Abstract
Matrix Gla protein (MGP) expression is increased in cardiac hypertrophy, but the precise mechanisms regulating its expression are unknown. Here we characterized the effect of pressure overload and myocardial infarction in vivo as well as mechanical stretch and hypertrophic agonists in vitro on MGP expression. When angiotensin II (Ang II) was administered by osmotic minipumps, left ventricular (LV) MGP mRNA levels increased significantly from 6 h to 2weeks, whereas intravenous arginine8-vasopressin increased LV MGP mRNA levels within 4 h. During post-infarction remodeling process, MGP mRNA levels were elevated at 24 h (1.3-fold, p<0.05) and the maximal increase was observed at 4 weeks (2.8-fold, p<0.01). Ang II increased MGP mRNA levels 20% (p<0.05) in neonatal rat cardiac myocytes and 40% (p<0.05) in cardiac fibroblasts, whereas endothelin-1 decreased MGP mRNA levels 30% (p<0.01) in myocytes and had no effect in fibroblasts. Cyclic mechanical stretch resulted in reduction of MGP gene expression in both cardiac myocytes and fibroblasts. These results demonstrate that MGP is rapidly upregulated in response to cardiac overload well before the development of LV hypertrophy and post-infarction remodeling process. Our results also suggest that Ang II may be involved in mediating load-induced activation of MGP expression.
Introduction
Pressure overload and myocardial infarction (MI) result in pathological remodeling of the myocardium (Citation1). This re-organization of the tissue aims to preserve cardiac output and function, but eventually it leads to maladaptive changes and heart failure. Remodeling of the heart involves alterations in cell size, number and shape as well as in extracellular matrix (ECM) composition. At the molecular level, remodeling is characterized by genetic re-programming such as activation of fetal gene program involving re-expression of e.g. β-myosin heavy chain and atrial natriuretic peptide, and later increased expression of type I collagen and other ECM components (Citation1). In addition to its structural function, ECM has an active role in regulating cardiac remodeling by mediating cell signaling through its ability to bind and modulate the action of various signaling molecules (e.g. growth factors and cytokines) as well as transmembrane proteins (e.g. integrins). Therefore, ECM is involved in regulating many cellular functions such as cell differentiation, proliferation, growth and apoptosis (Citation2,Citation3).
Matrix Gla protein (MGP) is an ECM protein that inhibits calcification in arteries and cartilage (Citation4). MGP is expressed in many tissues, highest mRNA levels being observed in heart, lung, kidney (Citation5) and cartilage (Citation6) and protein levels in the bone (Citation5). In addition to osteoblasts (Citation7) and chondrocytes (Citation8), various other cell types have been shown to express MGP, e.g. endothelial cells (Citation9), smooth muscle cells (Citation10), fibroblasts (Citation11) and macrophages (Citation12). MGP contains glutamic acid (Glu) residues, which are post-translationally modified by vitamin K-dependent γ-glutamyl carboxylase (Citation13) to γ-carboxyglutamic acid (Gla). This modification is needed for the calcium binding properties of the protein (Citation13) and it is thought that only the γ-carboxylated Gla form of MGP is functional (Citation14). Additionally, MGP can be post-translationally modified by phosphorylation of its serine residues, possibly affecting its secretion to ECM (Citation15).
In addition to extensive studies on arterial calcification, there are DNA-microarray studies connecting increased left ventricular (LV) MGP gene expression with acute and chronic pressure overload and myocardial hypertrophy in mice (Citation16,Citation17) and humans (Citation18). LV MGP gene expression is also increased in heart failure in mice (Citation19) and rats (Citation20). The ECM binding properties of MGP suggests that besides calcification inhibition it has other regulatory roles in cell-matrix interactions (Citation21), e.g. regulation of cell migration and proliferation or cell-matrix adhesion. However, little is known about the precise mechanisms regulating the cardiac specific induction of MGP gene expression. The present study was designed to characterize the MGP expression and regulation in the rat heart in vivo and neonatal rat myocyte and fibroblast cell cultures in vitro.
Methods
Animals
Male 2-month-old Sprague–Dawley (SD) rats weighting 250–300 g and male 20-month-old spontaneously hypertensive rats (SHR) of the Okamoto-Aoki strain and age-matched Wistar–Kyoto (WKY) rats from the colony of the Center of Experimental Animals at the University of Oulu, Finland, were used. SHR strain was originally obtained from Mollegaards Avslaboratorium, Skensved, Denmark. All rats were kept in individual plastic cages with free access to tap water and normal rat chow. A 12-h light and 12-h dark environmental light cycle was maintained. The experimental design was approved by the Animal Care and Use Committee of the University of Oulu.
Experimental design in conscious normotensive rats
The SD rats were anesthetized and instrumented for vehicle and drug infusions as previously described (Citation22,Citation23). The experiments were started in conscious animals by measurement of mean arterial pressure (MAP) and heart rate before arginine8-vasopressin (AVP) at a dose of 0.05 μg/kg/min or vehicle (0.9% NaCl) was infused intravenously. MAP rose rapidly and reached maximum values within 15 min during AVP infusion associated with a significant decrease in heart rate (Citation24). MAP and heart rate remained unchanged in the vehicle-treated animals. To validate the experimental system, we analyzed the effect of AVP infusion on the gene expression of Egr-1 and brain natriuretic peptide (BNP), which represent genetic hallmarks of the load-induced cardiac hypertrophic program. A significant increase in LV Egr-1 mRNA levels was seen already at 30min of AVP infusion, and at 4 h, both Egr-1 and BNP mRNA levels were markedly elevated (Citation24).
In separate series of experiments, angiotensin II (Ang II, 33 μg/kg/h), Ang II type 1 (AT1) receptor antagonist losartan, their combination or vehicle (0.9% NaCl) was infused through subcutaneously implanted osmotic minipumps (Alzet) for 2weeks (Citation25). Using this model of experimental hypertension, administration of Ang II increases MAP within 3 h and MAP remains significantly elevated throughout the 2-week period (Citation26). BNP gene expression increases within 6 h of Ang II infusion and remains elevated up to 2weeks (Citation24).
Myocardial infarction
Experimental MI was produced by ligation of the left anterior descending coronary artery (LAD) as previously described (Citation27). The sham-operated rats underwent the same surgical procedure without the ligation of LAD.
Cell culture
Neonatal rat ventricular myocytes and fibroblasts were prepared from 2–4-day-old SD rats as previously described (Citation28). Briefly, after digestion of ventricular tissue with collagenase (2 mg/ml), cell suspension was pre-plated for 30–45 min, and the attached cells were further cultured for two passages (divided at a ratio of 1:5 in each passage) in the presence of 10% fetal bovine serum (FBS) to ensure proliferation of fibroblasts over the other cell types. The non-attached cells, i.e. myocyte-enriched cells were plated at the density of 2×105/cm2 on cell culture plates (Falcon) or on flexible-bottomed collagen I-coated elastomere plates (Bioflex; Flexcell International) for stretch experiments and then cultured overnight with DMEM/F-12 medium containing 10% FBS and thereafter in complete serum-free medium (CSFM).
Fibroblasts were propagated in 10% FBS containing DMEM/F-12 medium until 24 h before the experiment, when medium was replaced with CSFM. Cells on Bioflex plates were exposed to cyclic mechanical stretch. Frequency of cyclic stretch was 0.5 Hz with pulsation of 10–25% elongation of cells from 1 min to 24 h. Cells were stretched by applying a cyclic vacuum suction under Bioflex plates with computer-controlled equipment (FX-3000; Flexcell International). As a genetic marker of the stretch-induced cardiac hypertrophic program, BNP mRNA levels were increased from 4 to 24 h of myocyte stretching (Citation29) and the rapid and transient induction of c-fos mRNA levels (Citation28) was used as a marker of fibrotic response (Citation30).
In a separate series of experiments, fibroblasts and myocyte-enriched cells were plated at the density of 2×105/cm2 on Falcon wells, and the cells were treated with Ang II (100 nM) or endothelin-1 (ET-1, 100 nM) for 24 h. Ang II and ET-1 were added to culture medium on the third day of the culture. After experiments, the cells were washed twice with PBS (phosphate buffered saline) and quickly frozen at -70°C.
Isolation and analysis of RNA
Total RNA was isolated by the guanidine thiocyanate- CsCl method (Citation31). For Northern Blot analyses, 20 μg samples of RNA from LV tissue were separated on agarose–formaldehyde gel electrophoresis and transferred to MAGNA nylon membrane (Osmonics Inc.). PCR-amplified probes corresponding following genes: rat BNP, c-fos, Egr-1, rat MGP and rat 18S were random primer labeled with [α32PdCTP, and the membranes were hybridized and washed as described previously (Citation24). The membranes were exposed to Phosphor screens (GE Healthcare Life Sciences). Radioactivity was measured with Molecular Imager FX Pro Plus equipment (Bio-Rad Laboratories) using Quantity One software (Bio-Rad). The signals were normalized to 18S in each sample.
Extraction of total proteins
The cardiac tissue samples were broken in liquid nitrogen and homogenized in lysis buffer consisting of 20mmol/l Tris–HCl (pH 7.5), 10mM NaCl, 0.1mM EDTA, 0.1mM EGTA, 1mM β-glycerophosphate, 1mM Na3VO4, 2mM benzamidine, 1mM phenylmethylsulfoxide (PMSF), 50mM NaF, 1mM dithiothreitol and 10 μg/ml each of leupeptin, pepstatin and aprotinin. The tissue homogenates were centrifuged at 2000 rpm in +4°C for 1 min. To separate the total protein fraction, 5×NEB was added to the tissue homogenate following by centrifucation at 12500 rpm for 20 min. The supernatant was frozen in liquid nitrogen and stored in -70°C until assayed. Protein concentrations were determined by protein assay (Bio-Rad).
Western blotting
For Western blot, 30 μg of protein was loaded onto an SDS-PAGE gel and transferred to nitrocellulose filters. The membranes were blocked in non-fat milk and incubated with anti-MGP antibody 52.1C5D (Alexis Corporation) at a 1:1000 dilution in 1% milk-TBS-Tween for 2 h. After incubation with secondary anti-mouse antibody (Cell Signaling Technology) at the dilution of 1:2000, the protein amount was detected by ECL Plus (GE Healthcare Life Sciences).
Statistics
For statistical analysis, data was first tested for normality by using the Kolmogorov-Smirnov test for normality. For normally distributed variables, unpaired Student's t-test or one-way analysis of variance (ANOVA) followed by LSD (least significant difference) post hoc for multiple comparisons was performed. For data that was not normally distributed, statistical analysis was performed by using a non-parametric Mann–Whitney U-test. A value of p<0.05 was considered statistically significant. Results are expressed as mean±SEM.
Results
Pressure overload increases LV MGP expression
The effect of pressure overload on LV MGP expression was studied by infusing Ang II for 6h, 12h and 2weeks in normotensive rats. During Ang II treatment, LV MGP mRNA levels increased at 6 h (1.8-fold, p<0.05) and remained elevated during the whole follow-up period, the highest induction being observed at 2weeks (). There were no significant changes in the total MGP protein levels during the Ang II infusion, as studied by Western blot (). Simultaneous infusion of AT1-receptor antagonist losartan completely abolished the activation of MGP gene expression induced by Ang II at 2weeks in the left ventricle ().
Figure 1. Effect of angiotensin II (Ang II) administration on left ventricular (A) matrix Gla protein (MGP) mRNA levels and (B) protein levels in rats. (C) Effect of Ang II administration and Ang II receptor type 1 blockade by losartan (los) on left ventricular MGP mRNA levels at 6 h in rats. Results are expressed as a ratio of MGP mRNA to 18S mRNA as determined by Northern blot analysis. White columns, vehicle; black columns, Ang II. Results are mean±SEM, n=4–8. *p<0.05, **p<0.01 and ***p<0.001 vs vehicle (Student's t-test, Mann-Whitney U-test or one-way analysis of variance for multiple comparisons). (D) Effect of arginine8-vasopressin (AVP)-infusion on left ventricular matrix Gla protein (MGP) mRNA levels in rats. Results are expressed as a ratio of MGP mRNA to 18S mRNA as determined by Northern blot analysis. White columns, vehicle; black columns, AVP. Results are mean±SEM, n=7–9. *p<0.05 vs vehicle (Student's t-test or Mann–Whitney U-test).
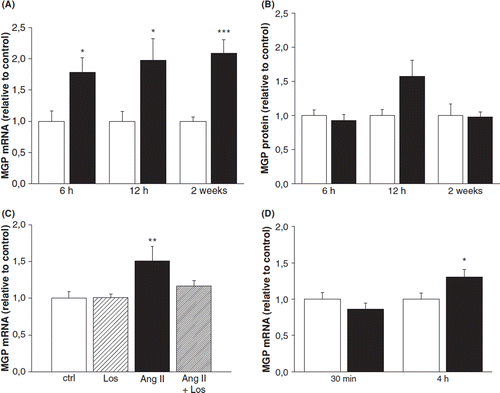
To further study the effect of acute pressure overload on MGP expression, we infused intravenously AVP for 30 min and 4 h in conscious normotensive rats. AVP infusion for 30 min had no effect on LV MGP gene expression, whereas at 4 h MGP mRNA levels were 1.3-fold (p<0.05) higher compared with vehicle-infused rats (). At 4 h there was no change in MGP protein levels (data not shown).
Cardiac remodeling after experimental myocardial infarction modulates MGP gene expression
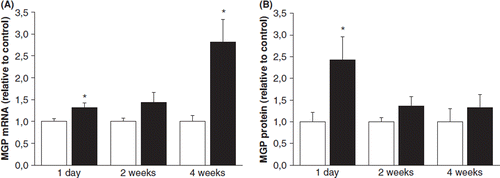
To study the effect of the early and late phase of post-infarction cardiac remodeling process on MGP gene expression, ligation of LAD was performed in rats. In response to experimental MI, MGP mRNA levels were elevated 1.3-fold (p<0.05) at 1 day and 2.8-fold (p<0.05) at 4 weeks (). A significant increase in the total MGP protein levels was seen at day 1 (2.4-fold, p<0.05) following ligation of LAD ().
Figure 2. Effect of experimental myocardial infarction on left ventricular matrix Gla protein (MGP) (A) mRNA levels and (B) protein levels in rats. Results are expressed as a ratio of MGP mRNA to 18S mRNA as determined by Northern blot analysis. White columns, sham; black columns, myocardial infarction. Results are mean±SEM, n=7–9. *p<0.05 vs sham (Student's t-test or Mann–Whitney U-test).
Expression of MGP protein in hypertrophied and failing hearts
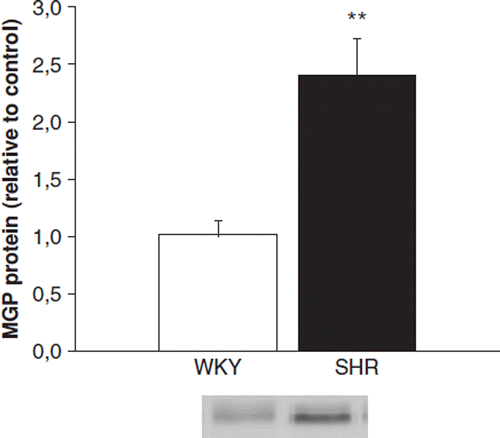
We next compared the MGP protein levels between 20-month-old SHR and WKY rats, because it has previously been observed that LV MGP mRNA levels are significantly higher in 20-month-old SHR compared with age-matched WKY (Citation20). In Western blot, the LV total MGP-protein levels were 2.4-fold (p<0.01) higher in old SHR compared with age-matched WKY ().
Figure 3. Western blot analysis showing matrix Gla protein (MGP) levels in 20-month-old Wistar-Kyoto (WKY) and spontaneously hypertensive rats (SHR). White columns, WKY; black columns, SHR. Results are mean±SEM, n=8. **p<0.01 vs WKY (Mann-Whitney U-test).
The effect of mechanical stretch on MGP gene expression in neonatal ventricular myocytes and fibroblasts
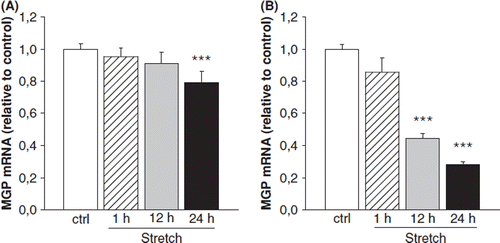
To further characterize the factors regulating cardiac MGP gene expression, we examined the effect of mechanical load on MGP gene expression by using an in vitro model of stretch in neonatal ventricular cardiomyocytes and fibroblasts, the two major cell types in myocardium. Mechanical stretch of neonatal ventricular myocytes had no effect on MGP mRNA levels after 1 h and 12 h, but reduced MGP gene expression by 20% (p<0.01) at 24 h (). Mechanical stretch of neonatal ventricular fibroblasts also reduced MGP mRNA levels (at 12 h; 60%, p<0.001; at 24 h, 70%, p<0.001) ().
Figure 4. The effect of mechanical stretch for 1, 12 and 24h on matrix Gla protein (MGP) gene expression in cultured neonatal rat ventricular (A) myocytes and (B) fibroblasts. Results are expressed as a ratio of MGP mRNA to 18S mRNA as determined by Northern blot analysis (n=13-18). Results are mean±SEM, from two to three independent cultures. ***p<0.001 vs control (one-way analysis of variance for multiple comparisons).
Angiotensin II increases MGP gene expression in neonatal ventricular myocytes and fibroblasts
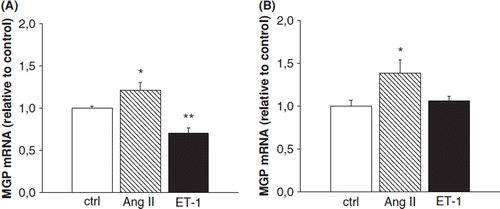
Finally, we treated neonatal rat ventricular myocytes and fibroblasts with two well-established hypertrophic agonists ET-1 and Ang II. In rat neonatal ventricular myocytes Ang II increased MGP mRNA levels significantly (1.2-fold, p<0.05), whereas ET-1 decreased the mRNA levels by 30% (p<0.01) compared with the cells treated with the vehicle (). In fibroblasts Ang II increased MGP mRNA levels 1.4-fold (p<0.05), whereas ET-1 had no effect on MGP mRNA levels ().
Figure 5. The effect of angiotensin II (Ang II) and endothelin-1 (Et-1) on matrix Gla protein (MGP) gene expression in cultured neonatal rat ventricular (A) myocytes and (B) fibroblasts. Results are expressed as a ratio of MGP mRNA to 18S mRNA as determined by Northern blot analysis (n=3-9). Results are mean±SEM, from two to three independent cultures. *p<0.05, **p<0.01 vs control (one-way analysis of variance for multiple comparisons).
Discussion
Previous studies have established MGP as an inhibitor of calcification and regulator of cell differentiation (Citation32). Importantly, MGP knock-out (KO) mice developed arterial calcification of the aorta, coronary arteries and aortic valves Citation4). It is thought that in arteries MGP inhibits calcification by binding calcium and blocking calcium crystal formation and/or by regulating cell differentiation via bone morphogenetic protein-2 inhibition (Citation14,Citation33). Both these actions require γ-carboxylation of MGP (Citation34) and local MGP expression, because circulating MGP does not affect the arterial calcification in MGP KO-mice (Citation35). Moreover, MGP has been associated with ECM binding, apoptosis as well as tissue growth and development (Citation21,Citation36,Citation37). The precise function of the protein in the heart is unknown, but because of the lack of myocardial calcification in the MGP KO mice, it is unlikely that the role of MGP in the heart is related to inhibition of calcification (Citation4). Also the mechanisms regulating cardiac MGP expression are not fully understood, although increased mRNA levels of MGP have been associated with variety of cardiac disorders in several DNA microarray studies (Citation16–20). Our present study identifies pressure overload and post-infarction remodeling process as important regulators of MGP expression in the heart.
A major observation of the present study is that cardiac MGP gene expression is rapidly upregulated in response to pressure overload. Our results show that the increase in blood pressure produced by AVP increases MGP expression within 4 h. This early induction was confirmed in angiotensin II-induced experimental hypertension showing increased MGP gene expression in the left ventricle of adult rat heart at 6 h. In addition, we observed that Ang II-induced upregulation of MGP gene expression in the left ventricle was abolished by AT1 receptor antagonist losartan, which most likely can be explained by the inhibition of Ang II-induced increase in blood pressure. The activation of MGP expression resembles the gene expression pattern of ECM regulatory factors such as transforming growth factor-β1 (TGF-β1), CCN1, connective tissue growth factor and thrombospondin-1 (Citation38–41) induced minutes or hours after cardiac overload, in contrast to structural components of ECM, such as type I collagen and fibronectin, whose gene expression is upregulated usually after few days of pressure overload (Citation38,Citation42,Citation43). Previously, increased MGP gene expression has been reported at day one after transverse aortic constriction (Citation17).
The rapid rise in MGP mRNA levels in response to pressure overload suggests that MGP may be involved in regulating cardiac hypertrophic process. Indeed, Mirotsou et al. found that MGP expression correlates with the level of LV hypertrophy and its expression pattern is similar to those of atrial natriuretic peptide and BNP in the model of aortic constriction in mice (Citation44). This finding was confirmed in both our models of pressure overload: the increase in MGP mRNA levels coincided with that of BNP (Citation24). It has also previously been shown that MGP mRNA levels increase with aging in SHR, whereas no increase was seen in WKY normotensive rats (Citation20). Because arterial calcification in rats increases with age and did not differ between the strains (Citation45), the change in MGP gene expression noted between SHR and WKY rats is unlikely associated with the vascular calcification. Instead, cardiac hypertrophy increases with aging in SHR (Citation20,Citation46), supporting the role of MGP in cardiac remodeling.
In agreement with the increased mRNA levels, the LV MGP protein levels were significantly higher in the SHR model of chronically elevated blood pressure and cardiac hypertrophy compared with normotensive rats. Yet, Ang II-induced pressure overload or late phase post-MI remodeling did not affect MGP protein levels despite the increased mRNA levels and cardiac hypertrophy, suggesting that MGP protein responds predominantly to the chronic pressure overload, like to that in SHR. Interestingly, however, an increase in MGP protein levels was observed at day one in response to experimental MI, and also polymorphisms of the human MGP gene have been associated with increased risk of MI (Citation47), both findings linking MGP to post-MI remodeling process. The inconsistency between MGP mRNA and protein levels is intriguing, but consistent with findings with other genes showing that mRNA levels do not necessarily correlate with protein levels of the gene and that transcript levels provide only little predictive value with respect to protein expression (Citation48,Citation49). This may be related to e.g. involvement of translational repression, post-transcriptional regulation of MGP gene expression and/or enhanced stability of MGP mRNA. Another possibility would be that the MGP protein is rapidly secreted and diffused into circulation. Nevertheless, if the functionality of MGP depends solely on its post-translational modifications, the abundance of the protein is not a useful indicator of its activity.
To examine the mechanisms of cardiac overload induced MGP gene expression, we tested the hypothesis that mechanical stretch acts directly on MGP mRNA levels by using an in vitro mechanical stretch model of neonatal cultured cardiomyocytes and fibroblasts as well as BNP as stretch-sensitive marker gene (Citation29). Interestingly, mechanical stretch decreased MGP gene expression in both rat cardiac myocytes and fibroblasts. Moreover, ET-1, known to be secreted and produced by stretch in cultured cardiomyocytes (Citation50), decreased MGP gene expression in myocytes and had no effect on MGP mRNA levels in fibroblasts. These results suggest that direct mechanical stretch or ET-1 are not involved in cardiac overload induced MGP gene expression. In contrast, Ang II increased MGP mRNA levels in both fibroblasts and myocytes in vitro, in agreement with the in vivo results. Because wall stretch is known to activate the hypertrophic response in part by increased local production or release of angiotensin II (Citation31,Citation50) these results suggest that Ang II may contribute to cardiac MGP gene expression in response to increased load.
Based on our present results, cardiac myocytes and fibroblasts seem to react distinctly to ET-1 in respect of MGP gene expression: ET-1 decreased MGP gene expression in myocytes, whereas it had no effect on MGP mRNA levels in fibroblasts. This is consistent with earlier studies showing that TGF-β and retinoic acid have differing effects on MGP expression in distinct cell types (Citation32,Citation36). Previously, it has been also postulated that MGP synthesis is restricted to distinct tissue specific cell types within each tissue and that rat cardiac myocytes are responsible for the immunoreactivity in the heart and in cell culture myocytes also secrete the protein (Citation5).
One of the main limitations of the present study is that our antibody does not distinguish between the Glu and Gla forms or phosphorylation of MGP, and therefore we could not assess the functionality of the existing MGP protein. Accordingly, we could not establish the precise role of MGP in the heart. Furthermore, the significance and the mechanisms of the variable mRNA and protein expression remain to be studied. However, our study shows that MGP seem to be involved in a common gene program underlying hypertrophic remodeling in vivo, regardless of how the hypertrophy was induced. Consequently, MGP together with other known markers of cardiac overload such as BNP constitute the molecular basis for cardiac hypertrophy. This transcriptional level information may be useful for prediction of a therapeutic response or diagnostics such as application of gene expression profiling to cardiovascular diseases.
In summary, the key finding of this study was that MGP gene expression is increased in response to pressure overload and myocardial infarction at the early phase of cardiac remodeling. Our results suggest that Ang II at least in part, but not ET-1 or direct stretch of myocytes and fibroblast, contributes to the upregulation of MGP in response to pressure overload. These results also imply that MGP may have a role in ECM remodeling beyond inhibition of calcification.
Acknowledgment
This study was supported by the Academy of Finland (Center of Excellence Funding), Sigrid Jusélius Foundation, the Finnish Foundation of Cardiovascular Research, Aarne Koskelo Foundation, Instrumentarium Research Foundation, Research and Science Foundation of Farmos, Maire Taponen Foundation, Astra-Zeneca and Paulo Foundation. We thank Kirsi Mustonen for statistical advice and Marja Arbelius, Pirjo Korpi, Kaisa Penttilä, Sirpa Rutanen and Kati Viitala for expert technical assistance.
References
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999; 79:215–262.
- Deschamps AM, Spinale FG. Matrix modulation and heart failure: New concepts question old beliefs. Curr Opin Cardiol. 2005; 20:211–216.
- Fedak PW, Verma S, Weisel RD, Li RK. Cardiac remodeling and failure From molecules to man (Part II). Cardiovasc Pathol. 2005; 14:49–60.
- Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al.. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997; 386:78–81.
- Fraser JD, Price PA. Lung, heart, and kidney express high levels of mRNA for the vitamin K-dependent matrix Gla protein. Implications for the possible functions of matrix Gla protein and for the tissue distribution of the gamma-carboxylase. J Biol Chem. 1988; 263:11033–11036.
- Hale JE, Fraser JD, Price PA. The identification of matrix Gla protein in cartilage. J Biol Chem. 1988; 263:5820–5824.
- Owen TA, Aronow MS, Barone LM, Bettencourt B, Stein GS, Lian JB. Pleiotropic effects of vitamin D on osteoblast gene expression are related to the proliferative and differentiated state of the bone cell phenotype: Dependency upon basal levels of gene expression, duration of exposure, and bone matrix competency in normal rat osteoblast cultures. Endocrinology. 1991; 128:1496–1504.
- Barone LM, Owen TA, Tassinari MS, Bortell R, Stein GS, Lian JB. Developmental expression and hormonal regulation of the rat matrix Gla protein (MGP) gene in chondrogenesis and osteogenesis. J Cell Biochem. 1991; 46:351–365.
- Boström K, Zebboudj AF, Yao Y, Lin TS, Torres A. Matrix GLA protein stimulates VEGF expression through increased transforming growth factor-beta1 activity in endothelial cells. J Biol Chem. 2004; 279:52904–52913.
- Shanahan CM, Weissberg PL, Metcalfe JC. Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ Res. 1993; 73:193–204.
- Cancela ML, Price PA. Retinoic acid induces matrix Gla protein gene expression in human cells. Endocrinology. 1992; 130:102–108.
- Shanahan CM, Cary NR, Metcalfe JC, Weissberg PL. High expression of genes for calcification-regulating proteins in human atherosclerotic plaques. J Clin Invest. 1994; 93:2393–2402.
- Shanahan CM, Proudfoot D, Farzaneh-Far A, Weissberg PL. The role of Gla proteins in vascular calcification. Crit Rev Eukaryot Gene Expr. 1998; 83:357–375.
- Demer LL, Tintut Y. Mineral exploration: Search for the mechanism of vascular calcification and beyond: The 2003 Jeffrey M. Hoeg Award lecture. Arterioscler Thromb Vasc Biol. 2003; 23:1739–1743.
- Schurgers LJ, Cranenburg EC, Vermeer C. Matrix Gla-protein: The calcification inhibitor in need of vitamin K. Thromb Haemost. 2008; 100:593–603.
- Mirotsou M, Dzau VJ, Pratt RE, Weinberg EO. Physiological genomics of cardiac disease: Quantitative relationships between gene expression and left ventricular hypertrophy. Physiol Genomics. 2006; 27:86–94.
- Weinberg EO, Mirotsou M, Gannon J, Dzau VJ, Lee RT, Pratt RE. Sex dependence and temporal dependence of the left ventricular genomic response to pressure overload. Physiol Genomics. 2003; 12:113–127.
- Hwang DM, Dempsey AA, Lee CY, Liew CC. Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics. 2000; 66:1–14.
- Blaxall BC, Spang R, Rockman HA, Koch WJ. Differential myocardial gene expression in the development and rescue of murine heart failure. Physiol Genomics. 2003; 15:105–114.
- Rysä J, Leskinen H, Ilves M, Ruskoaho H. Distinct upregulation of extracellular matrix genes in transition from hypertrophy to hypertensive heart failure. Hypertension. 2005; 45:927–933.
- Nishimoto SK, Nishimoto M. Matrix Gla protein C-terminal region binds to vitronectin. Co-localization suggests binding occurs during tissue development. Matrix Biol. 2005; 24:353–361.
- Magga J, Marttila M, Mäntymaa P, Vuolteenaho O, Ruskoaho H. Brain natriuretic peptide in plasma, atria, and ventricles of vasopressin- and phenylephrine-infused conscious rats. Endocrinology. 1994; 134:2505–2515.
- Romppanen H, Puhakka J, Foldes G, Szokodi II, Vuolteenaho O, Tokola H, et al.. Endothelin-1-independent and angiotensin II-independent induction of adrenomedullin gene expression. Hypertension. 2001; 37:84–90.
- Rysä J, Aro J, Ruskoaho H. Early left ventricular gene expression profile in response to increase in blood pressure. Blood Press. 2006; 15:375–383.
- Foldes G, Suo M, Szokodi I, Lako-Futo Z, deChatel R, Vuolteenaho O, et al.. Factors derived from adrenals are required for activation of cardiac gene expression in angiotensin II-induced hypertension. Endocrinology. 2001; 142:4256–4263.
- Suo M, Hautala N, Foldes G, Szokodi I, Toth M, Leskinen H, et al.. Posttranscriptional control of BNP gene expression in angiotensin II-induced hypertension. Hypertension. 2002; 39:803–808.
- Tenhunen O, Soini Y, Ilves M, Rysä J, Tuukkanen J, Serpi R, et al.. p38 Kinase rescues failing myocardium after myocardial infarction: Evidence for angiogenic and anti-apoptotic mechanisms. FASEB J. 2006; 20:1907–1909.
- Pikkarainen S, Tokola H, Kerkelä R, Ilves M, Mäkinen M, Orzechowski HD, et al.. Inverse regulation of preproendothelin-1 and endothelin-converting enzyme-1beta genes in cardiac cells by mechanical load. Am J Physiol Regul Integr Comp Physiol. 2006; 290:R1639–R1645.
- Pikkarainen S, Tokola H, Majalahti-Palviainen T, Kerkelä R, Hautala N, Bhalla SS, et al.. GATA-4 is a nuclear mediator of mechanical stretch-activated hypertrophic program. J Biol Chem. 2003; 278:23807–23816.
- van Wamel JE, Ruwhof C, van der Valk-Kokshoorn EJ, Schrier PI, van der Laarse A. Rapid gene transcription induced by stretch in cardiac myocytes and fibroblasts and their paracrine influence on stationary myocytes and fibroblasts. Pflugers Arch. 2000; 439:781–788.
- Magga J, Vuolteenaho O, Tokola H, Marttila M, Ruskoaho H. Involvement of transcriptional and posttranscriptional mechanisms in cardiac overload-induced increase of B-type natriuretic peptide gene expression. Circ Res. 1997; 81:694–702.
- Farzaneh-Far A, Weissberg PL, Proudfoot D, Shanahan CM. Transcriptional regulation of matrix Gla protein. Z Kardiol. 2001; 90Suppl 3:38–42.
- Boström KI. Cell differentiation in vascular calcification. Z Kardiol. 2000; 89Suppl 2:69–74.
- Sweatt A, Sane DC, Hutson SM, Wallin R. Matrix Gla protein (MGP) and bone morphogenetic protein-2 in aortic calcified lesions of aging rats. J Thromb Haemost. 2003; 1:178–185.
- Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; Different roles of two gla-containing proteins. J Cell Biol. 2004; 165:625–630.
- Proudfoot D, Shanahan CM. Molecular mechanisms mediating vascular calcification: Role of matrix Gla protein. Nephrology (Carlton). 2006; 11:455–461.
- Gilbert KA, Rannels SR. Matrix Gla protein modulates branching morphogenesis in fetal rat lung. Am J Physiol Lung Cell Mol Physiol. 2004; 286:L1179–L1187.
- Villarreal FJ, Dillmann WH. Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am J Physiol. 1992; 262:H1861–H1866.
- Hilfiker-Kleiner D, Kaminski K, Kaminska A, Fuchs M, Klein G, Podewski E, et al.. Regulation of proangiogenic factor CCN1 in cardiac muscle: Impact of ischemia, pressure overload, and neurohumoral activation. Circulation. 2004; 109:2227–2233.
- Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008; 19:133–144.
- Mustonen E, Aro J, Puhakka J, Ilves M, Soini Y, Leskinen H, et al.. Thrombospondin-4 expression is rapidly upregulated by cardiac overload. Biochem Biophys Res Commun. 2008; 373:186–191.
- Chapman D, Weber KT, Eghbali M. Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circ Res. 1990; 67:787–794.
- Crawford DC, Chobanian AV, Brecher P. Angiotensin II induces fibronectin expression associated with cardiac fibrosis in the rat. Circ Res. 1994; 74:727–739.
- Mirotsou M, Dzau VJ, Pratt RE, Weinberg EO. Physiological genomics of cardiac disease: Quantitative relationships between gene expression and left ventricular hypertrophy. Physiol Genomics. 2006; 27:86–94.
- Kieffer P, Robert A, Capdeville-Atkinson C, Atkinson J, Lartaud-Idjouadiene I. Age-related arterial calcification in rats. Life Sci. 2000; 66:2371–2381.
- Pfeffer JM, Pfeffer MA, Mirsky I, Braunwald E. Regression of left ventricular hypertrophy and prevention of left ventricular dysfunction by captopril in the spontaneously hypertensive rat. Proc Natl Acad Sci U S A. 1982; 79:3310–3314.
- Herrmann SM, Whatling C, Brand E, Nicaud V, Gariepy J, Simon A, et al.. Polymorphisms of the human matrix gla protein (MGP) gene, vascular calcification, and myocardial infarction. Arterioscler Thromb Vasc Biol. 2000; 20:2386–2393.
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999; 19:1720–1730.
- Tuomisto TT, Riekkinen MS, Viita H, Levonen AL, Yla-Herttuala S. Analysis of gene and protein expression during monocyte-macrophage differentiation and cholesterol loading-cDNA and protein array study. Atherosclerosis. 2005; 180:283–291.
- Komuro I. Molecular mechanism of mechanical stress-induced cardiac hypertrophy. Jpn Heart J. 2000; 41:117–129.