial assessments in MCAD patients are guided by symptoms and objective findings. No biomarkers have been validated as useful guides for monitoring or therapy. Occasional reassessment of serum tryptase, especially upon recognition of significant worsening of baseline symptoms, may help identify transformation warranting fundamental shift in therapeutic strategy. Some have advised surveillance marrow biopsies every 1–5 years in SM to pre-emptively identify emerging associated hematologic malignancy (Citation30), but deferral of marrow reassessment until clinical progression is felt reasonable by others.
Future development
Advancing understanding of genetic and epigenetic etiologies of MCAD will facilitate development of new therapies. Human MCs have been reported recently to express inhibitory receptors (and thus potential therapeutic targets) that could allow for induction of apoptosis and inhibition of activation, like Tumor Necrosis Factor Related Apoptosis Inducing Ligand receptor (TRAIL-R) and inhibitory receptors CD300a and Siglec-8 (Citation69). Tryptase, chymase, and histamine H3 receptor blockers are in development (Citation70,Citation71). Mefloquine was recently found to induce MC apoptosis in vitro (Citation72). Interactions between MCs and the intestinal microbiome, and related therapeutic opportunities, warrant investigation (Citation35). Next-generation sequencing and genome-wide association studies may facilitate identification of disease-related operator and/or regulator genes. Studies are needed to demonstrate which of the MC-related diseases listed in Supplementary Table 4 are, at their core, variants of MCAD. Identification of MCAD-predisposing states and proximate disease-causing triggers/events may abet prevention, reducing health care and welfare system expenditures.
Conclusion
The new top-level designation “MCAD” conveys new understanding that all MC disease involves inappropriate MCA. Mastocytosis caps a metaphorical MCAD iceberg and additionally features inappropriate MC proliferation and clustering, while the bulk of the iceberg is assorted forms of MCAS which, without neoplasia, is more difficult to clinically identify. Different patterns of constitutively activating mutations in KIT and other MC regulatory elements likely drive different clinical presentations, and therapeutic response profiles, seen in different primary MCAD patients. Diagnostic testing for mastocytosis, typically spurred by idiopathic flushing and anaphylaxis or serum tryptase persistently >20 ng/ml, focuses on finding aberrant MC proliferation in tissue, while evaluation for MCAS (in a patient with chronic symptoms of aberrant MC mediator release but little to no tryptase elevation) seeks other elevated MC mediators. Lifespan for most MCAD patients is normal (if morbid until accurate diagnosis and effective therapy). A wide array of palliative therapies targeting mediator production/action helps manage aberrant MCA (significantly less so for MC proliferation), but – except for KITD816V’s general (but not absolute) prediction of imatinib resistance – predictors of successful personalized therapy are not yet available and sequential/methodical trials of many therapies are often needed to find optimal therapy. Most MCAD patients eventually identify helpful therapy, substantially alleviating symptoms present (sometimes incapacitatingly so) for years to decades. Advancing understanding of the molecular genetic/epigenetic roots of the disease likely will facilitate improvement in therapeutic strategies.
Disclosure statement
The authors report that they have no conflicts of interest.
Funding information
Symptom prevalences shown in were determined in a data collection project funded by a research award to LBA from The Mastocytosis Society.
MCAD_Review_for_Major_Journal_v18_for_AoM_-_Supplementary_Material.docx
Download MS Word (479.3 KB)References
- Horny HP, Metcalfe DD, Bennett JM, Bain BJ, Akin C, Escribano L, et al. Mastocytosis. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumors of hematopoietic and lymphoid tissues. 4th ed, pp. 54–63. Lyon: International Agency for Research on Cancer, 2008.
- Akin C, Metcalfe DD. The biology of Kit in disease and the application of pharmacogenetics. J Allergy Clin Immunol. 2004;114:13–19.
- D’Ambrosio C, Akin C, Wu Y, Magnuson MK, Metcalfe DD. Gene expression analysis in mastocytosis reveals a highly consistent profile with candidate molecular markers. J Allergy Clin Immunol. 2003;112:1162–70.
- Orfao A, Garcia-Montero AC, Sanchez L, Escribano L. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12–30.
- Mayerhofer M, Gleixner KV, Hoelbl A, Florian S, Hoermann G, Aichberger KJ, et al. Unique effects of KIT D816V in BaF3 cells: induction of cluster formation, histamine synthesis, and early mast cell differentiation antigens. J Immunol. 2008;180:5466–76.
- Roberts LJ, Oates JA. Biochemical diagnosis of systemic mast cell disorders. J Invest Dermatol. 1991;96:19S–25.
- Sonneck K, Florian S, Müllauer L, Wimazal F, Födinger M, Sperr WR, et al. Diagnostic and subdiagnostic accumulation of mast cells in the bone marrow of patients with anaphylaxis: monoclonal mast cell activation syndrome. Int Arch Allergy Immunol. 2007;142:158–64.
- Akin C, Scott LM, Kocabas CN, Kushnir-Sukhov N, Brittain E, Noel P, et al. Demonstration of an aberrant mast-cell population with clonal markers in a subset of patients with “idiopathic anaphylaxis”. Blood. 2007;110:2331–3.
- Molderings GJ, Kolck UW, Scheurlen C, Brüss M, Homann J, Von Kügelgen I. Multiple novel alterations in Kit tyrosine kinase in patients with gastrointestinally pronounced systemic mast cell activation disorder. Scand J Gastroenterol. 2007;42:1045–53.
- Molderings GJ, Meis K, Kolck UW, Homann J, Frieling T. Comparative analysis of mutation of tyrosine kinase Kit in mast cells from patients with systemic mast cell activation syndrome and healthy subjects. Immunogenetics. 2010;62:721–7.
- Molderings GJ. The genetic basis of mast cell activation disease-looking through a glass darkly. Crit Rev Oncol Hematol. 2015;93:75–89.
- Ibelgaufts H. “MastCells” in COPE: Cytokines and Cells Online Pathfinder Encyclopaedia, 2015. Available at: http://www.cells-talk.com/index.php/page/copelibrary?key=mast%20cells (accessed 10 March 2016).
- Amon U, Hartmann K, Horny H-P, Nowak A. Mastocytosis-an update. J Dtsch Dermatol Ges. 2010;8:695–711.
- Jennings S, Russell N, Jennings B, Slee V, Sterling L, Castells M, et al. The Mastocytosis Society survey on mast cell disorders: patient experiences and perceptions. J Allergy Clin Immunol Pract. 2014;2:70–6.
- Afrin LB. Polycythemia from mast cell activation syndrome: lessons learned. Am J Med Sci. 2011;342:44–9.
- Afrin LB. Mast cell activation disorder masquerading as pure red cell aplasia. Int J Hematol. 2010;91:907–8.
- Marshall A, Kavanagh RT, Crisp AJ. The effect of pamidronate on lumbar spine bone density and pain in osteoporosis secondary to systemic mastocytosis. Br J Rheumatol. 1997;36:393–6.
- Theoharides TC. Mast cells in irritable bowel syndrome and ulcerative colitis: function not numbers is what makes all the difference. Dig Dis Sci. 2014;59:897–8.
- Frieling T, Meis K, Kolck UW, Homann J, Hülsdonk A, Haars U, et al. Evidence for mast cell activation in patients with therapy-resistant irritable bowel syndrome. Z Gastroenterol. 2011;49:191–4.
- Rafii M, Firooznia H, Golimbu C, Balthazar E. Pathologic fracture in systemic mastocytosis: radiographic spectrum and review of the literature. Clin Orthop Relat Res. 1983;180:260–7.
- Crivellato E, Ribatti D. The mast cell: an evolutionary perspective. Biol Rev Camb Philos Soc. 2010;85:347–60.
- Poglio S, De Toni-Costes F, Arnaud E, Laharrague P, Espinosa E, Casteilla L, et al. Adipose tissue as a dedicated reservoir of functional mast cell progenitors. Stem Cells. 2010;28:2065–72.
- Gri G, Frossi B, D'Inca F, Danelli L, Betto E, Mion F, et al. Mast cell: an emerging partner in immune interaction. Front Immunol. 2012;3:120.
- Theoharides TC, Conti P. Mast cells: the Jekyll and Hyde of tumor growth. Trends Immunol. 2004;25:235–41.
- Lundequist A, Pejler G. Biological implications of preformed mast cell mediators. Cell Mol Life Sci. 2011;68:965–75.
- Dvorak AM. Piecemeal degranulation of basophils and mast cells is effected by vesicular transport of stored secretory granule contents. Chem Immunol Allergy. 2005;85:135–84.
- Theoharides TC, Kempuraj D, Tagen M, Conti P, Kalogeromitros D. Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol Rev. 2007;217:65–78.
- Escribano L, Orfao A, Díaz-Agustin B, Villarrubia J, Cerveró C, López A, et al. Indolent systemic mast cell disease in adults: immunophenotypic characterization of bone marrow mast cells and its diagnostic implications. Blood. 1998;91:2731–6.
- Garcia-Montero AC, Jara-Acevedo M, Teodosio C, Sanchez ML, Nunez R, Prados A, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108:2366–72.
- Valent P, Akin C, Escribano L, Födinger M, Hartmann K, Brockow K, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37:435–53.
- Haenisch B, Nöthen MM, Molderings GJ. Systemic mast cell activation disease: the role of molecular genetic alterations in pathogenesis, heritability and diagnostics. Immunology. 2012;137:197–205.
- Molderings GJ, Haenisch B, Bogdanow M, Fimmers R, Nöthen MM. Familial occurrence of systemic mast cell activation disease. PLoS One. 2013;8:e76241–24098785.
- Haenisch B, Fröhlich H, Herms S, Molderings GJ. Evidence for contribution of epigenetic mechanisms in the pathogenesis of systemic mast cell activation disease. Immunogenetics. 2014;66:287–97.
- Nurmi K, Methuen T, Mäki T, Lindstedt KA, Kovanen PT, Sandler C, et al. Ethanol induces apoptosis in human mast cells. Life Sci. 2009;85:678–84.
- Afrin LB, Khoruts A. Mast cell activation disease and microbiotic interactions. Clin Ther. 2015;37:941–53.
- Berezowska S, Flaig MJ, Ruëff F, Walz C, Haferlach T, Krokowski M, et al. Adult-onset mastocytosis in the skin is highly suggestive of systemic mastocytosis. Mod Pathol. 2014;27:19–29.
- Akin C, Valent P, Metcalfe DD. Mast cell activation syndrome: proposed diagnostic criteria. J Allergy Clin Immunol. 2010;126:1099–1104e4.
- Picard M, Giavina-Bianchi P, Mezzano V, Castells M. Expanding spectrum of mast cell activation disorders: monoclonal and idiopathic mast cell activation syndromes. Clin Ther. 2013;35:548–62.
- Cardet JC, Castells MC, Hamilton MJ. Immunology and clinical manifestations of non-clonal mast cell activation syndrome. Curr Allergy Asthma Rep. 2013;13:10–18.
- Afrin LB, Molderings GJ. A concise, practical guide to diagnostic assessment for mast cell activation disease. World J Hematol. 2014;3:1–17. Available at: http://www.wjgnet.com/2218-6204/journal/v3/i1/index.htm.
- Cohen SS, Skovbo S, Vestergaard H, Kristensen T, Møller M, Bindslev-Jensen C, et al. Epidemiology of systemic mastocytosis in Denmark. Br J Haematol. 2014;166:521–8.
- Van Doormaal JJ, Arends S, Brunekreeft KL, van der Wal VB, Sietsma J, van Voorst Vader PC, et al. Prevalence of indolent mastocytosis in a Dutch region. J Allergy Clin Immunol. 2013;131:1429–31.e1.
- Lucas HJ, Brauch CM, Settas L, Theoharides TC. Fibromyalgia – new concepts of pathogenesis and treatment. Int J Immunopathol Pharmacol. 2006;19:5–10.
- Blanco I, Béritze N, Argüelles M, Cárcaba V, Fernández F, Janciauskiene S, et al. Abnormal overexpression of mastocytes in skin biopsies of fibromyalgia patients. Clin Rheumatol. 2010;29:1403–12.
- Lim K-H, Tefferi A, Lasho TL, Finke C, Patnaik M, Butterfield JH, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009;113:5727–36.
- Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, Garcia-Montero A, Núñez R, Almeida J, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol. 2009;124:514–21.
- Erben P, Schwaab J, Metzgeroth G, Horny HP, Jawhar M, Sotlar K, et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis. Ann Hematol. 2014;93:81–8.
- Sperr WR, Escribano L, Jordan JH, Schernthaner GH, Kundi M, Horny HP, et al. Morphologic properties of neoplastic mast cells: delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk Res. 2001;25:529–36.
- Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC, et al. Definitions, criteria, and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol. 2012;157:215–25.
- Sánchez-Muñoz L, Alvarez-Twose I, García-Montero AC, Teodosio C, Jara-Acevedo M, Pedreira CE, et al. Evaluation of the WHO criteria for the classification of patients with mastocytosis. Mod Pathol. 2011;24:1157–68.
- Butterfield JH, Li C-Y. Bone marrow biopsies for the diagnosis of systemic mastocytosis: is one biopsy sufficient? Am J Clin Pathol. 2004;121:264–7.
- Molderings GJ, Brettner S, Homann J, Afrin LB. Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options. J Hematol Oncol. 2011;4:10. doi:10.1186/1756-8722-4-10.
- Pardanani A, Chen D, Abdelrahman RA, Reichard KK, Zblewski D, Wood AJ, et al. Clonal mast cell disease not meeting WHO criteria for diagnosis of mastocytosis: clinicopathologic features and comparison with indolent mastocytosis. Leukemia. 2013;27:2091–4.
- Biesiekierski JR, Newnham ED, Irving PM, Barrett JS, Haines M, Doecke JD, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol. 2011;106:508–14.
- Molderings GJ, Raithel M, Kratz F, Azemar M, Haenisch B, Harzer S, et al. Omalizumab treatment of systemic mast cell activation disease: experiences from four cases. Intern Med. 2011;50:1–5.
- Afrin LB. Mast cell activation syndrome masquerading as agranulocytosis. Mil Med. 2012;177:113–17.
- Galli SJ, Costa JJ. Mast-cell-leukocyte cytokine cascades in allergic inflammation. Allergy. 1995;50:851–62.
- Gotlib J, George TI, Corless C, Linder A, Ruddell A, Akin C, et al. The Kit tyrosine kinase inhibitor midostaurine (PKC412) exhibits a high response rate in aggressive systemic mastocytosis (ASM): interim results of a phase II trial. Blood. 2007;110:3536.
- Valent P, Sperr WR, Akin C. How I treat patients with advanced systemic mastocytosis. Blood. 2010;116:5812–7.
- Butterfield JH. Interferon treatment for hypereosinophilic syndromes and systemic mastocytosis. Acta Haematol. 2005;114:26–40.
- Ustun C, Reiter A, Scott BL, Nakamura R, Damaj G, Kreil S, et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol. 2014;32:3264–74.
- de Olano DG, Twose IA, Esteban López MI, Sánchez-Muñoz L, de Durana MD, Vega A, et al. Safety and effectiveness of immunotherapy in patients with indolent systemic mastocytosis presenting with Hymenoptera venom anaphylaxis. J Allergy Clin Immunol. 2008;121:519–26.
- Del Mastro A, Petraroli A, Magliacane D, Squeglia V, Pelosi C, Gravante C, et al. Evaluation of the response to anti-mediator therapy in patients with mastocytosis after 2-years of follow-up. Allergy. 2013;68:691.
- Woidacki K, Jensen F, Zenclussen AC. Mast cells as novel mediators of reproductive processes. Front Immunol. 2013;4:29–23419968.
- Matito A, Alvarez-Twose I, Morgado JM, Sánchez-Muñoz L, Orfao A, Escribano L. Clinical impact of pregnancy in mastocytosis: a study of the Spanish Network on Mastocytosis (REMA) in 45 cases. Int Arch Allergy Immunol. 2011;156:104–11.
- Worobec AS, Akin C, Scott LM, Metcalfe DD. Mastocytosis complicating pregnancy. Obstet Gynecol. 2000;95:391–5.
- Seidel H, Molderings GJ, Oldenburg J, Meis K, Kolck UW, Homann J, et al. Bleeding diathesis in patients with mast cell activation disease. Thromb Haemost. 2011;106:987–9.
- Schatz M, Zeiger RS, Harden K, Hoffman CC, Chilingar L, Petitti D. The safety of asthma and allergy medications during pregnancy. J Allergy Clin Immunol. 1997;100:301–6.
- Migalovich-Sheikhet H, Friedman S, Mankuta D, Levi-Schaffer F. Novel identified receptors on mast cells. Front Immunol. 2012;3:238–22876248.
- He A, Shi GP. Mast cell chymase and tryptase as targets for cardiovascular and metabolic diseases. Curr Pharm Des. 2013;19:1114–25.
- Passani MB, Blandina P. Histamine receptors in the CNS as targets for therapeutic intervention. Trends Pharmacol Sci. 2011;32:242–9.
- Paivandy A, Calounova G, Zarnegar B, Ohrvik H, Melo FR, Pejler G. Mefloquine, an anti-malaria agent, causes reactive oxygen species-dependent cell death in mast cells via a secretory granule-mediated pathway. Pharmacol Res Perspect. 2014;2:e00066.
- Afrin L. Presentation, diagnosis, and management of mast cell activation syndrome. In: Murray D, ed. Mast cells: phenotypic features, biological functions, and role in immunity, pp. 155–231. Happauge, NY: Nova Science Publishers, 2013.