Abstract
Atherothrombotic disease is highly prevalent in Western countries and is associated with morbidity, mortality, and a significant economic burden. The primary pathophysiological mechanism of acute ischemic events in patients with atherothrombotic disease is complex but involves thrombotic occlusion in response to rupture or erosion of atherosclerotic lesions. Current treatments for long-term secondary prevention in patients with established atherothrombotic disease, such as those with prior myocardial infarction, ischemic stroke/transient ischemic attack, or symptomatic peripheral artery disease, include therapies aimed at preventing rupture/erosion of atherosclerotic lesions (life-style modification and blood pressure reduction, in addition to statins and angiotensin II-active agents) and thrombus formation (primarily antiplatelet agents, such as aspirin, thienopyridines (clopidogrel, prasugrel, ticlopidine), and, to a lesser degree, anticoagulants). Despite the proven benefits and broad use of these therapies, the long-term rates of mortality and recurrent ischemic events remain high. This residual risk can be attributed to the fact that atherothrombosis continues in the presence of current treatments; because these agents each inhibit relatively specific pathways, atherosclerosis, thrombus formation, and other processes may progress. These considerations suggest that novel therapies with a different mechanism of action may provide additional reductions in morbidity and mortality beyond those observed with current agents.
Key messages
Despite the clinical benefit achieved with life-style modifications, blood pressure reduction, statins, angiotensin II-active agents, and antiplatelet agents, the residual risk for recurrent acute events in patients with established atherothrombotic disease remains substantial. Observational registry data confirm the high residual risk of patients with documented atherothrombotic disease and highlight the need for more effective therapies.
Residual risk for thrombotic events is likely related to progression of atherosclerosis in the presence of statins and angiotensin II-active agents, lack of more complete inhibition of platelet activation in the presence of aspirin and clopidogrel, and other factors yet to be identified.
Current antiplatelet agents target thromboxane A2 and adenosine diphosphate and do not interfere with a multitude of other platelet activation pathways that may contribute to thrombotic events. These agents are also associated with bleeding risk.
These considerations underscore a critical need for novel therapies that provide more comprehensive platelet inhibition when used with current oral antiplatelet therapy for greater protection against thrombotic events, preferably without an incremental bleeding risk.
| Abbreviations | ||
| ACAT | = | acyl-coenzyme A:cholesterol acyltransferase |
| ACC | = | American College of Cardiology |
| ACE | = | angiotensin-converting enzyme |
| ACS | = | acute coronary syndromes |
| ADP | = | adenosine diphosphate |
| AHA | = | American Heart Association |
| ARB | = | angiotensin receptor blocker |
| BP | = | blood pressure |
| CAC | = | coronary artery calcification |
| CAD | = | coronary artery disease |
| CETP | = | cholesteryl ester transfer protein |
| CHD | = | coronary heart disease |
| CRP | = | C-reactive protein |
| CV | = | cardiovascular |
| DTI | = | direct thrombin inhibitor |
| ESRD | = | end-stage renal disease |
| FXa | = | factor Xa |
| GP | = | glycoprotein |
| HF | = | heart failure |
| HPS | = | Heart Protection Study |
| INR | = | International normalized ratio |
| LDL-C | = | low-density lipoprotein cholesterol |
| Lp-PLA2 | = | lipoprotein-associated PLA2 |
| LV | = | left ventricular |
| LVH | = | left ventricular hypertrophy |
| MI | = | myocardial infarction |
| PAD | = | peripheral artery disease |
| PLA2 | = | phospholipase A2 |
| PWV | = | pulse wave velocity |
| SMCs | = | smooth muscle cells |
| sPLA2 | = | soluble PLA2 |
| TIA | = | transient ischemic attack |
| TLRs | = | Toll-like receptors |
| TxA2 | = | thromboxane A2 |
Introduction
Atherothrombotic disease is the leading cause of morbidity and mortality in the United States (Citation1) and the Western world. For patients with atherothrombotic disease, such as those with a history of myocardial infarction (MI), ischemic stroke/transient ischemic attack (TIA), or peripheral artery disease (PAD), several pharmacological options are available, in addition to long-term life-style modification and blood pressure (BP) control. These include statins and angiotensin II-active agents, which reduce the likelihood of atherosclerotic plaque progression and rupture/erosion to prevent events, and antithrombotic agents, which inhibit platelet activation that may lead to occlusive thrombus-related events. While chronic life-style modification and BP control in conjunction with the use of these agents have been associated with a significant reduction in adverse events, the long-term residual risk for recurrent ischemia-related events remains substantial (Citation1–6). The purpose of this review is to summarize recent epidemiological data for atherothrombotic disease risk, the pathophysiology of atherothrombosis, and the available pharmacological treatment options (including results of key clinical trials), which are useful when added to life-style modification and BP reduction for secondary prevention of atherothrombotic disease. This information should provide a useful framework for discussion of novel therapies aimed at reducing the residual risk.
Morbidity, mortality, and cost of atherothrombotic disease in the United States
Atherothrombotic disease is highly prevalent and is associated with significant morbidity, mortality, and economic consequences. The estimated prevalence of MI in the United States is 8,100,000 (Citation7,Citation8), and the annual incidence of MI in 2008 is estimated at 920,000 (600,000 new and 320,000 recurrent cases) (Citation8). In 2004, over 150,000 Americans died of MI (Citation8). Among individuals aged ≥40 years with a first MI, 33% of men and 43% of women are estimated to die within 5 years. MI is also associated with significant morbidity: among individuals aged 40–69 years with a first MI, an estimated 4% of men and 6% of women will develop a stroke, and 7% of men and 12% of women will develop heart failure (HF).
The estimated prevalence of stroke in the United States is 5,800,000 (Citation7–9), and the annual incidence of stroke in 2008 is estimated at 780,000 (600,000 new and 180,000 recurrent cases) (Citation7–9). Ischemic stroke accounts for 87% of all strokes (Citation8). In 2004, over 150,000 Americans died of stroke, and about one half of men and women with a first stroke will die within 5 years. Furthermore, 15%–30% of stroke survivors are permanently disabled (Citation8). Approximately 13% of men and 22% of women between 40 and 69 years of age who have experienced stroke will have a recurrent stroke. Approximately 15% of all strokes are preceded by a TIA, and almost one half of those with TIAs will have stroke, MI, or vascular death in the next 10 years (Citation8).
The estimated prevalence of PAD in the United States is 8,000,000 (Citation10,Citation11), and the annual mortality rate for patients with lower-extremity PAD estimated from epidemiological studies is 4%–6% (Citation12–16). A prospective study estimated that patients with large-vessel PAD are at 3-fold higher risk of all-cause mortality, a >6-fold higher risk for fatal coronary artery disease (CAD), and a 6-fold higher risk for cardiovascular (CV) death (Citation12). These patients have a 40% higher risk of stroke compared with those without PAD (Citation17,Citation18). In addition, patients with either asymptomatic or symptomatic PAD have impaired quality of life due to a progressive loss of lower-extremity function over time (Citation13,Citation19).
The American Heart Association (AHA) estimated direct and indirect costs of all cardiovascular disease (CVD) for 2008 to be $448.5 billion (Citation8). Of the total cost, the estimated direct and indirect cost of coronary heart disease (CHD) is $156.4 billion, and the estimated direct and indirect cost of stroke is $65.5 billion. These costs are anticipated to rise as our population ages and the epidemics of obesity, metabolic syndrome, and diabetes continue.
Pathobiology of atherothrombosis and rationale for medical therapy
Atherosclerotic plaque development
Atherosclerosis develops within the intima of large-and medium-sized arteries and can be triggered by behavioral, environmental, biochemical, or genetic factors (Citation20). Studies in animal models and humans document that various factors are associated with dysfunction of endothelial cells, vascular smooth muscle cells (SMCs), and their precursor cells, which are important for understanding the limitations of vascular repair. These functional alterations lead to initial structural lesions (fatty streaks) and then raised lesions (plaques) that are relatively small and do not result in significant narrowing of the blood vessel lumen (Citation20,Citation21). The disease begins very early in life, as fatty streaks have been identified in the aortae of aborted fetuses and children <2 years old dying of accidental causes (Citation22–24). These stable, asymptomatic, and often undetectable plaques may progress to unstable, high-risk, disruption-prone lesions as a result of multiple local and systemic factors (Citation20). A developing non-occlusive but unstable plaque is composed of inflammatory cells, including leukocytes, SMCs, platelet aggregates, inflammatory cytokines, growth factors, and lipid-laden foam cells (Citation20,Citation25). An expanding lipid core of unstable lesions is separated from the arterial lumen by a fibrous cap, which is composed primarily of SMCs and collagen (Citation20,Citation21,Citation25). Rupture or erosion of these lesions exposes circulating blood to a highly thrombogenic environment, ultimately leading to occlusion of the lumen by platelet-rich thrombi. Formation of platelet-rich thrombi in response to rupture or erosion of atherosclerotic plaques leads to partial or complete obstruction of blood flow in the affected arteries, resulting in insufficient oxygen supply (ischemia) that manifests clinically as an MI, unstable angina, stroke/TIA, or symptomatic PAD (Citation20,Citation26).
Thrombosis
Platelets play a critical role in atherothrombotic disease, as they are the primary constituent of occlusive thrombi at the sites of ruptured or eroded plaques. Collagen and von Willebrand factor in the extracellular matrix of plaques facilitate initial platelet recruitment (Citation27,Citation28). Soluble mediators (e.g. thromboxane A2 (TxA2) and adenosine diphosphate (ADP)) released from the initial adherent platelets in response to contact with collagen promote additional platelet recruitment (Citation29). Thrombin, generated by tissue factor in ruptured or eroded plaques, facilitates formation of fibrin-rich clots that stabilize the protective platelet monolayer. Adherent platelets stabilized by fibrin-rich clots help prevent blood loss in response to vascular trauma and are also known as hemostatic plugs; under pathological conditions, however, multiple factors released from adherent platelets and/or generated at sites of plaque rupture or erosion stimulate subsequent platelet activation and aggregation (Citation20,Citation21,Citation25). Platelets can be activated by multiple factors, including thrombin, TxA2, ADP, collagen, serotonin, and others, which collectively amplify and sustain platelet activation (Citation25). A key feature of platelet activation is conversion of glycoprotein (GP) IIb/IIIa receptors on the platelet surface into an active form that binds fibrinogen and leads to platelet aggregation via cross-linking of GP IIb/IIIa receptors on adjacent platelets by fibrinogen bridges (Citation30). Multiple platelet aggregation reactions ultimately result in generation of occlusive platelet-rich thrombi that are likely responsible for many of the acute clinical manifestations of atherothrombotic disease (e.g. ST-elevation MI, non-ST-elevation MI, unstable angina, and ischemic stroke/TIA). Fibrin-rich clots generated by thrombin may also contribute to occlusive thrombosis.
Inflammation
Platelets interact with the vascular endothelium and link inflammation, thrombosis, and atherogenesis (Citation31,Citation32). Platelet participation in inflammation contributes to the development of atherosclerotic lesions and atherothrombosis, which is characterized by interactions between platelets, endothelial cells, and leukocytes. For example, activated platelets release CD40 ligand, promoting endothelial inflammation via production or recruitment of reactive oxygen species, adhesion molecules, chemokines, and tissue factor (Citation31). In addition, secretion of reactive oxygen species by activated platelets enhances platelet recruitment to a growing thrombus (Citation33 and induces enhanced lipid peroxidation of cell membrane phospholipids and circulating low-density lipoprotein (LDL). Furthermore, platelets express multiple Toll-like receptors (TLRs), which are mediators of innate immune response (Citation34). TLR2 and TLR4 are involved in the pathogenesis of CAD, establishing a link between the progression of coronary atherosclerosis and the immune response to endogenously generated inflammatory ligands (Citation35).
Taking into account the complex pathobiology of atherothrombotic disease, the goals of pharmacological therapy for prevention of acute ischemic events are to: 1) slow progression of atherosclerosis (reduce development of new lesions vulnerable to rupture or erosion) with life-style modification and statins (and possibly angiotensin II-active agents); 2) reduce the risk of rupture or erosion of existing atherosclerotic lesions (with a statin or with angiotensin II-active agents); and 3) inhibit thrombotic occlusion, primarily with antiplatelet therapy (e.g. aspirin and ADP receptor antagonists) and, to a lesser degree, with anticoagulants (Citation21,Citation25). Inflammation may represent a pathophysiological mechanism for thrombosis that may furnish novel therapeutic approaches.
Secondary prevention of atherothrombotic events: evidence from clinical trials
Life-style modification
The large international INTERHEART study (Citation36 identified nine risk factors that collectively accounted for >90% of acute MI risk. Risk predictors included life-style factors (such as smoking, low fruit and vegetable consumption, low exercise levels, lack of alcohol consumption), comorbidities (hypertension, diabetes, abdominal obesity, abnormal lipid profiles), as well as psycho-social factors (Citation36).
Multiple randomized trials have shown that life-style modifications can result in weight loss, improved fitness, and significant reduction in BP, each of which is important in reducing risk for atherothrombotic disease. The combination of group life-style modification counseling and pharmacotherapy resulted in approximately double the average weight loss of life-style modification or pharmacotherapy alone (Citation6). The benefits of a Mediterranean-style diet (defined as a diet rich in fruits, vegetables, bread, other forms of cereal, potatoes, beans, nuts, and seeds, with olive oil as an important fat source; low-to-moderate consumption of dairy products, fish, and poultry; little red meat; and low-to-moderate wine consumption) have also been well documented (Citation4). Available evidence from these studies supports life-styles that adopt strategies to lose weight, stop cigarette smoking, engage in regular moderate exercise and relaxation, and regularly consume light-to-moderate alcohol and (to a lesser extent) fatty fish in order to reduce platelet reactivity and coagulability, and to promote fibrinolysis. The overall effects can be expected to translate into an improved CV prognosis or other beneficial clinical outcome in healthy individuals and those with CV risk factors or established CHD (Citation5). Nevertheless, a high residual risk for secondary ischemic events, despite life-style modifications, is evident from the results of recent clinical practice registries, which justifies the need for adding pharmacotherapy.
Lipid-lowering therapy
Multiple trials have documented the benefits of statins for secondary prevention of CHD () (Citation37–48). Early trials with simvastatin (Citation37) and pravastatin (Citation38,Citation47) demonstrated 25% to 35% reductions in low-density lipoprotein cholesterol (LDL-C), which were accompanied by reductions in morbidity and mortality ().
Table I. Primary outcomes in selected randomized secondary prevention trials with statins.
Recent trials have assessed the benefit of more aggressive lipid-lowering for secondary prevention in patients with CHD (). The PROVE-IT trial (Citation40) demonstrated a 16% reduction in the primary composite outcome with atorvastatin 80 mg versus pravastatin 40 mg once daily (qd) in patients with acute coronary syndromes (ACS) (). The TNT trial (Citation44,Citation48) demonstrated a direct correlation between a decrease in LDL-C and a reduction in secondary events in subjects with stable CHD (). Phase Z of the A-Z trial (Citation41) showed no significant difference between an early and intensive simvastatin regimen versus a delayed and less intensive simvastatin regimen in patients with ACS, possibly due to low event rates and high rates of non-compliance (). The IDEAL trial (Citation45) compared low-dose simvastatin versus high-dose atorvastatin in patients with prior MI. After 4 years, intensive LDL-C lowering did not significantly reduce CV or all-cause mortality (), potentially due to a smaller than expected difference in LDL-C levels between the groups over the study duration (25% decrease in LDL-C versus ≈35% in earlier studies), a shorter follow-up duration than anticipated, and an increase in high-density lipoprotein cholesterol levels with simvastatin (which may compensate for LDL-C reductions with atorvastatin).
The benefits of long-term statin therapy in secondary prevention were also demonstrated by the UK Heart Protection Study (HPS), which included patients with prior MI, cerebrovascular disease, or PAD (Citation3). In addition to demonstrating a reduction in the primary outcome of all-cause mortality (), simvastatin also reduced rates of first major vascular event in the total population, as well as in patients with prior MI, cerebrovascular disease, or peripheral vascular disease (). Simvastatin also reduced the relative risk of total stroke and ischemic stroke by 25% and 30%, respectively (Citation3).
Figure 1. Event rates in patient subgroups allocated simvastatin versus placebo in the Heart Protection Study (HPS) (Citation3). A: The rate of the primary end-point of all-cause mortality was significantly lower in patients allocated simvastatin (12.9%) versus placebo (14.7%) in the total study population in the HPS (12% relative risk reduction, 88% residual risk; P=0.003). B: The rate of the first major vascular event was significantly lower in patients receiving simvastatin (19.8%) than in those receiving placebo (25.2%) in the total study population (21% relative risk reduction, 79% residual risk; P<0.001), as well as in patients with prior myocardial infarction (MI) (23.5% versus 29.4%; 20% relative risk reduction, 80% residual risk; P<0.001), patients with cerebrovascular disease (18.7% versus 23.6%; 21% relative risk reduction, 79% residual risk; P<0.001), and patients with peripheral vascular disease (PVD) (24.7% versus 30.5%; 19% relative risk reduction, 81% residual risk; P<0.001). Green arrows indicate relative risk reduction between treatment arms. Red arrows indicate residual risk for recurrent events.
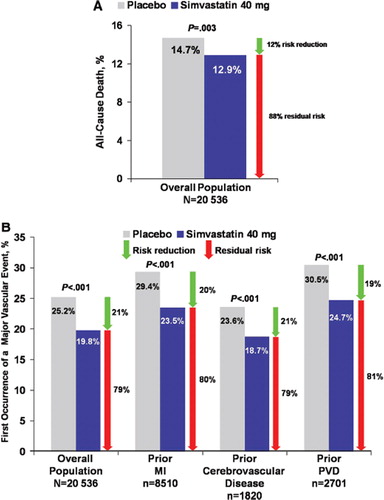
Beyond secondary prevention, large observational studies and small prospective trials of statins have suggested improved cardiac function and decreased cardiac death in patients with HF (Citation49–51). However, large prospective randomized trials have not confirmed these observations. Rosuvastatin did not demonstrate a reduction in mortality in patients with ischemic HF in CORONA or in patients with chronic HF in GISSI-HF () (Citation42,Citation43). These results suggest that once HF is established, statins may not be able to improve the underlying disease. Any potential benefit of statins may be offset by any detrimental effects of lowering LDL-C in this patient population.
Statins are recommended for patients with ischemic stroke (Citation9), although there is somewhat less evidence to support their benefits for secondary prevention of stroke. While simvastatin reduced the risk of stroke and TIA in the HPS (Citation3), it did not reduce the incidence of stroke in patients with pre-existing cerebrovascular disease (Citation3). The SPARCL trial (Citation39), however, demonstrated a benefit for high-dose atorvastatin (). Importantly, a small increase in hemorrhagic strokes was observed with atorvastatin. A considerably delayed initiation of statin therapy in the HPS (patients were enrolled 4.3 years after the index event) compared with immediate statin initiation in SPARCL may be one potential reason for the difference in study outcomes. In addition to timing, the use of different statins may also have contributed to the observed differences in the outcomes between the two trials.
Few studies have examined the benefits of statins in patients with PAD. The HPS (Citation3) indicated that simvastatin reduced the relative risk of secondary events in the subgroup of patients with PAD by 25%. Statins are recommended for all patients with PAD to achieve target LDL-C levels of <100 mg/dL, whereas in patients with lower-extremity PAD who are at high risk for thrombotic events the LDL-C goal is <70 mg/dL (Citation13).
The beneficial effects of statins on atherothrombotic disease have been confirmed by several recent meta-analyses, including those in patients with ACS (Citation52), stroke (Citation53), and PAD (Citation54). The JUPITER trial suggests that statins may also have clinical benefit in lower-risk individuals, as mentioned above.
Angiotensin II-active agents
Early trials of angiotensin-converting enzyme (ACE) inhibitors in patients with HF suggested a reduction in non-fatal MIs (Citation55), and subsequent studies evaluated their efficacy in secondary and primary prevention among patients without HF or left ventricular (LV) dysfunction. The ACE inhibitor quinapril did not demonstrate a benefit in reduction of ischemic events in patients with ischemic heart disease and preserved LV function in the QUIET trial (Citation56). In contrast, the HOPE trial demonstrated a 22% relative risk reduction in CV death, MI, or stroke with ramipril 10 mg over 5 years (P<0.001). The beneficial effect of ramipril was evident in both the secondary and primary prevention cohorts (Citation57). Similarly, in the EUROPA trial, treatment with perindopril was associated with a 20% relative risk reduction in CV death, MI, or cardiac arrest (P=0.0003) during the 4.2-year study period, with consistent benefits among patients with or without prior MI or revascularization (Citation58). However, the PEACE trial did not demonstrate a beneficial effect of trandolapril, possibly due to a lower-risk profile of the study population, a high rate of coronary revascularizations prior to enrollment, and more intensive management of risk factors (Citation59). A meta-analysis of QUIET, HOPE, EUROPA, and PEACE (pooled n=31,555) found that ACE inhibition significantly reduced the risk of all-cause mortality, CV mortality, acute MI, and stroke (P<0.001 for each), supporting the benefit of ACE inhibition for secondary prevention in patients with preserved LV function (Citation60).
In TRANSCEND, the angiotensin receptor blocker (ARB) telmisartan did not reduce CV death, MI, stroke, or hospitalizations in patients with CVD or diabetes with end-stage organ damage who were intolerant to ACE inhibitors (Citation61). A modest reduction in the secondary outcome of CV death, MI, or stroke was observed. The ONTARGET trial demonstrated that the combination of the ACE inhibitor ramipril and the ARB telmisartan provided no incremental benefit over either therapy alone in patients with vascular disease or high-risk diabetes, while resulting in a higher rate of adverse events, particularly renal dysfunction (Citation62). Among patients with ischemic stroke, telmisartan initiated soon after ischemic stroke and continued for 2.5 years did not significantly lower the rate of recurrent stroke or major CV events in the PROFESS trial (Citation63). These findings are in contrast to the PROGRESS trial, which demonstrated reduced risk of recurrent stroke and other CV events in patients randomized to receive active treatment with the ACE inhibitor perindopril plus the diuretic indapamide (Citation64). (Flexibility regarding the use of combination therapy or perindopril monotherapy was allowed in patients allocated to receive active treatment for whom the treating physician was concerned about intensive blood pressure lowering.)
Collectively, the results of these trials suggest the potential benefit of angiotensin II-active agents may be attenuated in patients who are treated with other evidence-based therapies because intensive pharmacological management may reduce residual risk for recurrent ischemic events. Current AHA/American College of Cardiology (ACC) guide-lines for secondary prevention recommend ACE inhibitors for all patients with LV ejection fraction ≤40% and for those with hypertension, diabetes, or chronic kidney disease. ACE inhibitors may be considered for all other patients (Citation65).
Glycemic control
The findings from ADVANCE, ACCORD, and UKPDS have contributed to the understanding of the utility of intensive glycemic control in the prevention of vascular disease. The benefit of intensive glycemic control (Citation66), but not BP control (Citation67), on microvascular disease, renal and metabolic complications, MI, and death was evident in UKPDS during 10 years of post-trial monitoring. Both UKPDS and ADVANCE (Citation68) demonstrated that tighter glucose control prevented microvascular but not macrovascular complications during the period of randomized comparison. Neither trial showed evidence of an increase in mortality, a finding that was observed with intensive glucose control in ACCORD (Citation69). All three trials similarly observed that good or nearly normal glycemic control does not reduce major CV events in the short term.
The UKPDS reports suggest that in patients with type 2 diabetes, intensive glucose control may have a ‘legacy effect’, i.e. clinical benefit remains long after the cessation of therapy, whereas the same benefit was not observed with tight BP control. However, the increase in death rates observed in ACCORD indicates that it is not simply a matter of ‘lower is better’ for glucose control. Taken together, these studies indicate that it is not appropriate to manage risk factors and conditions individually. The results reinforce the importance of utilizing a collective approach to risk management and to maintenance of good glycemic control, not only for the prevention of diabetic complications but also for protection against the development of CVD in the long term.
β-Adrenergic system blockers
The benefit of β-blockers in ischemia and hypertension has been demonstrated in several trials, and subsequent studies showed benefit in chronic HF, despite prior contraindication. β-Blockers have been considered valuable for secondary prevention in patients after unstable angina and MI. However, the CV benefit of β-blockers has been challenged by subsequent randomized trials and meta-analyses. The COMMIT trial, which separately assessed the efficacy and safety of early β-blocker therapy (intravenous then oral metoprolol) and antiplatelet therapy (adding clopidogrel to aspirin) in suspected acute MI, found that allocation to metoprolol did not reduce the rate of death, reinfarction, or cardiac arrest, but significantly increased the risk of cardiogenic shock (P<0.00001) (Citation70). A meta-analysis of nine trials of patients with hypertension found that heart rate reduction (as achieved with β-blocker therapy) was significantly associated with greater risk of all-cause mortality, CV mortality, MI, and HF (all P<0.0001) (Citation72). Furthermore, a meta-analysis of 33 trials of patients having non-cardiac surgery found that perioperative β-blocker therapy did not reduce the risk of all-cause mortality, CV mortality, or HF, but decreased the risk of MI. However, the risks of stroke and perioperative bradycardia and hypotension were significantly increased (Citation71). The disparity between older and more recent trials assessing the CV benefit of β-blockers could stem from the evolution of treatment intensity during this time frame. As patients are now treated with multiple agents, the benefit of β-blockers is not as manifest. Currently, the strongest evidence for the clinical benefit of β-blockers is in patients with HF (Citation73), and AHA/ACC guide-lines strongly recommend the use of these agents before discharge for secondary prevention in those patients with compensated HF or LV systolic dysfunction (Citation74).
Anticoagulants
The anticoagulant warfarin has been widely used for prophylaxis and treatment of deep vein thrombosis and pulmonary embolism, as well as for prevention of ischemic stroke in patients with atrial fibrillation. However, warfarin is rarely used for secondary prevention in patients with atherothrombosis, because clinical trials have not demonstrated a clear benefit over aspirin alone. Furthermore, warfarin is associated with bleeding risk, numerous drug and food interactions, and requires frequent blood testing for monitoring.
The CARS trial (Citation75) indicated that the combination of a low fixed dose of warfarin (1 mg or 3 mg) with aspirin 80 mg resulted in a higher rate of spontaneous major bleeding versus aspirin 160 mg alone, but was not associated with a reduction in ischemic events in patients with prior MI. However, the doses of warfarin used in CARS were not anticoagulant in most subjects, which limits the interpretation of these findings. The CHAMP trial (Citation76) similarly compared the efficacy of aspirin 81 mg plus warfarin (INR (International normalized ratio) 1.5 to 2.1) versus aspirin 162 mg monotherapy in patients with prior MI, demonstrating no difference in ischemic events and significantly higher rates of both major and minor bleeding with combination therapy. Warfarin did not demonstrate prevention of recurrent ischemic events in patients with prior ischemic stroke as monotherapy at an INR of 1.4–2.8 versus aspirin in the WARSS trial (Citation77) or in patients with symptomatic PAD as an add-on to aspirin at an INR of 2.0–3.0 in the WAVE trial (Citation78). Hemorrhagic stroke and bleeding risk were significantly higher in the warfarin-plus-aspirin arm in WAVE. The SPIRIT trial, which evaluated a more intensive anticoagulation regimen (INR 3.0–4.5) versus aspirin in patients with TIA or minor stroke, was halted early because of an excess of major bleeding (particularly intracranial bleeding) in patients receiving warfarin (Citation79). In contrast to these findings, a meta-analysis of ten smaller, mostly European trials (Citation80) in patients with recent ACS showed that the combination of warfarin plus aspirin resulted in reduced rates of MI, ischemic stroke, and revascularization versus aspirin alone. However, major bleeding risk was 2.5-fold higher with warfarin plus aspirin versus aspirin alone (Citation80).
The clinical experience with warfarin has prompted the development of several novel anticoagulants that aim to improve upon its risk/benefit profile (reviewed in (Citation81)). The coagulation pathway provides many potential therapeutic targets, including factor Xa (FXa) and thrombin. Numerous oral, direct FXa inhibitors are in various stages of clinical development, including rivaroxaban, apixaban, LY517717, YM150, DU-176b, and betrixaban. Dabigatran is the only oral direct thrombin inhibitor (DTI) in late-stage development. On-going trials will assess whether these agents can mitigate bleeding risk without the need for frequent monitoring or dose adjustment.
Antiplatelet agents
Secondary prevention with antiplatelet therapy has been associated with significant reductions in ischemic events among patients with documented atherothrombotic disease. A comprehensive meta-analysis of 287 studies in patients with prior MI, prior stroke/TIA, or PAD (Citation2) confirmed a 25% relative risk reduction in secondary events (non-fatal MI, non-fatal stroke, vascular death) in patients treated with antiplatelet therapy (either aspirin alone or the combination of aspirin with another antiplatelet agent). However, regular use of aspirin ≤300 mg qd has been associated with a 2-fold increased risk of gastrointestinal bleeding (Citation82).
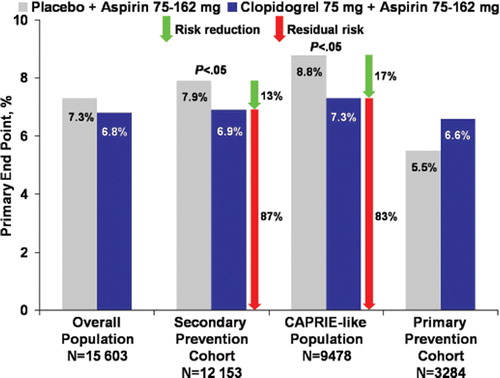
The platelet ADP P2Y12 receptor antagonist clopidogrel has also demonstrated beneficial effects in secondary prevention. The CAPRIE trial (Citation1) evaluated clopidogrel 75 mg versus aspirin 325 mg in patients with established atherothrombotic disease (MI, ischemic stroke, or objectively established PAD). Vascular death, MI, or ischemic stroke occurred in 5.8% of patients receiving aspirin and 5.3% of those receiving clopidogrel (P=0.043). Subgroup analysis showed a relative risk reduction of 23.8% (P=0.003) with clopidogrel among patients with PAD, whereas no significant difference between clopidogrel and aspirin was apparent in patients with prior ischemic stroke or MI. Bleeding rates were not significantly different between the two treatment arms. The CHARISMA trial (Citation83) compared the efficacy of clopidogrel plus aspirin versus aspirin monotherapy in patients who had either CVD, cerebrovascular disease, or PAD (secondary prevention cohort) or multiple risk factors (primary prevention cohort). There was no significant difference in the incidence of CV death, MI, or stroke between the treatment arms () (Citation83). Severe bleeding was not different between treatment arms, although moderate bleeding was higher in the clopidogrel-plus-aspirin arm (P=0.004). Subgroup analyses indicated that treatment with clopidogrel plus aspirin appeared more effective than aspirin alone in the secondaryprevention cohort (6.9% versus 7.9%, respectively; P=0.046), whereas a trend toward harm was evident in the primary prevention cohort (6.6% versus 5.5%, respectively; P=0.20) (). The primary outcome was reduced from 8.8% with aspirin to 7.3% with aspirin plus clopidogrel (P=0.01) in the ‘CAPRIE-like’ group of patients with prior ischemic events () (Citation84). Results from post hoc analyses should be interpreted with caution and require additional studies for confirmation of findings.
Figure 2. Primary end-point rate in the CHARISMA trial (Citation83,Citation84). No significant difference was obtained in the primary end-point rate (composite of myocardial infarction, stroke, or death from cardiovascular causes) between placebo+aspirin versus clopidogrel+aspirin groups in the overall study population and in the primary prevention cohort (Citation83). Significant reductions in favor of clopidogrel+aspirin were evident in the secondary prevention cohort (Citation83) and in the CAPRIE-like population (Citation84). Green arrows indicate relative risk reduction between treatment arms. Red arrows indicate residual risk for recurrent events.
Other trials have documented the benefit of antiplatelet therapy in patients with prior stroke or TIA. Early trials of the phosphodiesterase inhibitor dipyridamole, alone or in combination with aspirin, gave conflicting results (Citation85–87). The ESPS-2 trial demonstrated a relative 18% reduction in stroke with aspirin alone, 16% with extended-release dipyridamole alone, and 37% with the combination, suggesting an additive effect (Citation88). However, several criticisms of this trial, including low aspirin dose, high patient withdrawal rate, and low compliance rate, have led to reservations about its results. The ESPRIT trial demonstrated that the combination of extended-release dipyridamole (ERDP) plus aspirin significantly reduced the relative risk for death from all vascular causes, non-fatal stroke, non-fatal MI, or major bleeding complications by 20% versus aspirin alone (Citation89). Dual antiplatelet therapy with aspirin and clopidogrel did not provide significant clinical benefit compared with clopidogrel alone in patients with recent ischemic stroke or TIA in the MATCH trial. The addition of aspirin to clopidogrel caused a significantly greater frequency of life-threatening and major bleeding compared with clopidogrel alone (Citation88). The antiplatelet comparison within the PROFESS trial compared the benefit of aspirin plus ERDP versus clopidogrel in patients with recent stroke followed for 2.5 years (Citation90). Similar rates of recurrent stroke were observed in the two treatment arms (8.8% versus 9.0%). Higher rates of hemorrhagic events were observed in patients receiving aspirin plus ERDP versus clopidogrel (4.1% versus 3.6%), including intracranial hemorrhage (1.4% versus 1.0%). Most recently, the ACTIVE trial evaluated the benefit of aspirin plus clopidogrel in patients with atrial fibrillation who were not candidates for vitamin K antagonist therapy (Citation91). Dual antiplatelet therapy was associated with a significant 11% reduction in the relative risk of major vascular events versus aspirin alone, which was mainly attributed to a reduction in the risk of stroke (2.4% versus 3.3%; relative risk 0.72; P<0.001). Major bleeding occurred more frequently with clopidogrel plus aspirin versus aspirin alone (2.0% versus 1.3%; P<0.001). Current AHA/ACC guide-lines for stroke prevention recommend aspirin, the combination of aspirin and extended-release dipyridamole, or clopidogrel for patients with ischemic stroke or TIA, with the combination of aspirin and extended-release dipyridamole suggested over aspirin alone (Citation9).
Residual risk
Despite the unambiguous clinical benefit achieved with life-style modification, BP control, statins, angiotensin II-active agents, and antiplatelet agents, the residual risk for recurrent acute events in patients with established atherothrombotic disease remains substantial () (Citation2,Citation52). This residual risk is likely related to progression of atherosclerosis in the presence of statins and angiotensin II-active agents, lack of more complete inhibition of platelet activation in the presence of aspirin and clopidogrel, and other factors yet to be identified.
Figure 3. Residual atherothrombotic risk: rates of cardiovascular (CV) death and myocardial infarction (MI) persisting with active treatments from meta-analyses of trials of secondary prevention with statins (Citation52), angiotensin-converting enzyme (ACE) inhibitors (Citation60), β-blockers (Citation71), antiplatelets (Citation2), and warfarin (Citation80). Treatment and comparator arms consisted of the following: high-dose versus standard-dose statins; ACE inhibitor versus placebo; β-blocker versus placebo or other antihypertensive agents; antiplatelet therapy versus control; and warfarin+aspirin versus aspirin. *Event rate is shown for coronary death/MI for statin meta-analysis (individual rates for MI not available). OR = odds ratio.
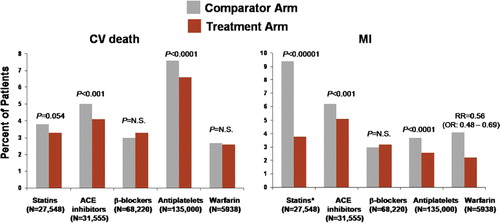
It is currently unknown how much additional reduction of this residual risk could be achieved with improved approaches to limit atherosclerosis progression or platelet-mediated thrombosis. Recent attempts to further limit atherosclerosis progression with novel approaches, such as inhibition of cholesteryl ester transfer protein (CETP) (Citation92,Citation93), inhibition of acyl-coenzyme A:cholesterol acyltransferase (ACAT) (Citation94), and inhibition of intestinal cholesterol absorption (Citation95), have generally been unsuccessful. Further attempts at CETP inhibition with the novel agent anacetrapib have shown favorable changes in lipid profiles in patients with dyslipidemia, without safety concerns in early clinical trials (Citation96), and a phase 3 trial is on-going (Clinicaltrials.gov identifier: NCT00685776). The addition of niacin to a statin has been shown to slow the progression of atherosclerosis (Citation97), and an on-going trial is comparing the effects of niacin versus a cholesterol absorption inhibitor, ezetimibe, on progression of atherosclerosis in patients receiving a statin (Clinicaltrials.gov identifier: NCT00687076). However, a recent study with niacin/laropiprant designed to assess the effect on atherosclerosis progression was prematurely terminated (Clinicaltrials.gov identifier: NCT00384293). It should be noted that the studies that evaluated the effect of statins on progression of atherosclerosis have produced equivocal results (Citation92,Citation93,Citation95,Citation98,Citation99). Nevertheless, novel lipid-modifying approaches have so far not been demonstrated to further reduce adverse CV events beyond statin alone (Citation100), and a large on-going trial is evaluating the effect of adding ezetimibe to a statin on CV outcomes (Clinicaltrials.gov identifier: NCT00202878). The effect of adding niacin to a statin in secondary prevention has not been evaluated to date. It is also interesting to speculate that improving therapeutic regimens to include treating the inflammatory component of CV disease may also improve patient outcomes, as mentioned above.
Response variability (also referred to as platelet resistance) to antiplatelet therapy is considered to be a potentially important limitation of antiplatelet therapy and may contribute to residual risk for thrombotic events. Although a standardized definition and methodology for measurement of low responsiveness to antiplatelet therapy has not been established, sufficient evidence supports the concept that persistence of enhanced platelet reactivity despite the use of aspirin (Citation101) and/or clopidogrel (Citation102) is associated with adverse clinical outcomes. A number of factors may influence response to antiplatelet therapy, including drug dose or absorption, patient compliance, and genetic polymorphisms (Citation102). Recent analyses suggest that genetic polymorphisms of the cytochrome P (CYP) 450 enzymes, which are required for the conversion of clopidogrel into an active metabolite, can significantly modulate individual response to clopidogrel and are important determinants of prognosis (Citation103–105). Whether new antiplatelet agents exhibit less variability or no variability in platelet responsiveness and the association with clinical outcomes will be interesting to note as these agents progress through clinical development.
Novel approaches to limit platelet-mediated thrombosis also offer promise. Ticagrelor (AZD6140) is a third-generation P2Y12 receptor antagonist that is currently in phase 3 trials (Clinicaltrials.gov identifiers NCT00385138, NCT00305162, and NCT00391872). Phase 3 trials of cangrelor, a novel P2Y12 receptor antagonist, were recently halted due to a lack of efficacy in an interim analysis. Prasugrel is approved in Europe and the US. Prasugrel, which demonstrated increased efficacy and bleeding versus clopidogrel in TRITON, also exhibits less variability in platelet responsiveness (Citation106). Current antiplatelet agents and those in development inhibit the TxA2 or ADP platelet activation pathways but do not interfere with a multitude of other platelet activation pathways that may contribute to thrombotic events. In addition to significant residual risk for ischemic events, dual antiplatelet therapy is associated with increased bleeding. These considerations underscore the critical need for novel therapies that provide more comprehensive platelet inhibition for greater protection against thrombotic events, preferably without an incremental bleeding risk.
Surrogate markers
Available data from epidemiological studies indicate that traditional risk factors do not fully explain the residual risk for thrombotic events or treatment responses (Citation107). There is a need for the development and clinical implementation of new markers of atherothrombotic disease which are intended to predict future risk of thrombotic events, particularly in high-risk patients. For example, measurement of urinary 11-dehydro-thromboxane B2, a stable metabolite of TxA2 can be used to assess the degree of inhibition of TxA2 production by aspirin (and hence aspirin resistance) (Citation108). An analysis of HOPE revealed that increasing quartiles of urinary 11-dehydro-thromboxane B2 were associated with increased risk of MI, stroke, or CV death (odds ratio 1.8; 95% confidence interval 1.2–2.9; P<0.009 for first versus fourth quartile), suggesting the predictive value of this metabolite (Citation109).
Vascular calcification has also been shown to have predictive value. The presence and extent of coronary artery calcification (CAC) is associated with atherosclerotic plaque burden (Citation110). Elevated CAC scores have been shown to predict future cardiac events in both asymptomatic (Citation111–115) and symptomatic (Citation116,Citation117) individuals. Current guidelines state that asymptomatic individuals at intermediate risk for CHD ‘may be reasonable candidates’ for CAC testing, whereas in symptomatic patients, exclusion of CAC score may be an effective risk stratification tool before invasive procedures or hospital admission (Citation118).
Aortic stiffness is associated with changes in the arterial wall resulting in increased systolic BP and may also reflect coronary artery lesions (Citation119). As measured by aortic pulse wave velocity (PWV), aortic stiffness has predictive value for all-cause and CV mortality, MI, and stroke in a wide spectrum of patients, including those with end-stage renal disease (ESRD) (Citation120), diabetes (Citation121), hypertension (Citation122,Citation123), and healthy individuals (Citation124–126). Whether a reduction in PWV independently predicts a reduction in CV events has been suggested in patients with ESRD (Citation127) but remains to be established in patients at lower CV risk. Left ventricular hypertrophy (LVH) is an additional predictor of CV events which is associated with hypertension severity. Increasing LVH is associated with increases in risk of stroke, CHD, and heart failure, and reducing LVH with antihypertensive medications lowers CV risk (Citation128). However, different classes of antihypertensive drugs reduce LVH to varying degrees which may not correlate with reduction in BP (Citation129).
Chronic inflammation plays a key role in plaque instability and subsequent occlusive thrombosis. Elevated levels of inflammatory mediators have predictive value for vascular events (reviewed in Libby et al. (Citation130), and several of these molecules are under clinical investigation as therapeutic targets. For example, human A2 phospholipases (PLA2), such as lipoprotein-associated PLA2 (Lp-PLA2), independently predict risk of CHD and ischemic stroke (Citation131). Several novel inhibitors of Lp-PLA2 and secretory PLA2 (sPLA2) are under development (Citation132,Citation133). The oral agent darapladib showed a reduction in atheroma growth in patients with documented coronary disease receiving standard of care (Citation133). Of note, darapladib did not lower LDL-C (Citation133), whereas an 8% reduction in LDL-C was noted with the sPLA2 inhibitor A-002 (Citation132), suggesting disparate effects on LDL clearance. STABILITY, a large on-going phase 3 trial, is assessing the benefit of darapladib on CV events in patients with CHD (Citation134), while A-002 is in phase 2 testing in combination with atorvastatin (Citation135). C-reactive protein (CRP) is arguably the most promising inflammatory biomarker based on results of multiple population-based studies showing that CRP levels predict future CV events, diabetes, and hypertension in patients with and without CVD (reviewed by Ridker (Citation136). These studies have suggested that the relative predictive power of CRP is at least as strong as other established risk factors such as LDL-C, BP, and smoking (Citation136). The results of the recent JUPITER trial, which demonstrated the benefit of rosuvastatin in patients without hyperlipidemia but with elevated CRP, support the notion that CRP levels may be useful in stratifying patient risk and suggest that statin treatment may be beneficial in otherwise healthy individuals (Citation46). Current guide-lines recommend the optional use of CRP measurement in patients at intermediate risk (i.e. 10%–20% risk of CHD over 10 years) to help guide considerations for further evaluation or therapy (Citation137).
Registry analyses document unmet needs in current clinical practice
The global REACH registry offered a unique opportunity to asses the prevalence of risk factors, the use of various medications, and clinical outcomes around the world, including North America (Citation138). Patients eligible for enrollment included those with documented atherothrombotic disease (CAD, cerebrovascular disease, or PAD) as well as those with ≥3 risk factors for atherothrombosis. Over 68,000 patients in 44 countries were followed for 1 year from 2003 to 2004. North American participants (n=25,999), of whom 75% had symptomatic atherothrombotic disease, were characterized by a high prevalence of hypertension (86.4%), hypercholesterolemia (82.7%), and high body mass index (≥25; 77.8%), as well as current or former smoking (58.1%) and diabetes (50.6%). The vast majority of North American patients (88.4%) were managed by either general/family practitioners or internists, and only 7.4% were managed by cardiologists. Statins and aspirin were used in 77.1% and 71.4% of North American patients, respectively, whereas only a minority (14.2%) received oral anticoagulants. Use of dual antiplatelet therapy in North American patients was also low (14.2%), suggesting opportunities for improvement in patient care. In the global cohort, the combined incidence of CV death, MI, stroke, or hospitalization for an atherothrombotic event at 1 year was almost 3-fold higher in patients with established atherothrombotic disease than in those with multiple risk factors (14.4% versus 5.3%, respectively). In the North American cohort that included 75% of patients with established atherothrombosis, the 1-year composite rate of CV death, MI, stroke, or hospitalization for atherothrombosis was 11.6%. Furthermore, the annual incidence of CV death, MI, or stroke exceeded the anticipated 3% event rate in all regions including North America (3.7%). These observational data confirm the high residual risk of patients with documented atherothrombotic disease reported in clinical trials and highlight the need for more effective therapies.
Conclusion
Although death rates due to CVD continue to decline, atherothrombotic disease remains a major public health issue; its prevalence can be expected to rise as our population ages and the incidence of diabetes/metabolic syndrome continues to increase. Life-style modifications, BP control, statins, angiotensin II-active agents, and antiplatelet therapy—current cornerstones of secondary prevention—target key pathophysiological processes responsible for acute ischemic events, namely atherosclerotic plaque rupture/erosion and platelet-mediated thrombosis. Clinical trials with pharmacological agents added to life-style modification have demonstrated significant reductions in morbidity and mortality over those achieved with attempts at life-style modification alone. Despite these proven benefits, residual risk for secondary ischemic events remains substantial, as thrombotic events continue to occur even in the presence oflife-style modification and pharmacotherapy. Therefore, there is a need for novel agents, perhaps those directed at other pathways, that can further reduce ischemic events by providing more comprehensive inhibition of platelet-mediated thrombosis, without increasing the risk of bleeding. In addition, the approach to care will likely require a risk/benefit assessment in high-risk patients in order to balance safety with preventing potentially fatal outcomes.
Acknowledgements
Dr Pepine reports serving as a consultant for Schering-Plough. Dr Pepine did not receive payment for the development of this manuscript, and no outside sources were involved in the development, review, or final approval of its content.
Dr Pepine would like to thank Gina Fusaro, PhD, and Joshua Barbach, MA, for providing editorial assistance during the preparation of this manuscript. This assistance was funded by Schering-Plough.
Related Research Data
References
- A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet. 1996;348:1329–39.
- Antithrombotic Trialists' Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22.
- Kris-Etherton P, Eckel RH, Howard BV, St Jeor S, Bazzarre TL. AHA Science Advisory: Lyon Diet Heart Study. Benefits of a Mediterranean-style, National Cholesterol Education Program/American Heart Association Step I Dietary Pattern on Cardiovascular Disease. Circulation. 2001;103:1823–5.
- Lee KW, Lip GY. Effects of lifestyle on hemostasis, fibrinolysis, and platelet reactivity: a systematic review. Arch Intern Med. 2003;163:2368–92.
- Wadden TA, Berkowitz RI, Womble LG, Sarwer DB, Phelan S, Cato RK, . Randomized trial of lifestyle modification and pharmacotherapy for obesity. N Engl J Med. 2005;353:2111–20.
- Ferguson JJ. The role of oral antiplatelet agents in atherothrombotic disease. Am J Cardiovasc Drugs. 2006;6:149–57.
- Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, . Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146.
- Sacco RL, Adams R, Albers G, Alberts MJ, Benavente O, Furie K, . Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Circulation. 2006;113:e409–49.
- Allison MA, Ho E, Denenberg JO, Langer RD, Newman AB, Fabsitz RR, . Ethnic-specific prevalence of peripheral arterial disease in the United States. Am J Prev Med. 2007;32:328–33.
- Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation. 2004;110:738–43.
- Criqui MH, Langer RD, Fronek A, Feigelson HS, Klauber MR, McCann TJ, . Mortality over a period of 10 years in patients with peripheral arterial disease. N Engl J Med. 1992;326:381–6.
- Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, . ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation. 2006;113:e463–654.
- Howell MA, Colgan MP, Seeger RW, Ramsey DE, Sumner DS. Relationship of severity of lower limb peripheral vascular disease to mortality and morbidity: a six-year follow-up study. J Vasc Surg. 1989;9:691–6; discussion 6–7.
- McKenna M, Wolfson S, Kuller L. The ratio of ankle and arm arterial pressure as an independent predictor of mortality. Atherosclerosis. 1991;87:119–28.
- McDermott MM, Feinglass J, Slavensky R, Pearce WH. The ankle-brachial index as a predictor of survival in patients with peripheral vascular disease. J Gen Intern Med. 1994;9:445–9.
- Hirsh J, O'Donnell M, Eikelboom JW. Beyond unfractionated heparin and warfarin: current and future advances. Circulation. 2007;116:552–60.
- Leng GC, Lee AJ, Fowkes FG, Whiteman M, Dunbar J, Housley E, . Incidence, natural history and cardiovascular events in symptomatic and asymptomatic peripheral arterial disease in the general population. Int J Epidemiol. 1996;25:1172–81.
- McDermott MM, Liu K, Greenland P, Guralnik JM, Criqui MH, Chan C, . Functional decline in peripheral arterial disease: associations with the ankle brachial index and leg symptoms. JAMA. 2004;292:453–61.
- Wasserman EJ, Shipley NM. Atherothrombosis in acute coronary syndromes: mechanisms, markers, and mediators of vulnerability. Mt Sinai J Med. 2006;73:431–9.
- Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–8.
- Napoli C, D'Armiento FP, Mancini FP, Postiglione A, Witztum JL, Palumbo G, . Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100:2680–90.
- Napoli C, de Nigris F, Welch JS, Calara FB, Stuart RO, Glass CK, . Maternal hypercholesterolemia during pregnancy promotes early atherogenesis in LDL receptor-deficient mice and alters aortic gene expression determined by microarray. Circulation. 2002;105:1360–7.
- Napoli C, Witztum JL, de Nigris F, Palumbo G, D'Armiento FP, Palinski W. Intracranial arteries of human fetuses are more resistant to hypercholesterolemia-induced fatty streak formation than extracranial arteries. Circulation. 1999;99:2003–10.
- Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–94.
- Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–75.
- Sakariassen KS, Nievelstein PF, Coller BS, Sixma JJ. The role of platelet membrane glycoproteins Ib and IIb-IIIa in platelet adherence to human artery subendothelium. Br J Haematol. 1986;63:681–91.
- Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28:403–12.
- Brass LF. Thrombin and platelet activation. Chest. 2003;124:Suppl:18–25S.
- Kulkarni S, Dopheide SM, Yap CL, Ravanat C, Freund M, Mangin P, . A revised model of platelet aggregation. J Clin Invest. 2000;105:783–91.
- Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–84.
- May AE, Seizer P, Gawaz M. Platelets: inflammatory firebugs of vascular walls. Arterioscler Thromb Vasc Biol. 2008;28:s5–10.
- Krotz F, Sohn HY, Gloe T, Zahler S, Riexinger T, Schiele TM, . NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002;100:917–24.
- Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005;83:196–8.
- Satoh M, Ishikawa Y, Minami Y, Takahashi Y, Nakamura M. Role of Toll like receptor signaling pathway in ischemic coronary artery disease. Front Biosci. 2008;13:6708–15.
- Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, . Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–52.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344:1383–9.
- Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–57.
- Amarenco P, Bogousslavsky J, Callahan A 3rd, Goldstein LB, Hennerici M, Rudolph AE, . High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549–59.
- Cannon CP, Braunwald E, Mc Cabe CH, Rader DJ, Rouleau JL, Belder R, . Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504.
- de Lemos JA, Blazing MA, Wiviott SD, Lewis EF, Fox KA, White HD, . Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292:1307–16.
- Gissi HFI, Tavazzi L, Maggioni AP, Marchioli R, Barlera S, Franzosi MG, . Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1231–9.
- Kjekshus J, Apetrei E, Barrios V, Bohm M, Cleland JG, Cornel JH, . Rosuvastatin in older patients with systolic heart failure. N Engl J Med. 2007;357:2248–61.
- La Rosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, . Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–35.
- Pedersen TR, Faergeman O, Kastelein JJ, Olsson AG, Tikkanen MJ, Holme I, . High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA. 2005;294:2437–45.
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto A, Jr, Kastelein JJ, . Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207.
- Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, . The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–9.
- Waters DD, La Rosa JC, Barter P, Fruchart JC, Gotto A Jr, Carter R, . Effects of high-dose atorvastatin on cerebrovascular events in patients with stable coronary disease in the TNT (treating to new targets) study. J Am Coll Cardiol. 2006;48:1793–9.
- Levy WC. Observational studies of statins in systolic heart failure. Heart Fail Clin. 2008;4:201–8.
- Stypmann J, Schubert A, Welp H, Schulte H, Assmann G, Breithardt G, . Atorvastatin therapy is associated with reduced levels of N-terminal prohormone brain natriuretic peptide and improved cardiac function in patients with heart failure. Clin Cardiol. 2008;31:478–81.
- Vrtovec B, Okrajsek R, Golicnik A, Ferjan M, Starc V, Schlegel TT, . Atorvastatin therapy may reduce the incidence of sudden cardiac death in patients with advanced chronic heart failure. J Card Fail. 2008;14:140–4.
- Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol. 2006;48:438–45.
- O'Regan C, Wu P, Arora P, Perri D, Mills EJ. Statin therapy in stroke prevention: a meta-analysis involving 121,000 patients. Am J Med. 2008;121:24–33.
- Aung PP, Maxwell HG, Jepson RG, Price JF, Leng GC. Lipid-lowering for peripheral arterial disease of the lower limb. Cochrane Database Syst Rev. 2007(4): CD000123.
- Yusuf S, Pepine CJ, Garces C, Pouleur H, Salem D, Kostis J, . Effect of enalapril on myocardial infarction and unstable angina in patients with low ejection fractions. Lancet. 1992;340:1173–8.
- Pitt B, O'Neill B, Feldman R, Ferrari R, Schwartz L, Mudra H, . The QUinapril Ischemic Event Trial (QUIET): evaluation of chronic ACE inhibitor therapy in patients with ischemic heart disease and preserved left ventricular function. Am J Cardiol. 2001;87:1058–63.
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–53.
- Fox KM. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet. 2003;362:782–8.
- Braunwald E, Domanski MJ, Fowler SE, Geller NL, Gersh BJ, Hsia J, . Angiotensin-converting-enzyme inhibition in stable coronary artery disease. N Engl J Med. 2004;351:2058–68.
- Saha SA, Molnar J, Arora RR. Tissue ACE inhibitors for secondary prevention of cardiovascular disease in patients with preserved left ventricular function: a pooled meta-analysis of randomized placebo-controlled trials. J Cardiovasc Pharmacol Ther. 2007;12:192–204.
- Yusuf S, Teo K, Anderson C, Pogue J, Dyal L, Copland I, . Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet. 2008;372:1174–83.
- Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, . Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–59.
- Yusuf S, Diener HC, Sacco RL, Cotton D, Ounpuu S, Lawton WA, . Telmisartan to prevent recurrent stroke and cardiovascular events. N Engl J Med. 2008;359:1225–37.
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet. 2001;358:1033–41.
- Smith S, Jr, Allen J, Blair SN, Bonow RO, Brass LM, Fonarow GC, . AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update: endorsed by the National Heart, Lung, and Blood Institute. Circulation. 2006;113:2363–72.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–89.
- Holman RR, Paul SK, Bethel MA, Neil HA, Matthews DR. Long-term follow-up after tight control of blood pressure in type 2 diabetes. N Engl J Med. 2008;359:1565–76.
- Patel A, Mac Mahon S, Chalmers J, Neal B, Billot L, Woodward M, . Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–72.
- Gerstein HC, Miller ME, Byington RP, Goff DC, Jr, Bigger JT, Buse JB, . Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–59.
- Chen ZM, Pan HC, Chen YP, Peto R, Collins R, Jiang LX, . Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622–32.
- Bangalore S, Wetterslev J, Pranesh S, Sawhney S, Gluud C, Messerli FH. Perioperative beta blockers in patients having non-cardiac surgery: a meta-analysis. Lancet. 2008;372:1962–76.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol. 2008;52:1482–9.
- Campbell DJ, Aggarwal A, Esler M, Kaye D. beta-blockers, angiotensin II, and ACE inhibitors in patients with heart failure. Lancet. 2001;358:1609–10.
- Anderson JL, Adams CD, Antman EM, Bridges CR, Califf RM, Casey DE, Jr, . ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol. 2007;50:e1–157.
- Randomised double-blind trial of fixed low-dose warfarin with aspirin after myocardial infarction. Coumadin Aspirin Reinfarction Study (CARS) Investigators. Lancet. 1997;350:389–96.
- Fiore LD, Ezekowitz MD, Brophy MT, Lu D, Sacco J, Peduzzi P. Department of Veterans Affairs Cooperative Studies Program Clinical Trial comparing combined warfarin and aspirin with aspirin alone in survivors of acute myocardial infarction: primary results of the CHAMP study. Circulation. 2002;105:557–63.
- Mohr JP, Thompson JL, Lazar RM, Levin B, Sacco RL, Furie KL, . A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke. N Engl J Med. 2001;345:1444–51.
- Anand S, Yusuf S, Xie C, Pogue J, Eikelboom J, Budaj A, . Oral anticoagulant and antiplatelet therapy and peripheral arterial disease. N Engl J Med. 2007;357:217–27.
- A randomized trial of anticoagulants versus aspirin after cerebral ischemia of presumed arterial origin. The Stroke Prevention in Reversible Ischemia Trial (SPIRIT) Study Group. Ann Neurol. 1997;42:857–65.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med. 2005;143:241–50.
- Turpie AG. New oral anticoagulants in atrial fibrillation. Eur Heart J. 2008;29:155–65.
- Garcia Rodriguez LA, Hernandez-Diaz S, de Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiologic studies. Br J Clin Pharmacol. 2001;52:563–71.
- Bhatt DL, Fox KA, Hacke W, Berger PB, Black HR, Boden WE, . Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354:1706–17.
- Bhatt DL, Flather MD, Hacke W, Berger PB, Black HR, Boden WE, . Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol. 2007;49:1982–8.
- Persantine Aspirin Trial in cerebral ischemia. Part II: Endpoint results. The American-Canadian Co-Operative Study group. Stroke. 1985;16:406–15.
- The European Stroke Prevention Study (ESPS). Principal end-points. The ESPS Group. Lancet. 1987;2:1351–4.
- Bousser MG, Eschwege E, Haguenau M, Lefaucconnier JM, Thibult N, Touboul D, . ‘AICLA’ controlled trial of aspirin and dipyridamole in the secondary prevention of athero-thrombotic cerebral ischemia. Stroke. 1983;14:5–14.
- Diener HC, Cunha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci. 1996;143:1–13.
- Halkes PH, van Gijn J, Kappelle LJ, Koudstaal PJ, Algra A. Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): randomised controlled trial. Lancet. 2006;367:1665–73.
- Sacco RL, Diener HC, Yusuf S, Cotton D, Ounpuu S, Lawton WA, . Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008;359:1238–51.
- Connolly SJ, Pogue J, Hart RG, Hohnloser SH, Pfeffer M, Chrolavicius S, . Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med. 2009;360:2066–78.
- Kastelein JJ, van Leuven SI, Burgess L, Evans GW, Kuivenhoven JA, Barter PJ, . Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007;356:1620–30.
- Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, . Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356:1304–16.
- Tardif JC, Gregoire J, L'Allier PL, Anderson TJ, Bertrand O, Reeves F, . Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation. 2004;110:3372–7.
- Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, . Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–43.
- Krishna R, Anderson MS, Bergman AJ, Jin B, Fallon M, Cote J, . Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370:1907–14.
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110:3512–7.
- Crouse J, 3rd, Raichlen JS, Riley WA, Evans GW, Palmer MK, O'Leary DH, . Effect of rosuvastatin on progression of carotid intima-media thickness in low-risk individuals with subclinical atherosclerosis: the METEOR Trial. JAMA. 2007;297:1344–53.
- Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet. 2001;357:577–81.
- Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, . Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
- Krasopoulos G, Brister SJ, Beattie WS, Buchanan MR. Aspirin ‘resistance’ and risk of cardiovascular morbidity: systematic review and meta-analysis. BMJ. 2008;336:195–8.
- Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Alfonso F, Macaya C, Bass TA, . Variability in individual responsiveness to clopidogrel: clinical implications, management, and future perspectives. J Am Coll Cardiol. 2007;49:1505–16.
- Collet JP, Hulot JS, Pena A, Villard E, Esteve JB, Silvain J, . Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet. 2009;373:309–17.
- Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, . Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360:354–62.
- Simon T, Verstuyft C, Mary-Krause M, Quteineh L, Drouet E, Meneveau N, . Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med. 2009;360:363–75.
- Jernberg T, Payne CD, Winters KJ, Darstein C, Brandt JT, Jakubowski JA, . Prasugrel achieves greater inhibition of platelet aggregation and a lower rate of non-responders compared with clopidogrel in aspirin-treated patients with stable coronary artery disease. Eur Heart J. 2006;27:1166–73.
- Brown TM, Bittner V. Biomarkers of atherosclerosis: clinical applications. Curr Cardiol Rep. 2008;10:497–504.
- Catella F, Healy D, Lawson JA, Fitz Gerald GA. 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc Natl Acad Sci U S A. 1986;83:5861–5.
- Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–5.
- Budoff MJ, Gul KM. Expert review on coronary calcium. Vasc Health Risk Manag. 2008;4:315–24.
- Anand DV, Lim E, Hopkins D, Corder R, Shaw LJ, Sharp P, . Risk stratification in uncomplicated type 2 diabetes: prospective evaluation of the combined use of coronary artery calcium imaging and selective myocardial perfusion scintigraphy. Eur Heart J. 2006;27:713–21.
- Arad Y, Goodman KJ, Roth M, Newstein D, Guerci AD. Coronary calcification, coronary disease risk factors, C-reactive protein, and atherosclerotic cardiovascular disease events: the St. Francis Heart Study. J Am Coll Cardiol. 2005;46:158–65.
- Raggi P, Cooil B, Shaw LJ, Aboulhson J, Takasu J, Budoff M, . Progression of coronary calcium on serial electron beam tomographic scanning is greater in patients with future myocardial infarction. Am J Cardiol. 2003;92:827–9.
- Shaw LJ, Raggi P, Schisterman E, Berman DS, Callister TQ. Prognostic value of cardiac risk factors and coronary artery calcium screening for all-cause mortality. Radiology. 2003;228:826–33.
- Vliegenthart R, Oudkerk M, Song B, van der Kuip DA, Hofman A, Witteman JC. Coronary calcification detected by electron-beam computed tomography and myocardial infarction. The Rotterdam Coronary Calcification Study. Eur Heart J. 2002;23:1596–603.
- Georgiou D, Budoff MJ, Kaufer E, Kennedy JM, Lu B, Brundage BH. Screening patients with chest pain in the emergency department using electron beam tomography: a follow-up study. J Am Coll Cardiol. 2001;38:105–10.
- Keelan PC, Bielak LF, Ashai K, Jamjoum LS, Denktas AE, Rumberger JA, . Long-term prognostic value of coronary calcification detected by electron-beam computed tomography in patients undergoing coronary angiography. Circulation. 2001;104:412–7.
- Greenland P, Bonow RO, Brundage BH, Budoff MJ, Eisenberg MJ, Grundy SM, . ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain: a report of the American College of Cardiology Foundation Clinical Expert Consensus Task Force (ACCF/AHA Writing Committee to Update the 2000125 Expert Consensus Document on Electron Beam Computed Tomography). Circulation. 2007;115:402–26.
- Laurent S, Boutouyrie P. Arterial stiffness: a new surrogate end point for cardiovascular disease? J Nephrol. 2007;20:(Suppl 12):S45–50.
- Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation. 1999;99:2434–9.
- Cruickshank K, Riste L, Anderson SG, Wright JS, Dunn G, Gosling RG. Aortic pulse-wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation. 2002;106:2085–90.
- Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, . Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236–41.
- Laurent S, Katsahian S, Fassot C, Tropeano AI, Gautier I, Laloux B, . Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke. 2003;34:1203–6.
- Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, . Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation. 2006;113:657–63.
- Sutton-Tyrrell K, Najjar SS, Boudreau RM, Venkitachalam L, Kupelian V, Simonsick EM, . Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation. 2005;111:3384–90.
- Willum-Hansen T, Staessen JA, Torp-Pedersen C, Rasmussen S, Thijs L, Ibsen H, . Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation. 2006;113:664–70.
- Guerin AP, Blacher J, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness attenuation on survival of patients in end-stage renal failure. Circulation. 2001;103:987–92.
- Ruilope LM, Schmieder RE. Left ventricular hypertrophy and clinical outcomes in hypertensive patients. Am J Hypertens. 2008;21:500–8.
- Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med. 2003;115:41–6.
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43.
- Oei HH, van der Meer IM, Hofman A, Koudstaal PJ, Stijnen T, Breteler MM, . Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke: the Rotterdam Study. Circulation. 2005;111:570–5.
- Rosenson RS, Hislop C, McConnell D, Elliott M, Stasiv Y, Wang N, . Effects of 1-H-indole-3-glyoxamide (A-002) on concentration of secretory phospholipase A2 (PLASMA study): a phase II double-blind, randomised, placebo-controlled trial. Lancet. 2009;373:649–58.
- Serruys PW, Garcia-Garcia HM, Buszman P, Erne P, Verheye S, Aschermann M, . Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–82.
- The Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy Trial (STABILITY). NCT00799903. Available at: http://www.clinicaltrials.gov/ct2/show/NCT00799903 (accessed 26 February 2009).
- FRANCIS-ACS Trial: A Study of the Safety and Efficacy of A 002 in Subjects With Acute Coronary Syndromes. NCT00743925. Available at: //www.clinicaltrials.gov/ct2/show/NCT00743925 (accessed 26 February 2009).
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49:2129–38.
- Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon R, 3rd, Criqui M, . Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511.
- Steg PG, Bhatt DL, Wilson PW, D'Agostino R, SrOhman EM, Rother J, . One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA. 2007;297:1197–206.